3. Experimental
Melting points are uncorrected. NMR spectra are recorded using a 400 or 500 MHz NMR spectrometer and are run at 302 K unless otherwise indicated. 1H NMR values are reported as chemical shifts δ, relative integral, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad peak) and coupling constant (J). Chemical shifts are reported in parts per million (ppm) relative to CDCl3 (1H residual CHCl3 singlet δ 7.26 ppm, 13C CDCl3 triplet δ 77.23 ppm) or DMSO-d6 (1H residual DMSO-d5 pentet δ 2.49 ppm, 13C DMSO-d6 septet δ 39.7 ppm), and coupling constants are taken directly from the spectra. NMR assignments are made with the aid of 1H-1H COSY, HSQC, DEPT-135, and nOe difference proton NMR spectroscopy. Two-dimensional (2D)-NMR experiments are performed using standard software. Mass spectral data are acquired using positive-mode electrospray ionization (ESI+) and a high-resolution time-of-flight mass spectrometer.
Dimethyl 5-methoxy-1,3-benzenedicarboxylate (
11a). Dimethyl 5-hydroxyisophthalate (1.860 g, 8.9 mmol), methyl iodide (2.301 g, 16.2 mmol), and potassium carbonate (1.342 g, 10.4 mmol) were dissolved in acetonitrile (30 mL) and refluxed overnight. The solution was cooled, and the solvent was removed on a rotary evaporator. The residue was dissolved in ethyl acetate (50 mL) and then washed with water (2 × 30 mL) and saturated sodium bicarbonate (2 × 30 mL). The organic layer was dried over sodium sulfate and filtered, and the solvent was evaporated under reduced pressure to yield 5-methoxyisophthalic acid dimethyl ester (1.989 g, 8,88 mmol, quantitative) as a white solid, mp 111–112 °C (lit. mp [
26] 110–112 °C).
1H NMR (500 MHz, CDCl
3): δ 8.29 (t, 1H,
4JHH = 1.4 Hz, 2-H), 7.75 (d, 2H,
4JHH = 1.4 Hz, 4,6-H), 3.94 (s, 6H, 2 × CO
2CH
3), 3.89 (s, 3H, OCH
3).
13C NMR (125 MHz, CDCl
3): δ 166.3 (C=O), 159.9 (5-C), 128.3 (1,3-C), 123.1 (2-CH), 119.5 (4,6-CH), 55.9 (OMe), 52.5 (2 × ester OMe).
5-Methoxy-1,3-bis(hydroxymethyl)benzene (
12a). Lithium aluminum hydride (1.111 g, 29.3 mmol) was added to a solution of
11a (1.918 g, 8.56 mmol) in dry THF (100 mL), and the resulting mixture was stirred at room temperature overnight. Dilute hydrochloric acid (80 mL) was added dropwise to the solution, and the contents of the reaction flask were stirred for an additional 30 min. Ethyl acetate (200 mL) was added, and the aqueous layer was drawn off. The organic layer was washed with water (2 × 100 mL) and saturated sodium bicarbonate solution (2 × 100 mL). The ethyl acetate layer was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to yield
12a (1.439 g, 8.56 mmol, quantitative) as a white solid, mp 66–68 °C (lit. mp [
27] 66–67 °C).
1H NMR (500 MHz, CDCl
3): δ 6.94 (s, 1H, 2-H), 6.85 (br s, 2H, 4,6-H), 4.67 (s, 4H, 2 × CH
2), 3.82 (s, 3H, OCH
3), 1.76 (br s, 2H, 2 × OH).
13C NMR (125 MHz, CDCl
3): δ 160.4 (5-C), 143.3 (1,3-C). 117.7 (2-CH), 111.8 (4,6-CH), 65.4 (2 × CH
2OH), 55.5 (OCH
3).
5-Methoxybenzene-1,3-dicarbaldehyde (
8a). Dialcohol
12a (1.515 g, 9.02 mmol) was dissolved in dichloromethane (108 mL) and THF (72 mL). Pyridinium chlorochromate (5.979 g, 27.7 mmol) and silica gel (3.720 g, 61.9 mmol) were added to the solution, and the mixture was stirred at room temperature for 1 h. The contents of the reaction flask were immediately chromatographed twice on silica gel, eluting with dichloromethane. The desired column fractions were evaporated under reduced pressure to afford the dialdehyde (1.185 g, 7.22 mmol, 80%) as a white solid, mp 109–110 °C (lit. mp [
28] 108–109 °C).
1H NMR (500 MHz, CDCl
3): δ 10.05 (s, 2H, 2 × CHO), 7.96 (t, 1H,
4JHH = 1.4 Hz, 2-H), 7.65 (d, 2H,
4JHH = 1.4 Hz, 4,6-H), 3.93 (s, 3H, OCH
3).
13C NMR (125 MHz, CDCl
3): δ 191.0 (2 × CHO), 161.1 (5-C), 138.6 (1,3-C), 124.4 (2-CH), 119.6 (4,6-CH), 56.2 (OCH
3).
Dimethyl 5-ethoxy-1,3-benzenedicarboxylate (
11b). Dimethyl 5-hydroxy-isophthalate (1.680 g, 8.0 mmol), ethyl iodide (1.905 g, 12.2 mmol), and potassium carbonate (1.344 g, 9.7 mmol) were dissolved in acetonitrile (30 mL) and refluxed overnight. The solution was cooled, and the solvent was removed using a rotary evaporator. The resulting solid was dissolved in ethyl acetate (50 mL) and washed with water (2 × 30 mL) and saturated sodium bicarbonate (2 × 30 mL). The organic layer was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to yield 5-ethoxyisophthalic acid dimethyl ester (1.754 g, 7,57 mmol, 92%) as a white solid, mp 101–102 °C (lit. mp [
29] 102 °C).
1H NMR (500 MHz, CDCl
3): δ 8.25 (t, 1H,
4JHH = 1.4 Hz, 2-H), 7.73 (d, 2H,
4JHH = 1.4 Hz, 4,6-H), 4.11 (q, 2H,
3JHH = 7.0 Hz, OC
H2CH
3), 3.93 (s, 6H, 2 × OCH
3), 1.44 (t, 3H,
3JHH = 7.0 Hz, OCH
2C
H3).
13C NMR (125 MHz, CDCl
3): δ 166.4 (C=O), 159.2 (5-C), 131.9 (1,3-C), 123.0 (2-CH), 120.0 (4,6-CH), 64.3 (OCH
2), 52.5 (2 × OMe), 14.8 (OCH
2CH
3).
5-Ethoxy-1,3-bis(hydroxymethyl)benzene (
12b). Lithium aluminum hydride (1.018 g, 26.8 mmol) was added to a solution of
11b (2.018 g, 8.48 mmol) in dry THF (100 mL). The mixture was stirred at room temperature overnight, dilute hydrochloric acid (80 mL) was added dropwise to the solution, and the contents of the reaction flask were stirred for an additional 30 min. Ethyl acetate (200 mL) was added, and the aqueous layer was drawn off. The organic layer was washed with water (2 × 100 mL) and a saturated sodium bicarbonate solution (2 × 100 mL). The organic solution was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to yield the dialcohol (1.534 g, 8.43 mmol, 99%) as a white solid, mp 123–124 °C (lit. mp [
29] 124–125 °C).
1H NMR (500 MHz, CDCl
3): δ 6.88 (br s, 1H, 2-H), 6.79 (br s, 2H, 4,6-H), 4.59 (s, 4H, 2 × C
H2OH), 4.02 (q, 2H,
3JHH = 7.0 Hz, OC
H2CH
3), 2.53 (br s, 2H, 2 × OH), 1.39 (t, 3H,
3JHH = 7.0 Hz, OCH
2C
H3).
13C NMR (125 MHz, CDCl
3): δ 159.6 (5-C), 143.0 (1,3-C). 117.6 (2-CH), 112.3 (4,6-CH), 65.2 (2 × CH
2OH), 63.7 (O
CH
2CH
3), 15.0 (CH
2CH
3).
5-Ethoxybenzene-1,3-dicarbaldehyde (
8b). Dialcohol
12b (0.191 g, 1.05 mmol) was dissolved in dichloromethane (12 mL) and THF (8 mL). Pyridinium chlorochromate (0.656 g, 3.0 mmol) and silica gel (0.411 g, 6.8 mmol) were added to the solution, and the mixture was stirred at room temperature for 1 h. The contents of the reaction flask were immediately chromatographed twice on silica gel, eluting with dichloromethane. The desired column fractions were evaporated under reduced pressure to give the dialdehyde (0.175 g, 0.98 mmol, 94%) as a white solid, mp 207–208 °C (lit. mp [
29] 208–209 °C).
