

Room Temperature Diels–Alder Reactions of 4-Vinylimidazoles

Abstract
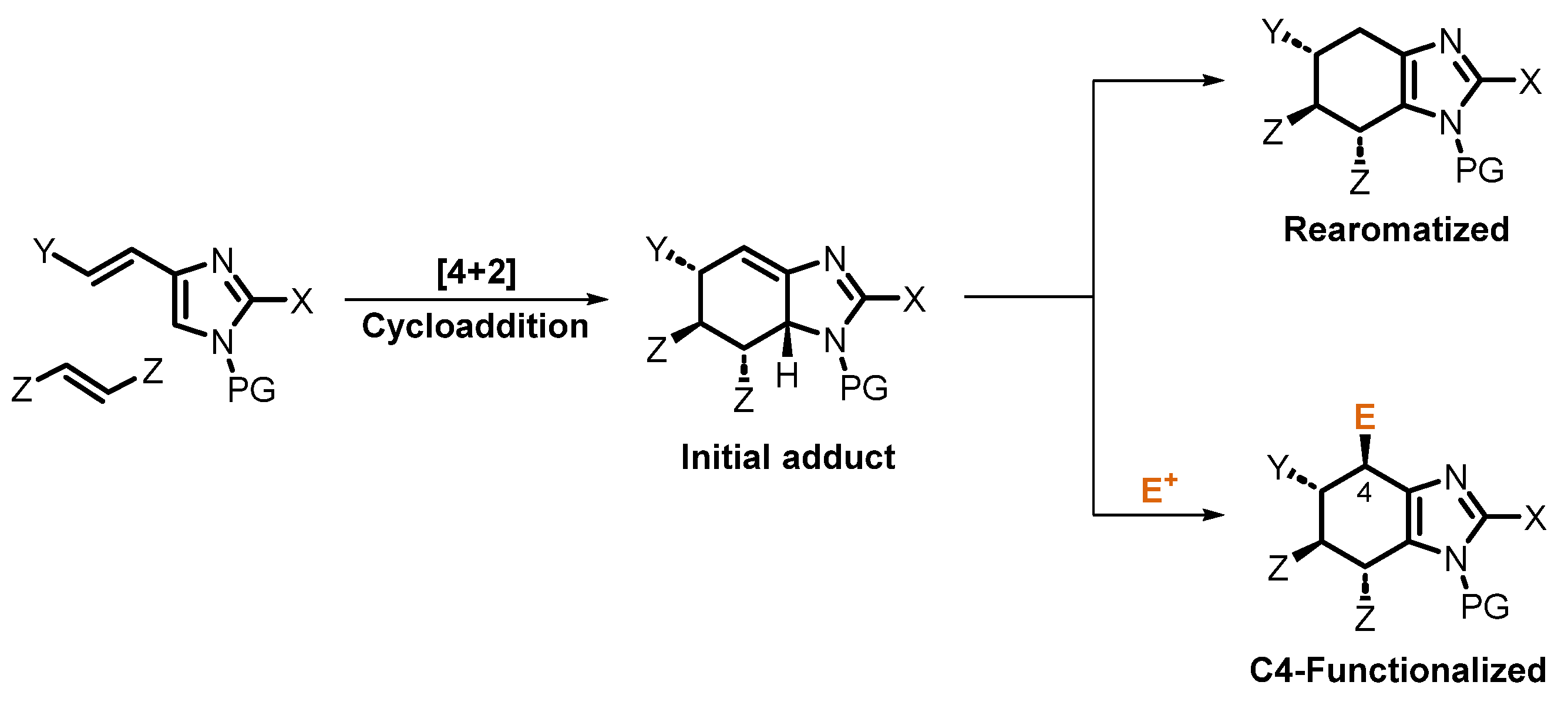
1. Introduction
2. Results and Discussion
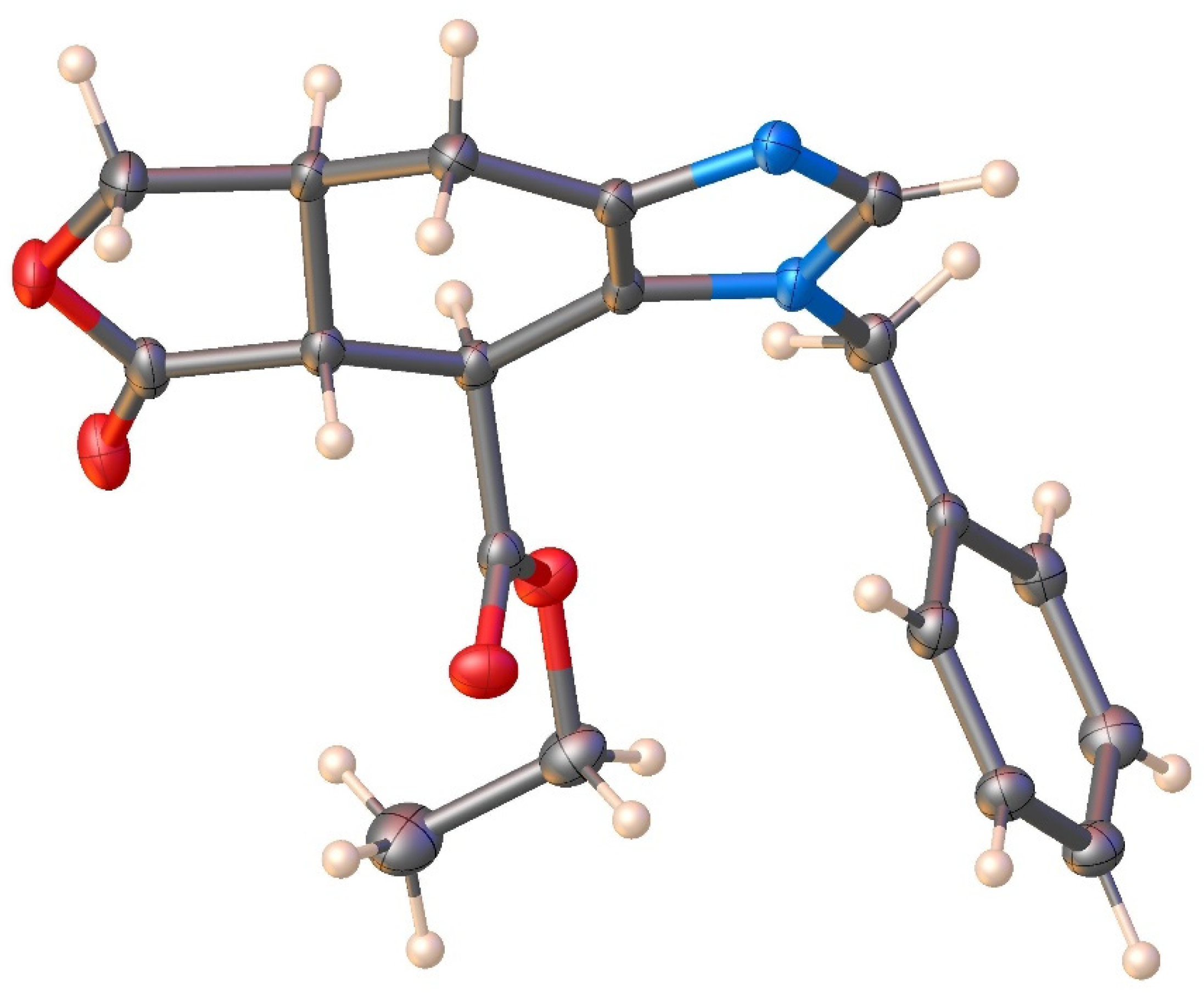