1H NMR (500 MHz, CDCl
3): δ 10.05 (s, 2H, 2 × CHO), 7.94 (s, 1H,
4JHH = 1.4 Hz, 2-H), 7.64 (s, 2H,
4JHH = 1.4 Hz, 4,6-H), 4.16 (q, 4H,
3JHH = 7.0 Hz, OCH
2), 1.47 (t, 3H,
3JHH = 7.0 Hz, CH
2C
H3).
13C NMR (125 MHz, CDCl
3): δ 191.1 (2 × CHO), 160.4 (5-C), 138.6 (1,3-C), 124.2 (2-CH), 120.1 (4,6-CH), 64.6 (OCH
2), 14.8 (CH
2CH
3).
5-Hydroxybenzene-1,3-dicarbaldehyde (
14). Lithium aluminum hydride (6.076 g, 160 mmol) was dissolved in dry THF (150 mL) in a 3-neck flask fitted with an addition funnel, stopper, and condenser. A solution of dimethyl 5-hydroxyisophthalate (10.501 g, 50.0 mmol) in dry THF (175 mL) was added dropwise to the solution, and the mixture was refluxed for 2 h and subsequently stirred at room temperature overnight. A mixture of ethanol (5 mL), ethyl acetate (10 mL), and brine (50 mL) was added dropwise. After the addition was complete, the contents of the flask were filtered, and the solid was washed with ethanol. The filtrate was evaporated under reduced pressure to yield 3,5-bis(hydroxymethyl)phenol (
13, 7.005 g, 45.5 mmol, 91%) as a white solid, mp 174–175 °C (lit. mp [
30] 175–176 °C). The crude solid (3.90 g, 25.3 mmol) and potassium dichromate (14.94 g, 50.8 mmol) were dissolved in DMSO (166 mL) and stirred at 100 °C for 4 h. The mixture was cooled, poured into a beaker of water (750 mL), and extracted with diethyl ether (5 × 200 mL). The aqueous layer was discarded, and the organic phase was washed with water (150 mL), dried over sodium sulfate, and filtered, and the solvent was removed on a rotary evaporator to give the dialdehyde (2.512 g, 16.7 mmol, 66%) as a light tan solid, mp 146–147 °C (lit. mp [
30] 146–147.5 °C).
1H NMR (500 MHz, CDCl
3): δ 10.04 (s, 2H, 2 × CHO), 7.95 (t, 1H,
4JHH = 1.4 Hz, 2-H), 7.63 (d, 2H,
4JHH = 1.4 Hz, 4,6-H), 6.23 (1H, br s, OH).
13C NMR (125 MHz, CDCl
3): δ 191.3 (2 × CHO), 157.5 (5-C), 138.8 (1,3-C), 124.5 (2-CH), 121.0 (4,6-CH).
5-Methoxycarbonylmethoxybenzene-1,3-dicarbaldehyde (8c). Dialdehyde 12 (301 mg, 2.00 mmol), potassium carbonate (357 mg, 2.6 mmol), and methyl bromoacetate (0.3 mL, 3.2 mmol) in acetone (25 mL) were stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was dissolved in ethyl acetate (20 mL). The solution was washed with water and saturated sodium bicarbonate solution. The organic layer was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to yield 8c (275 mg, 1.24 mmol, 62%) as a white solid, mp 97–99 °C. 1H NMR (500 MHz, CDCl3): δ 10.05 (s, 2H, 2 × CHO), 8.01 (t, 1H, 4JHH = 1.3 Hz, 2-H), 7.66 (d, 2H, 4JHH = 1.3 Hz, 4,6-H), 4.77 (s, 2H, CH2O), 3.83 (s, 3H, OCH3). 13C NMR (100 MHz, CDCl3): δ 190.6 (CHO), 168.4 (OCH2CO2CH3), 159.0 (5-C), 138.4 (1,3-C), 124.9 (2-CH), 119.9 (4,6-CH), 65.3 (OCH2CO2CH3), 52.4 (OCH2CO2CH3). HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C11H11O5+ 223.0601; Found 223.0601.
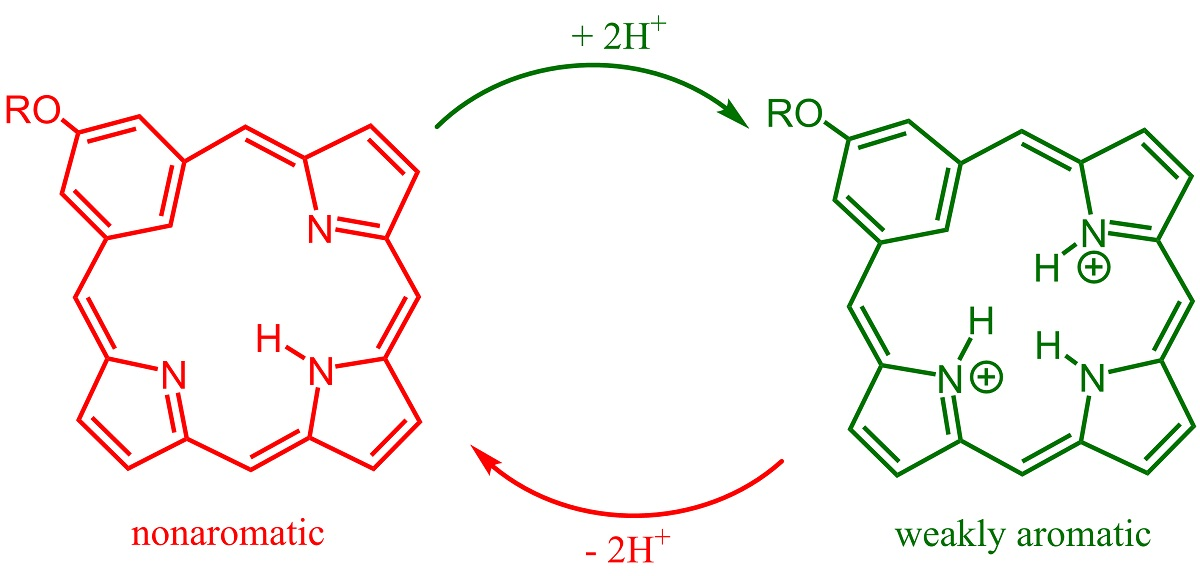
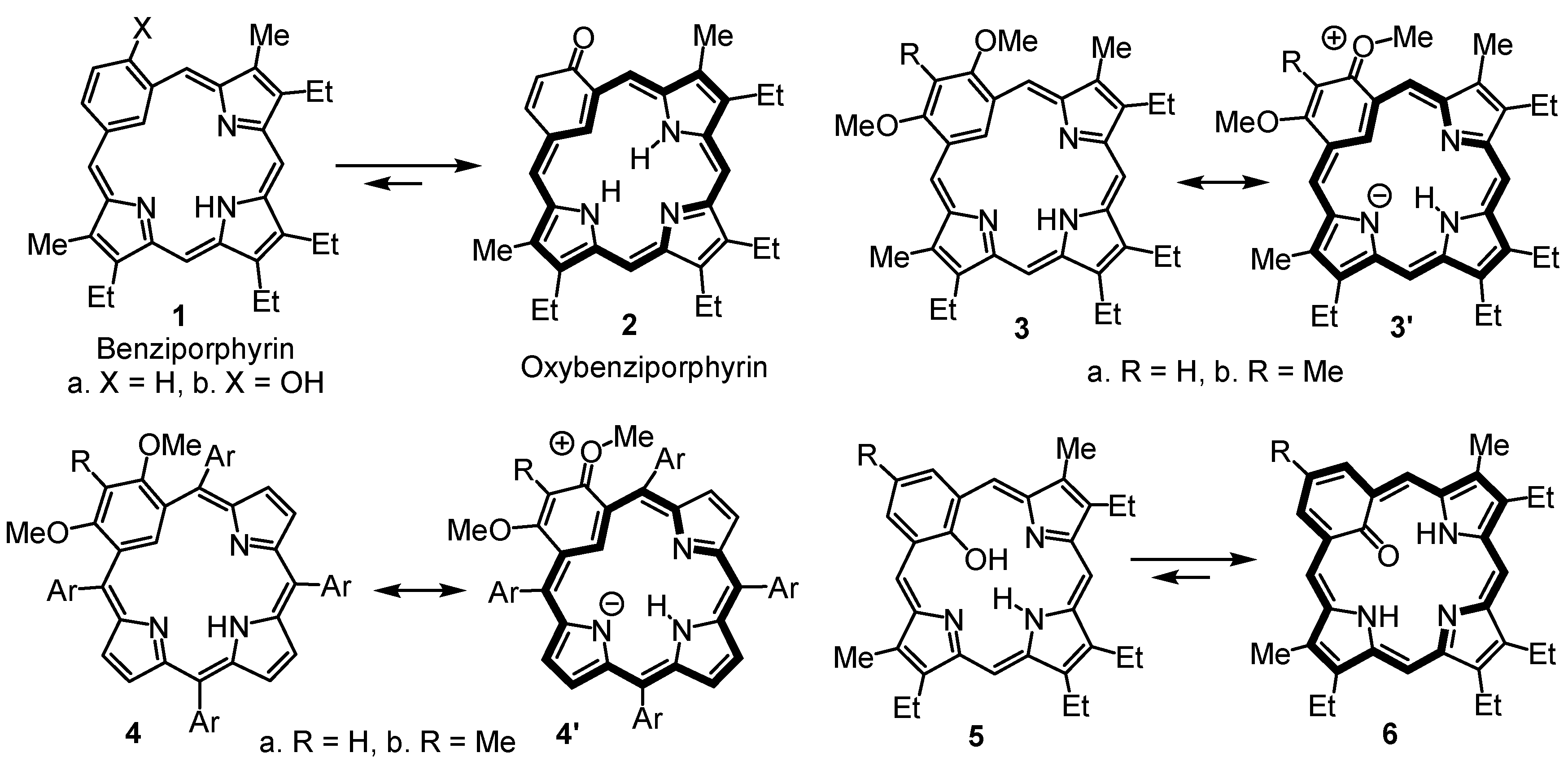
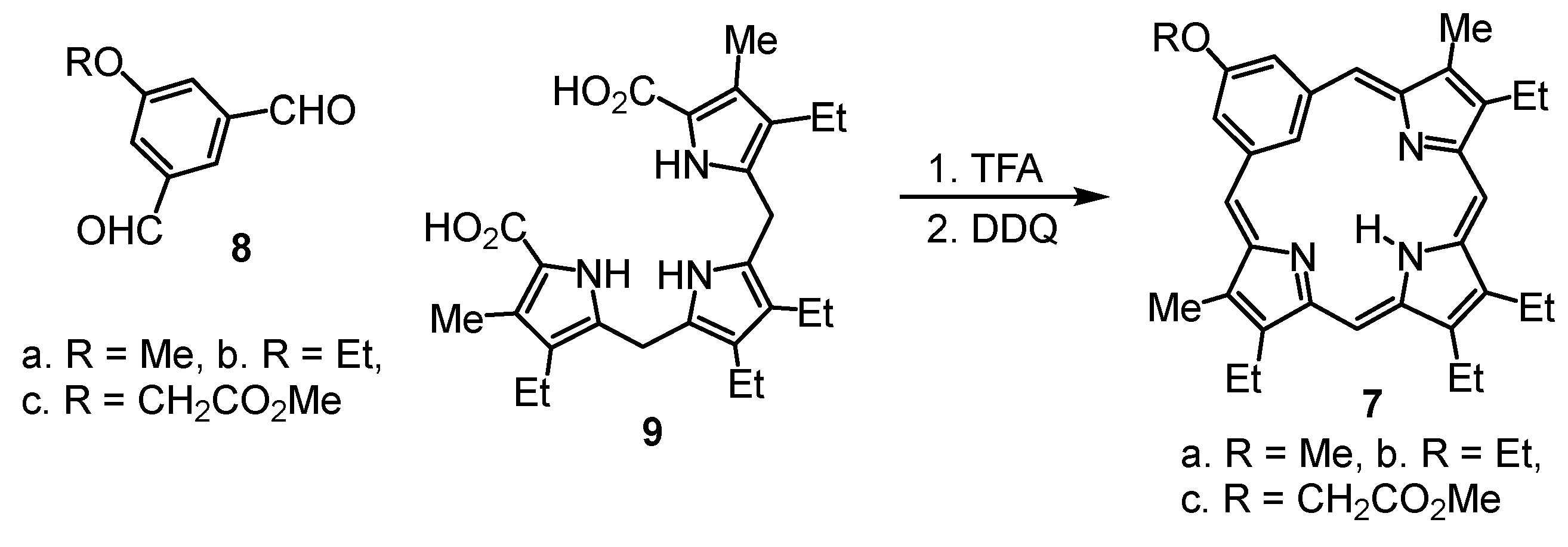
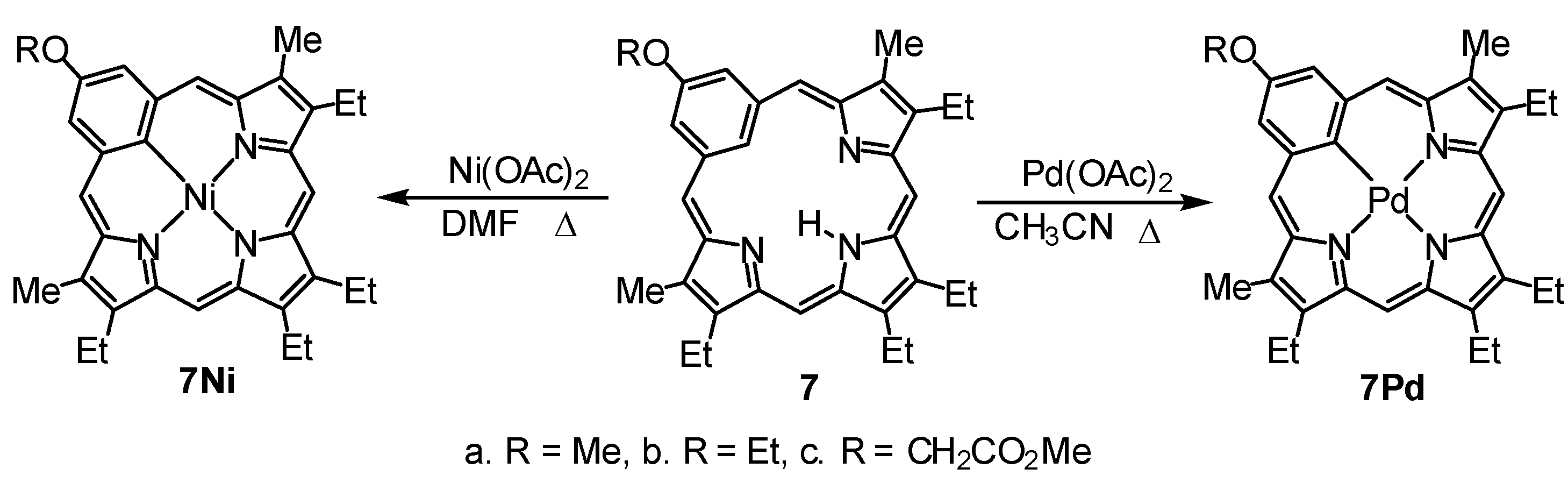
9,13,14,18-Tetraethyl-3-methoxy-8,19-dimethylbenziporphyrin (7a). In a pear-shaped flask, tripyrrane dicarboxylic acid 9 (99 mg, 0.22 mmol) was dissolved in TFA (1 mL) and stirred under nitrogen for 2 min. Dichloromethane (100 mL) was added, followed by 5-methoxybenzene-1,3-dicarbaldehyde (42 mg, 0.25 mmol). The flask was subsequently covered in aluminum foil to protect the reaction from ambient light, and the solution was stirred overnight under nitrogen. DDQ (81 mg, 0.357 mmol) was added, and the mixture was stirred for 1 h. The resulting solution was washed with water and saturated sodium bicarbonate solution (the aqueous layers were back-extracted with chloroform to ensure all of the product was collected). The combined organic layers were evaporated under reduced pressure, and the residue was purified on a grade 3 basic alumina column, eluting with dichloromethane. The product was collected as a dark-blue band. Recrystallization from chloroform-methanol afforded the benziporphyrin (37 mg, 0.075 mmol, 34%) as dark-purple crystals, mp > 300 °C. UV-vis (1% Et3N-CH2Cl2): λmax (log ε): 308 (4.73), 382 (4.84), 580 (sh, 3.46), 630 (3.62), 672 (3.65), 733 nm (3.44). UV-vis (1% TFA-CH2Cl2): λmax (log ε): 315 (4.63), 399 (4.78), 504 (3.91), 573 (3.36), 692 (sh, 3.53), 745 (3.07), 838 nm (sh, 3.61). 1H NMR (500 MHz, CDCl3): δ 9.21 (br s, 1H, NH), 7.86 (s, 1H, 22-H), 7.49 (d, 2H, 4JHH = 1.2 Hz, 2,4-H), 7.14 (s, 2H, 6,21-H), 6.48 (s, 2H, 11,16-H), 4.04 (s, 3H, OCH3), 2.82 (q, 4H, 3JHH = 7.6 Hz, 9,18-CH2CH3), 2.73 (q, 4H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 2.39 (s, 6H, 8,19-CH3), 1.33 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.25 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 1H NMR (500 MHz, TFA-CDCl3): 9.86 (br s, 1H, NH), 7.92 (s, 2H, 6,21-H), 7.75 (s, 2H, 2,4-H), 6.92 (s, 2H, 11,16-H), 5.06 (s, 1H, 22-H), 4.03 (s, 3H, OCH3), 2.92–2.87 (m, 8H, 9,13,14,18-CH2), 2.61 (s, 6H, 8,19-CH3), 1.33 (t, 6H, 3JHH = 7.6 Hz), 1.30 (t, 6H, 3JHH = 7.6 Hz) (4 × CH2CH3). 13C NMR (125 MHz, CDCl3): δ 169.3, 159.6, 157.4, 148.1, 141.3, 141.2, 140.5, 135.2, 122.6 (6,21-CH), 122.1 (2,4-CH), 118.7 (22-CH), 93.1 (11,16-CH), 55.7 (OCH3), 18.5 (13,14-CH2CH3), 18.1 (9,18-CH2CH3), 16.1 (13,14-CH2CH3), 15.3 (9,18-CH2CH3), 10.4 (8,19-CH3). 13C NMR (125 MHz, TFA-CDCl3): δ 163.1, 162.8, 154.7, 148.0, 146.6, 143.8, 141.6, 133.2, 128.4 (6,21-CH), 124.7 (2,4-CH), 101.7 (22-CH), 94.2 (11,16-CH), 56.3 (OCH3), 18.4 (13,14-CH2CH3), 18.1 (9,18-CH2CH3), 15.2 (13,14-CH2CH3), 14.3 (9,18-CH2CH3), 10.7 (8,19-CH3). HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C33H38N3O+ 492.3009; Found 492.3011.
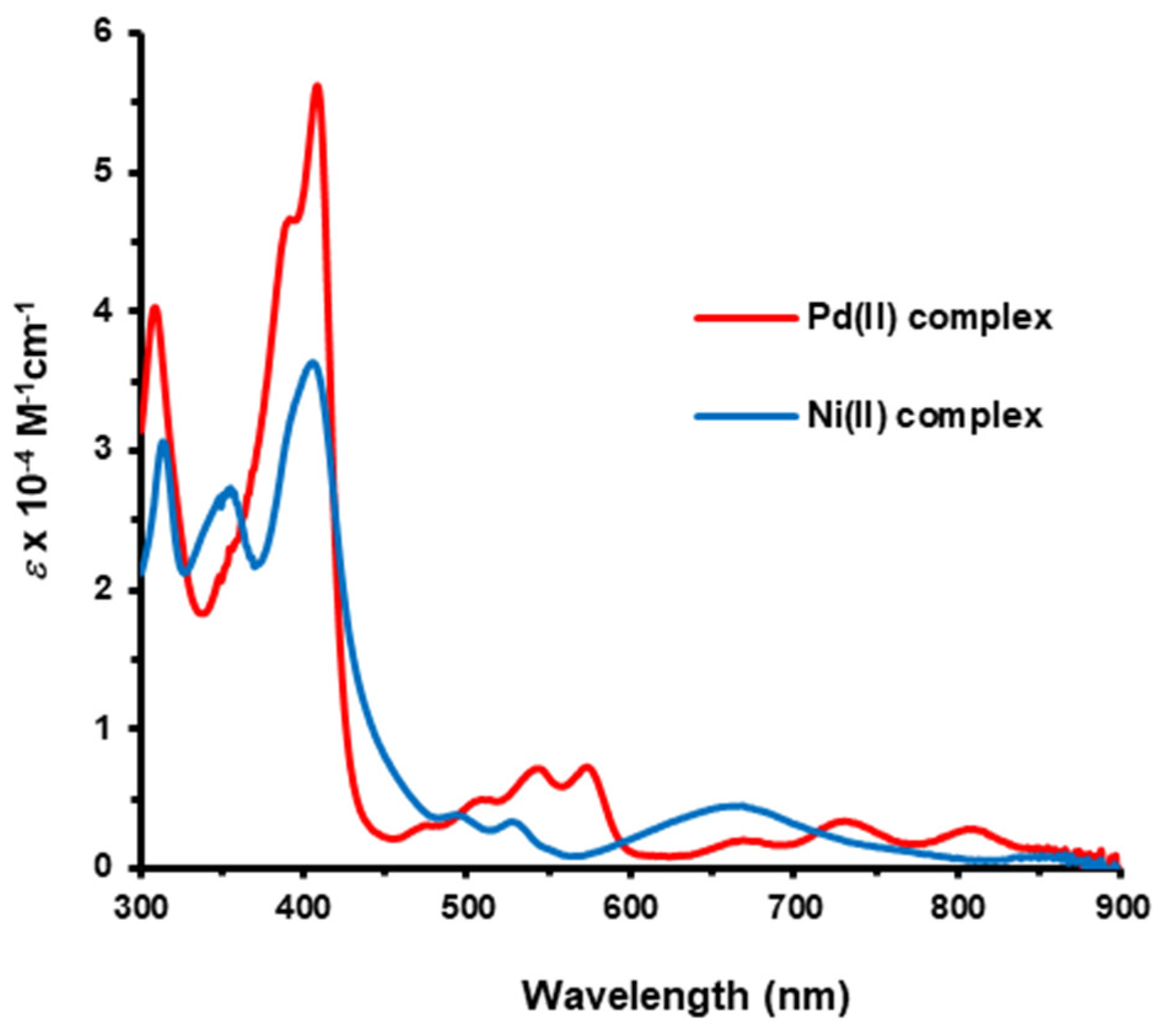
[9,13,14,18-Tetraethyl-3-methoxy-8,19-dimethylbenziporphyrinato]palladium(II) (7aPd). Methoxybenziporphyrin 7a (17 mg, 0.0346 mmol) and palladium(II) acetate (20 mg, 0.089 mmol) in acetonitrile (15 mL) were refluxed for 30 min. The solution was cooled to room temperature and then diluted with dichloromethane (35 mL). The solution was washed with water, and the solvent was removed using a rotary evaporator. The residue was chromatographed on a grade 3 basic alumina column, eluting with dichloromethane. A reddish-purple fraction was collected, and the solvent was removed under reduced pressure. The residue was recrystallized from chloroform-methanol to give the palladium complex (15 mg, 0.025 mmol, 72%) as a dark-purple solid, mp > 300 °C. UV-vis (CH2Cl2): λmax (log ε): 308 (4.61), 391 (sh, 4.69), 407 (4.75), 474 (sh, 3.48), 507 (sh, 3.67), 543 (3.82), 572 (3.84), 666 (3.31), 728 (3.55), 807 (3.50). 1H NMR (400 MHz, CDCl3): δ 7.68 (2H, s, 2,4-H), 7.57 (2H, s, 6,21-H), 7.25 (2H, s, 11,16-H), 4.10 (3H, s, OCH3), 3.01–2.92 (8H, overlapping quartets, 4 × CH2CH3), 2.59 (6H, s, 8,19-CH3), 1.43 (6H, t, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.37 (6H, t, 3JHH = 7.6 Hz, 9,18-CH2CH3). 13C NMR (100 MHz, CDCl3): δ 157.4, 157.2, 153.7, 145.3, 144.0, 141.1, 138.5, 133.4, 131.6, 126.8 (2,4-CH), 126.6 (6,16-CH), 95.5 (11,21-CH), 55.8 (OCH3), 18.9 (13,14-CH2CH3), 18.6 (9,18-CH2CH3), 16.6 (13,14-CH2CH3), 15.3 (9,18-CH2CH3). HRMS (ESI-TOF) m/z: M+ Calcd for C33H36N3OPd+ 506.1888; Found 596.1869.
[9,13,14,18-Tetraethyl-3-methoxy-8,19-dimethylbenziporphyrinato]nickel(II) (7aNi). Methoxybenziporphyrin 7a (19 mg, 0.0387 mmol) and nickel(II) acetate (40 mg, 0.16 mmol) in DMF (20 mL) were refluxed for 30 min. The solution was cooled to room temperature and diluted with chloroform (25 mL). The solution was washed with water, and the solvent was evaporated under reduced pressure. The residue was chromatographed on a grade 3 basic alumina column, eluting with chloroform. A dark-green band was collected, and the solvent was removed under reduced pressure. The residue was recrystallized from chloroform-methanol to give the nickel complex (16 mg, 0.029 mmol, 75%) as a dark-green solid, mp > 300 °C. UV-vis (CH2Cl2): λmax (log ε): 313 (4.32), 353 (4.27), 405 (4.39), 493 (sh, 3.43), 527 (3.38), 660 (3.51). 1H NMR (500 MHz, CDCl3) δ 7.63 (s, 2H, 2,4-H), 7.35 (s, 2H, 6,21-H), 7.08 (s, 2H, 11,16-H), 4.05 (s, 3H, OCH3), 2.91 (q, 4H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 2.84 (q, 4H, 3JHH = 7.6 Hz, 9,18-CH2CH3), 2.44 (s, 6H, 8,19-CH3), 1.37 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.30 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 13C NMR (125 MHz, CDCl3): δ 159.5, 157,8, 153.5, 146.7, 144.7, 141.4, 139.9, 136.7, 130.0, 125.8 (2,4-CH), 123.2 (6,21-CH), 95.3 (11,16-CH), 55.7 (OCH3), 18.50, 18.48 (13,14-CH2CH3), 16.5 (13,14-CH2CH3), 15.3 (9,18-CH2CH3), 10.4 (8,19-CH3). HRMS (ESI-TOF) m/z: M+ Calcd for C33H36N3NiO+ 548.2206; Found 548.2205.
3-Ethoxy-9,13,14,18-tetraethyl-8,19-dimethylbenziporphyrin (7b). In a pear-shaped flask, tripyrrane dicarboxylic acid 9 (106 mg, 0.234 mmol) was dissolved in TFA (1 mL) and stirred under nitrogen for 2 min. Dichloromethane (100 mL) was added, followed by dialdehyde 8b (42 g, 0.236 mol), and the mixture was stirred in the dark overnight under nitrogen. DDQ (73 mg, 0.321 mmol) was added, and the mixture was stirred for 1 h. The resulting solution was washed with water and saturated sodium bicarbonate, back extracting the aqueous layers with chloroform at each stage to ensure that all of the product was collected. The combined organic layers were evaporated under reduced pressure, and the residue was purified on a grade 3 basic alumina column, eluting with dichloromethane. The product was collected as a dark-blue band. Recrystallization from chloroform-methanol gave benziporphyrin 7b (52 mg, 0.103 mmol, 44%) as dark-purple crystals, mp > 300 °C. UV-vis (1% Et3N-CH2Cl2): λmax (log ε): 308 (4.77), 382 (4.89), 581 (sh, 3.37), 626 (3.59), 673 (3.68), 733 nm (3.48). UV-vis (5 equiv. TFA-CH2Cl2): λmax (log ε): 304 (4.65), 391 (4.87), 479 (3,74), 524 (3.71), 566 (3.73), 776 (3.90), 858 (4.15). UV-vis (1% TFA-CH2Cl2): λmax (log ε): 315 (4.65), 400 (4.82), 510 (3.92), 573 (3.36), 711 (sh, 3.57), 760 (3.70), 842 nm (sh, 3.60). 1H NMR (500 MHz, CDCl3): δ 9.16 (br s, 1H, NH), 7.80 (s, 1H, 22-H), 7.49 (br d, 2H, 4JHH = ca. 1 Hz, 2,4-H), 7.14 (s, 1H, 6,21-H), 6.48 (s, 2H, 11,16-H), 4.29 (q, 2H, 3JHH = 6.9 Hz, OCH2), 2.82 (q, 4H, 3JHH = 7.6 Hz, 13,14-CH2), 2.74 (q, 4H, 3JHH = 7.6 Hz, 9,18-CH2), 2.39 (s, 6H, 8,19-CH3), 1.53 (t, 3H, 3JHH = 6.9 Hz, OCH2CH3), 1.34 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.26 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 1H NMR (500 MHz, TFA-CDCl3): δ 9.42 (br s, 2H, 2 × NH), 8.03 (s, 2H, 6,21-H), 7.83 (br d, 2H, 2,4-H), 7.04 (s, 2H, 10,15-H), 4.77 (s, 1H, 22-H), 4.30 (q, 2H, 3JHH = 7.0 Hz, OCH2), 2.98–2.92 (m, 8H, 9,13,14,18-CH2), 2.66 (s, 6H, 8,19-CH3), 1.54 (t, 3H, 3JHH = 7.0 Hz, OCH2CH3), 1.37 (t, 6H, 3JHH = 7.7 Hz), 1.32 (t, 6H, 3JHH = 7.7 Hz) (4 × CH2CH3). 13C NMR (125 MHz, CDCl3): δ 169.2, 158.9, 157.3, 148.1, 141.3, 141.1, 140.5, 135.2, 122.9 (2,4-CH), 122.7 (6,21-CH), 118.5 (22-CH), 93.1 (11,16-CH), 64.0 (OCH2), 18.5 (13,14-CH2), 18.1 (9,18-CH2), 16.1 (13,14-CH2CH3), 15.3 (9,18-CH2CH3), 15.2 (OCH2CH3), 10.4 (8,19-CH3). 13C NMR (125 MHz, TFA-CDCl3): δ 162.7, 162.3, 155.0, 147.3, 146.3, 143.8, 141.3, 133.2, 128.0 (6,21-CH), 124.8 (2,4-CH), 101.9 (22-CH), 94.0 (11,16-CH), 64.8 (OCH2), 18.4 (13,14-CH2), 18.1 (9,18-CH2), 15.2 (13,14-CH2CH3), 14.8 (9,18-CH2CH3), 14.3 (OCH2CH3), 10.6 (8,19-CH3). HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C34H40N3O+ 506.3166; Found 506.3167.
[3-Ethoxy-9,13,14,18-tetraethyl-8,19-dimethylbenziporphyrinato]palladium(II) (7bPd). Ethoxybenziporphyrin 7b (17 mg, 0.0336 mmol) and palladium(II) acetate (17 mg, 0.076 mmol) in acetonitrile (15 mL) were refluxed for 30 min. The solution was cooled to room temperature and then diluted with dichloromethane (35 mL). The solution was washed with water, and the solvent was removed on a rotary evaporator. The residue was chromatographed on a grade 3 basic alumina column, eluting with dichloromethane. A reddish-purple fraction was collected, and the solvent was removed under reduced pressure. The residue was recrystallized from chloroform-methanol to give the palladium complex (15 mg, 0.025 mmol, 73%) as a dark-purple solid, mp > 300 °C. UV-vis (CH2Cl2): λmax (log ε): 308 (4.61), 391 (sh, 4.67), 408 (4.75), 474 (sh, 3.48), 507 (sh, 3.69), 542 (3.85), 573 (3.86), 666 (3.27), 731 (3.53), 806 (3.46). 1H NMR (500 MHz, CDCl3): δ 7.70 (s, 2H, 2,4-H), 7.57 (s, 1H, 6,21-H), 7.27 (s, 2H, 11,16-H), 4.37 (q, 2H, 3JHH = 7.0 Hz, OCH2), 3.01–2.93 (m, 8H, 9,13,14,18-CH2), 2.59 (s, 6H, 8,19-CH3), 1.57 (t, 3H, 3JHH = 7.0 Hz, OCH2CH3), 1.43 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.38 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 13C NMR (125 MHz, CDCl3): δ 157.1, 156.1, 153.7, 145.2, 144.0, 141.1, 138.5, 133.3, 131.5, 127.7 (2,4-CH), 126.6 (6,21-CH), 95.5 (11,16-CH), 64.0 (OCH2), 18.9, 18.6 (9,13,14,18-CH2), 16.6 (13,14-CH2CH3), 15.3 (OCH2CH3 and 9,18-CH2CH3), 10.5 (8,19-CH3). HRMS (ESI-TOF) m/z: M+ Calcd for C34H38N3OPd+ 610.2044; Found 610.2037.
[3-Ethoxy-9,13,14,18-tetraethyl-8,19-dimethylbenziporphyrinato]nickel(II) (7bNi). Ethoxybenziporphyrin 7b (20 mg, 0.0396 mmol) and nickel(II) acetate (32 mg, 0.129 mmol) in DMF (20 mL) were refluxed for 30 min. The mixture was cooled to room temperature and diluted with chloroform (20 mL). The solution was washed with water, and the solvent was evaporated under reduced pressure. The residue was chromatographed on a grade 3 basic alumina column, eluting with chloroform. A dark-green band was collected, and the solvent was removed under reduced pressure. The residue was recrystallized from chloroform-methanol to give the nickel complex (12 mg, 0.0213 mmol, 54%) as a dark-green solid, mp > 300 °C. UV-vis (CH2Cl2): λmax (log ε): 313 (4.48), 355 (4.43), 406 (4.56), 493 (sh, 3.58), 528 (3.52), 669 (3.65). 1H NMR (500 MHz, CDCl3): δ 7.63 (s, 2H, 2,4-H), 7.34 (s, 1H, 6,21-H), 7.09 (s, 2H, 11,16-H), 4.31 (q, 2H, 3JHH = 7.0 Hz, OCH2), 2.91 (q, 4H, 3JHH = 7.6 Hz, 12,13-CH2), 2.84 (q, 4H, 3JHH = 7.6 Hz, 9,18-CH2), 2.44 (s, 6H, 8,19-CH3), 1.53 (t, 3H, 3JHH = 7.0 Hz, OCH2CH3), 1.37 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.30 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 13C NMR (125 MHz, CDCl3): δ 159.5, 157.1, 153.5, 146.7, 144.7, 141.4, 139.9, 136.7, 130.0, 126.7 (2,4-CH), 123.2 (6,21-CH), 95.3 (11,16-CH), 64.0 (OCH2), 18.51, 18.49 (9,13,14,18-CH2), 16.5 (13,14-CH2CH3), 15.29 (9,18-CH2CH3), 15.27 (OCH2CH3), 10.4 (8,19-CH3). HRMS (ESI-TOF) m/z: M+ Calcd for C34H38N3NiO+ 562.2363; Found 562.2352.
9,13,14,18-Tetraethyl-3-(methoxycarbonylmethoxy)-8,19-dimethylbenziporphyrin (7c). In a pear-shaped flask, tripyrrane dicarboxylic acid 9 (202 mg, 0.446 mmol) was dissolved in TFA (2 mL) and stirred under nitrogen for 2 min. Dichloromethane (200 mL) was added, followed by dialdehyde 8c (104 mg, 0.468 mmol). The flask was covered in aluminum foil to protect the reaction from ambient light, and the solution was stirred overnight under nitrogen. DDQ (154 mg, 0.678 mmol) was added, and the mixture was stirred for 1 h. The resulting solution was washed with water and saturated sodium bicarbonate, back-extracting the aqueous layers with chloroform at each stage to ensure that all of the product was collected. The organic layers were evaporated under reduced pressure, and the residue was purified on a grade 3 basic alumina column, eluting with dichloromethane. The product was collected as a dark-blue band. Recrystallization from chloroform-methanol afforded benziporphyrin 7c (100 mg, 0.182 mmol, 41%) as dark-purple crystals, mp > 300 °C. UV-vis (1% Et3N-CH2Cl2): λmax (log ε): 308 (5.60), 380 (5.72), 541 (sh, 4.39), 583 (sh, 4.59), 629 (sh, 4.62), 677 (sh, 4.46), 742 (sh, 4.12). UV-vis (1% TFA-CH2Cl2): λmax (log ε): 313 (5.56), 398 (5.71), 494 (sh, 4.89), 535 (sh, 4.66), 575 (sh, 4.55), 723 (sh, 4.69), 836 (sh, 4.46). 1H NMR (500 MHz, CDCl3): δ 9.35 (br s, 1H, NH), 8.04 (br t, 1H, 22-H), 7.48 (d, 2H, 4JHH = 1.2 Hz, 2,4-H), 7.07 (s, 2H, 6,21-H), 6.45 (s, 2H, 11,16-H), 4.86 (s, 2H, CH2CO2Me), 3.85 (s, 3H, OCH3), 2.81 (q, 4H, 3JHH = 7.6 Hz, 12,13-CH2CH3), 2.72 (q, 4H, 3JHH = 7.6 Hz, 9,18-CH2CH3), 2.37 (s, 6H, 8,19-CH3), 1.33 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.25 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 1H NMR (400 MHz, TFA-CDCl3): δ 7.71 (s, 2H), 7.64 (s, 2H, 6,21-H), 6.70 (s, 2H, 11,16-H), 4.83 (s, 2H, CH2CO2Me), 3.85 (s, 3H, OCH3), 2.82 (q, 8H, 3JHH = 7.6 Hz, 4 × CH2CH3), 2.55 (s, 6H, 8,19-CH3), 1.32–1.26 (overlapping triplets, 12H, 4 × CH2CH3). 13C NMR (125 MHz, CDCl3): δ 169.7, 169.5, 157.9, 157.6, 148.3, 141.34, 141.26, 140.6, 135.4, 122.6 (2,4-CH), 122.3 (6,21-CH), 119.6 (22-CH), 93.1 (11,16-CH), 66.0 (OCH2), 52.6 (OCH3), 18.4 (13,14-CH2CH3), 18.1 (9,18-CH2CH3), 16.1 (13,14-CH2CH3), 15.3 (9,18-CH2CH3), 10.4 (8,19-CH3). 13C NMR (100 MHz, TFA-CDCl3): δ 169.3, 163.2, 161.1, 155.3, 147.4, 146.4, 141.1, 133.5, 127.6 (2,4-CH), 124.3 (6,21-CH), 102.6 (22-CH), 94.1 (11,16-CH), 65.9 (OCH2), 52.9 (OCH3), 18.4 (13,14-CH2CH3), 18.1 (9,18-CH2CH3), 15.2 (13,14-CH2CH3), 14.3 (9,18-CH2CH3), 10.6 (8,19-CH3). HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C35H40N3O3+ 550.3064; Found 550.3067.
[9,13,14,18-Tetraethyl-3-(methoxycarbonylmethoxy)-8,19-dimethylbenziporphyrinato]palladium(II) (7cPd). Benziporphyrin 7c (20 mg, 0.0364 mmol) and palladium(II) acetate (22 mg, 0.098 mmol) in acetonitrile (15 mL) were refluxed for 30 min. The solution was cooled to room temperature and diluted with dichloromethane (35 mL). The solution was washed with water, and the solvent was removed on a rotary evaporator. The residue was chromatographed on a grade 3 basic alumina column, eluting with dichloromethane. A reddish-purple fraction was collected, and the solvent was removed under reduced pressure. The residue was recrystallized from chloroform-methanol to give the palladium complex (12 mg, 0.0183 mmol, 50%) as a dark-purple solid, mp > 300 °C. UV-vis (CH2Cl2): λmax (log ε): 308 (4.47), 350 (sh, 4.21), 389 (sh, 4.49), 406 (4.63), 473 (sh, 3.33), 507 (sh, 3.52), 539 (3.65), 570 (3.63), 665 (3.21), 730 (3.41), 805 (3.31). 1H NMR (500 MHz, CDCl3): δ 7.67 (s, 2,4-H), 7.50 (s, 2H, 6,21-H), 7.22 (s, 2H, 11,16-H), 4.93 (s, 2H, CH2CO2Me), 3.87 (s, 3H, OCH3), 2.99–2.91 (m, 8H, 4 × CH2CH3), 2.57 (s, 6H, 8,19-CH3), 1.42 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.37 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 13C NMR (125 MHz, CDCl3): δ 169.9, 157.4, 155.8, 153.9, 145.4, 144.0, 141.2, 138.6, 133.6, 132.9, 127.1 (2,4-CH), 126.3 (6,21-CH), 95.6 (11,16-CH), 66.1 (OCH2), 52.6 (OCH3), 18.9 (13,14-CH2CH3), 18.6 (9,18-CH2CH3), 16.6 (13,14-CH2CH3), 15.3 (9,18-CH2CH3), 10.4 (8,19-CH3). HRMS (ESI-TOF) m/z: M+ Calcd for C35H38N3O3Pd+ 654.1943; Found 654.1934.
[9,13,14,18-Tetraethyl-3-(methoxycarbonylmethoxy)-8,19-dimethylbenziporphyrinato]nickel(II) (7cNi). Benziporphyrin 40c (19 mg, 0.0346 mmol) and nickel(II) acetate (40 mg, 0.161 mmol) in DMF (20 mL) were refluxed for 30 min. The solution was cooled to room temperature, diluted with chloroform (20 mL), and washed with water. The solvent was evaporated under reduced pressure, and the residue chromatographed on a grade 3 basic alumina column, eluting with chloroform. A dark-green band was collected, and the solvent was removed under reduced pressure. The residue was recrystallized from chloroform-methanol to give the nickel complex (16 mg, 0.026 mmol, 76%) as a dark-green solid, mp > 300 °C. UV-vis (CH2Cl2): λmax (log ε): 312 (4.49), 348 (4.45), 405 (4.56), 492 (sh, 3.66), 526 (3.59), 667 (3.69). 1H NMR (500 MHz, CDCl3): δ 7.61 (s, 2,4-H), 7.29 (s, 2H, 6,21-H), 7.05 (s, 2H, 11,16-H), 4.87 (s, 2H, CH2CO2Me), 3.85 (s, 3H, OCH3), 2.89 (q, 4H, 3JHH = 7.6 Hz, 13,14-CH2), 2.82 (q, 4H, 3JHH = 7.6 Hz, 9,18-CH2), 2.42 (s, 6H, 8,19-CH3), 1.36 (t, 6H, 3JHH = 7.6 Hz, 13,14-CH2CH3), 1.29 (t, 6H, 3JHH = 7.6 Hz, 9,18-CH2CH3). 13C NMR (125 MHz, CDCl3): δ 169.9, 159.8, 156.1, 153.7, 146.9, 144.8, 141.5, 140.0, 136.9, 131.6, 126.1 (2,4-CH), 123.0 (6,21-CH), 95.4 (11,16-CH), 66.1 (OCH2), 52.5 (OCH3), 18.48, 18.46 (4 × CH2CH3), 16.4 (13,14-CH2CH3), 15.2 (9,18-CH2CH3), 10.4 (8,19-CH3). HRMS (ESI-TOF) m/z: M+ Calcd for C35H38N3NiO3+ 606.2261; Found 606.2262.
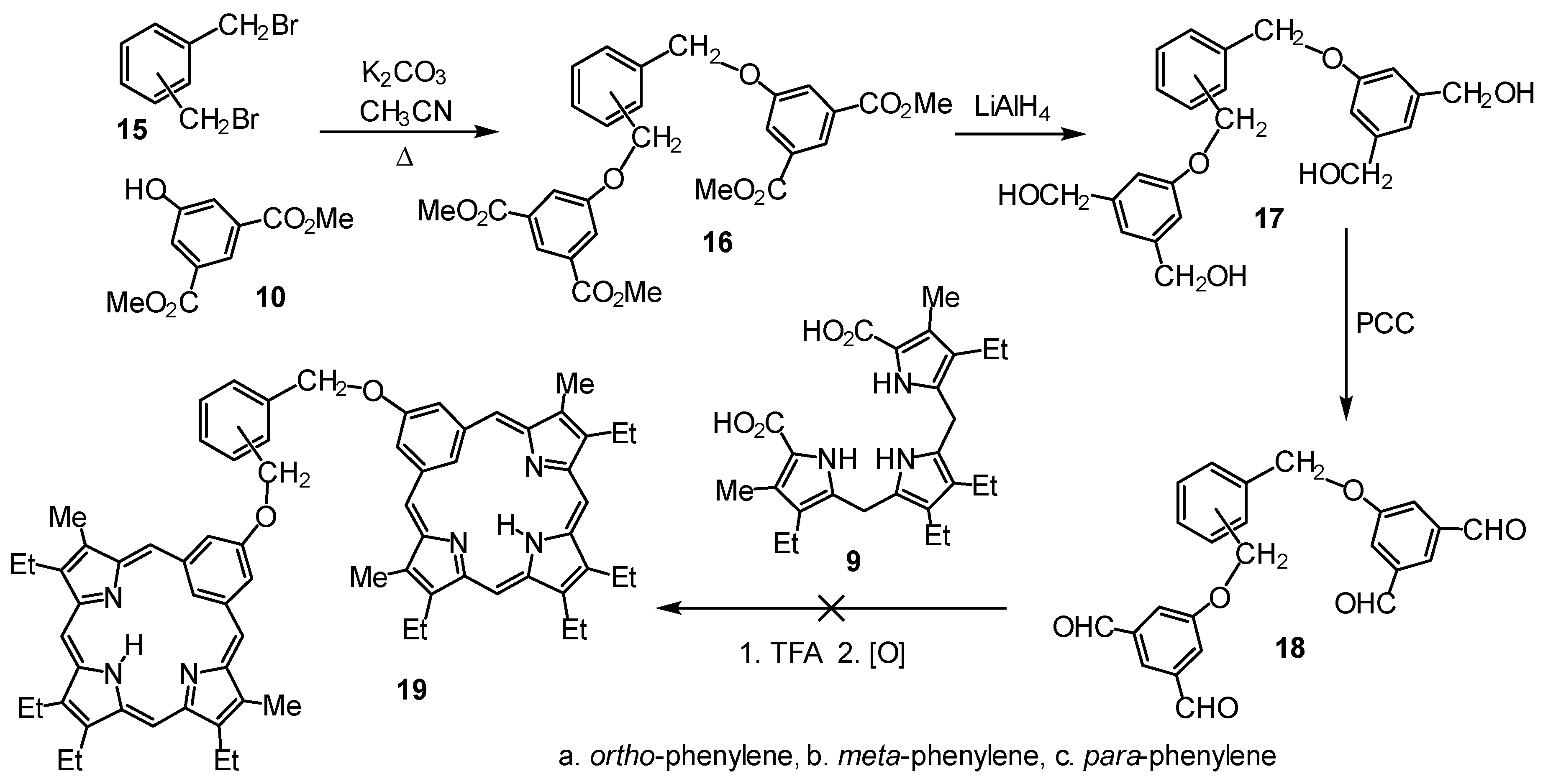
1,2-Bis[3,5-dimethoxycarbonylbenzyloxy]benzene (16a). Dimethyl 5-hydroxyisophthalate (2.415 g, 11.5 mmol), α,α’-dibromo-o-xylene (1.510 g, 5.72 mmol), and potassium carbonate (1.66 g, 12 mmol) in acetonitrile (100 mL) were refluxed overnight. The solvent was removed on a rotary evaporator, and the solid residue was dissolved in ethyl acetate (75 mL) and washed with water (2 × 30 mL) and saturated sodium bicarbonate (2 × 30 mL). The organic layer was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to yield 1,2-bis[3,5-dimethoxycarbonylbenzyloxy]benzene (2.90 g, 5.55 mmol, 97%) as a white solid, mp 150–151 °C. 1H NMR (400 MHz, CDCl3): δ 8.26 (t, 2H, 4JHH = 1.4 Hz, 4′,4″-H), 7.78 (d, 4H, 4JHH = 1.4 Hz, 2′,2″,6′,6″-H), 7.53–7.50 (m, 2H, 3,6-H), 7.42–7.39 (m, 2H, m, 4,5-H), 5.26 (s, 4H, 2 × CH2O), 3.92 (s, 12H, s, 4 × OCH3). 13C NMR (100 MHz, CDCl3): δ 166.2 (CO2CH3), 158.8, 134.8, 132.1, 129.7 (4,5-CH), 129.1 (3,6-CH), 123.6 (4′,4″-H), 120.3 (2′,2″,6′,6″-H), 69.0 (\1–\2CH2O), 52.6 (4 × OCH3). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C28H26NaO10+ 545.1418; Found 545.1415.
1,2-Bis[3,5-diformylbenzyloxy]benzene (18a). Lithium aluminum hydride (0.400 g, 10.5 mmol) was added to a solution of 1,2-bis[3,5-dimethoxycarbonylbenzyloxy]benzene (1.006 g, 1.93 mmol) in dry THF (100 mL). The mixture was stirred at room temperature overnight. Dilute hydrochloric acid (40 mL) was added dropwise to the solution, and the contents of the reaction flask were stirred for an additional 30 min. Ethyl acetate (200 mL) was added, and the aqueous layer was drawn off. The organic layer was washed with water (2 × 100 mL) and saturated sodium bicarbonate solution (2 × 100 mL). The organic layer was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to yield 1,2-bis[3,5-dihydroxymethylbenzyloxy]benzene (17a, 0.735 g, 1.79 mmol, 93%) as a white solid, mp 65–66 °C. 1H NMR (500 MHz, DMSO-d6): δ 7.53–7.50 (m, 2H, 4,5-H), 7.37–7.34 (m, 2H, 3,6-H), 6.86 (br s, 2H, 4′,4″-H), 6.83 (br s, 4H, 2′,2″,6′,6″-H), 5.19 (s, 4H, 2 × CH2O), 5.13 (t, 4H, 3JHH = 5.7 Hz, 4 × OH), 4.43 (8H, d, 3JHH = 5.7 Hz, 4 × CH2OH). 13C NMR (125 MHz, DMSO-d6): δ 158.4, 144.1, 135.5, 128.6 (3,6-CH), 128.1 (4,5-CH), 117.2 (4′,4″-CH), 111.1 (2′,2″,6′,6″-CH), 67.1 (2 × CH2O), 63.0 (4 × CH2OH). Tetraalcohol 17a (100 mg, 0.246 mmol) was dissolved in dichloromethane (12 mL) and THF (8 mL). Pyridinium chlorochromate (0.330 g, 1.5 mmol) and silica gel (0.342 g, 5.7 mmol) were added to the solution, and the mixture was stirred at room temperature for 1 h. The contents of the reaction flask were immediately chromatographed twice on silica gel, eluting with dichloromethane. The desired column fractions were evaporated under reduced pressure to yield tetraaldehyde 18a (72 mg, 0.179 mmol, 73%) as a white solid, mp 175–177 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.02 (s, 4H, 4 × CHO), 8.00 (t, 2H, 4JHH = 1.3 Hz, 4′,4″-H), 7.79 (d, 4H, 4JHH = 1.3 Hz, 2′,2″,6′,6″-H), 7.61–7.57 (m, 2H, 3,6-H), 7.44–7.40 (m, 2H, 4,5-H), 5.43 (s, 4H, 2 × CH2O). 13C NMR (100 MHz, DMSO-d6): δ 192.4 (CHO), 159.5, 138.3, 134.9, 129.3 (4,5-CH), 128.7 (3,6-CH), 123.3 (4′,4″-H), 120.4 (2′,2″,6′,6″-CH), 68.3 (CH2O). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C24H18NaO6+ 425.0996; Found 425.0999.
1,3-Bis[3,5-dimethoxycarbonylbenzyloxy]benzene (16b). Dimethyl 5-hydroxyisophthalate (735 mg, 3.50 mmol), α,α’-dibromo-m-xylene (4.60 mg, 1.74 mmol) and potassium carbonate (500 mg, 3.6 mmol) were dissolved in acetonitrile (30 mL) and refluxed overnight. The solution was cooled, and the solvent was removed on a rotary evaporator. The solid residue was dissolved in ethyl acetate (75 mL) and then washed with water (2 × 30 mL) and saturated sodium bicarbonate solution (2 × 30 mL). The organic layer was dried over sodium sulfate and filtered, and the solvent was evaporated under reduced pressure to afford tetraester 16b (902 mg, 1.73 mmol, 99%) as a white solid, mp 92–93 °C. 1H NMR (500 MHz, CDCl3): δ 8.28 (t, 2H, 4JHH = 1.4 Hz, 4′,4″-H), 7.82 (d, 4H, 4JHH = 1.4 Hz, 2′,2″,6′,6″-H), 7.53 (br s, 1H, 2-H), 7.43–7.41 (m, 3H, 4,5,6-H), 5.15 (s, 4H, CH2O), 3.92 (s, 12H, 4 × OCH3). 13C NMR (125 MHz, CDCl3): δ 166.2 (4 × C=O), 158.9, 136.9, 132.1, 129.2 (5-CH), 127.5 (4,6-CH), 126.7 (2-CH), 123.5 (4′,4″-CH), 120.3 (2′,2″,6′,6″-CH), 70.2 (2 × CH2O), 52.6 (4 × OCH3). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C28H26NaO10+ 545.1418; Found 545.1418.
1,3-Bis[3,5-diformylbenzyloxy]benzene (18b). Lithium aluminum hydride (100 mg, 2.63 mmol) was added to a solution of 16b (209 mg, 0.400 mmol) in dry THF (50 mL), and the resulting mixture was stirred at room temperature overnight. Dilute hydrochloric acid (30 mL) was added dropwise to the solution, and the contents of the reaction flask were stirred for an additional 30 min. Ethyl acetate (100 mL) was added, and the aqueous layer was drawn off. The organic layer was washed with water (2 × 50 mL) and a saturated sodium bicarbonate solution (2 × 50 mL). The organic solution was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to give 1,3-bis[3,5-dihydroxymethylbenzyloxy]benzene (17b, 147 mg, 0.358 mmol, 89%) as a white solid, mp 82–84 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.53 (s, 1H, 2-H), 7.42–7.38 (3H, s, 4,5,6-H), 6.86 (br s, 2H, 4′,4″-H), 6.84 (br s, 4H, 2′,2″,6′,6″-H), 5.14 (4H, t, 3JHH = 5.7 Hz, 2 × OH), 5.09 (s, 4H, 2 × CH2O), 4.45 (d, 8H, 3JHH = 5.7 Hz, 4 × CH2OH). 13C NMR (100 MHz, DMSO-d6): δ 158.5, 144.1, 137.7, 128.7 (5-CH), 127.1 (4,6-CH), 126.8 (2-CH), 117.1 (2′,2″,6′,6″-CH), 111.2 (4′,4″-CH), 69.2 (CH2O), 63.0 (CH2OH). Tetraalcohol 17b (100 mg, 0.244 mmol) was dissolved in dichloromethane (12 mL) and THF (8 mL), pyridinium chlorochromate (0.426 g, 2.0 mmol) and silica gel (0.208 g, 3.5 mmol) were added, and the mixture was stirred at room temperature for 1 h. The contents of the reaction flask were immediately run through a short silica gel column and eluted with dichloromethane. The solvent was evaporated, and the residue was purified on silica, eluting fist with dichloromethane and then with chloroform. The product fractions were evaporated under reduced pressure, and the residue was recrystallized from chloroform-hexanes to yield the tetraaldehyde (55 mg, 0.137 mmol, 56%) as a white solid, mp 158–159 °C. 1H NMR (500 MHz, CDCl3): δ 10.05 (s, 4H, 4 × CHO), 7.98 (t, 2H, 4JHH = 1.3 Hz, 4′,4″-H), 7.73 (d, 4H, 4JHH = 1.3 Hz, 2′,2″,6′,6″-H), 7.55 (br s, 1H, 2-H), 7.48–7.43 (m, 3H, 4,5,6-H), 5.22 (s, 4H, CH2O). 13C NMR (125 MHz, CDCl3): δ 190.9 (4 × C=O), 160.0, 138.7, 136.6, 129.5 (5-CH), 127.8 (4,6-CH), 126.7 (2-CH), 124.8 (4′,4″-CH), 120.4 (2′,2″,6′,6″-CH), 70.6 (2 × CH2O). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C24H18NaO6+ 425.0996; Found 425.1000.
1,4-Bis[3,5-dimethoxycarbonylbenzyloxy]benzene (16c). Dimethyl 5-hydroxyisophthalate (2.508 g, 11.9 mmol), α,α’-dibromo-p-xylene (1.066 g, 4.04 mmol), and potassium carbonate (1.70 g, 12.3 mmol) were refluxed in acetonitrile (100 mL) overnight. After the solution was cooled, the solvent was evaporated on a rotary evaporator. The residue was dissolved in ethyl acetate (50 mL) and washed with water (2 × 30 mL) and saturated sodium bicarbonate solution (2 × 30 mL). The organic layer was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to give 16c (1.42 g, 2.72 mmol, 67%) as a white solid, mp 197–200 °C. 1H NMR (400 MHz, CDCl3): δ 8.30 (t, 2H, 4JHH = 1.4 Hz, 4′,4″-H), 7.84 (d, 4H, 4JHH = 1.4 Hz, 2′,2″,6′,6″-H), 7.48 (s, 4H, 2,3,5,6-H), 5.16 (s, 4H, 2 × CH2O), 3.94 (12H, s, 4 × OCH3). 13C NMR (100 MHz, CDCl3): δ 166.3, 158.9, 136.4, 132.1, 128.1 (2,3,5,6-CH), 123.5 (4′,4″-CH), 120.4 (2′,2″,6′,6″-CH), 70.4 (CH2O), 52.6 (CO2CH3). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C28H26NaO10+ 545.1418; Found 545.1418.
1,4-Bis[3,5-diformylbenzyloxy]benzene (18c). Lithium aluminum hydride (0.200 g, 5.3 mmol) was added to a solution of 16c (0.472 g, 0.904 mmol) in dry THF (50 mL). The mixture was stirred at room temperature overnight. Dilute hydrochloric acid (30 mL) was added dropwise to the solution, and the contents of the reaction flask were stirred for an additional 30 min. Ethyl acetate (100 mL) was added, and the aqueous layer was drawn off. The organic layer was washed with water (2 × 50 mL) and saturated sodium bicarbonate solution (2 × 50 mL). The ethyl acetate layer was dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure to yield 1,4-bis[3,5-dihydroxymethylbenzyloxy]benzene (17c, 0.369 g, 0.90 mmol, 99%) as a white solid, mp 165–167 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.45 (s, 4H, 2,3,5,6-H), 6.85 (br s, 2H, 4′,4″-H), 6.82 (br s, 4H, 2′,2″,6′,6″-H), 5.13 (t, 4H, 3JHH = 5.7 Hz, 2 × OH), 5.08 (s, 4H, 2 × CH2O), 4.44 (d, 8H, 3JHH = 5.7 Hz, 4 × CH2OH). 13C NMR (100 MHz, DMSO-d6): δ 158.5, 144.1, 137.0, 127.8 (2,3,5,6-CH), 117.0 (4′,4″-CH), 111.1 (2′,2″,6′,6″-CH), 69.0 (CH2O), 63.0 (CH2OH). Tetraalcohol 17c (94 mg, 0.23 mmol), was dissolved in dichloromethane (12 mL) and THF (8 mL), pyridinium chlorochromate (0.426 g, 2.0 mmol) and silica gel (0.208 g, 3.5 mmol) were added, and the mixture was stirred at room temperature for 1 h. The contents of the reaction flask were immediately chromatographed twice on silica gel, eluting with dichloromethane. The desired column fractions were evaporated under reduced pressure to yield the tetraaldehyde (90 mg, 0.224 mmol, 97%) as a white solid, mp 195–198 °C. 1H NMR (500 MHz, DMSO-d6): δ 10.06 (s, 4H, CHO), 8.04 (br t, 2H, 4JHH = 1.2 Hz, 4′,4″-H), 7.83 (br d, 4H, 4JHH = 1.2 Hz, 2′,2″,6′,6″-H), 7.53 (s, 4H, 2,3,5,6-H), 5.30 (s, 4H, 2 × CH2O). 13C NMR (125 MHz, DMSO-d6): δ 192.5 (CHO), 159.5, 138.4, 136.3, 128.1 (2,3,5,6-CH), 123.1 (4′,4″-CH), 120.5 (2′,2″,6′,6″-CH), 69.8 (CH2O). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C24H18NaO6+ 425.0996; Found 425.1005.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









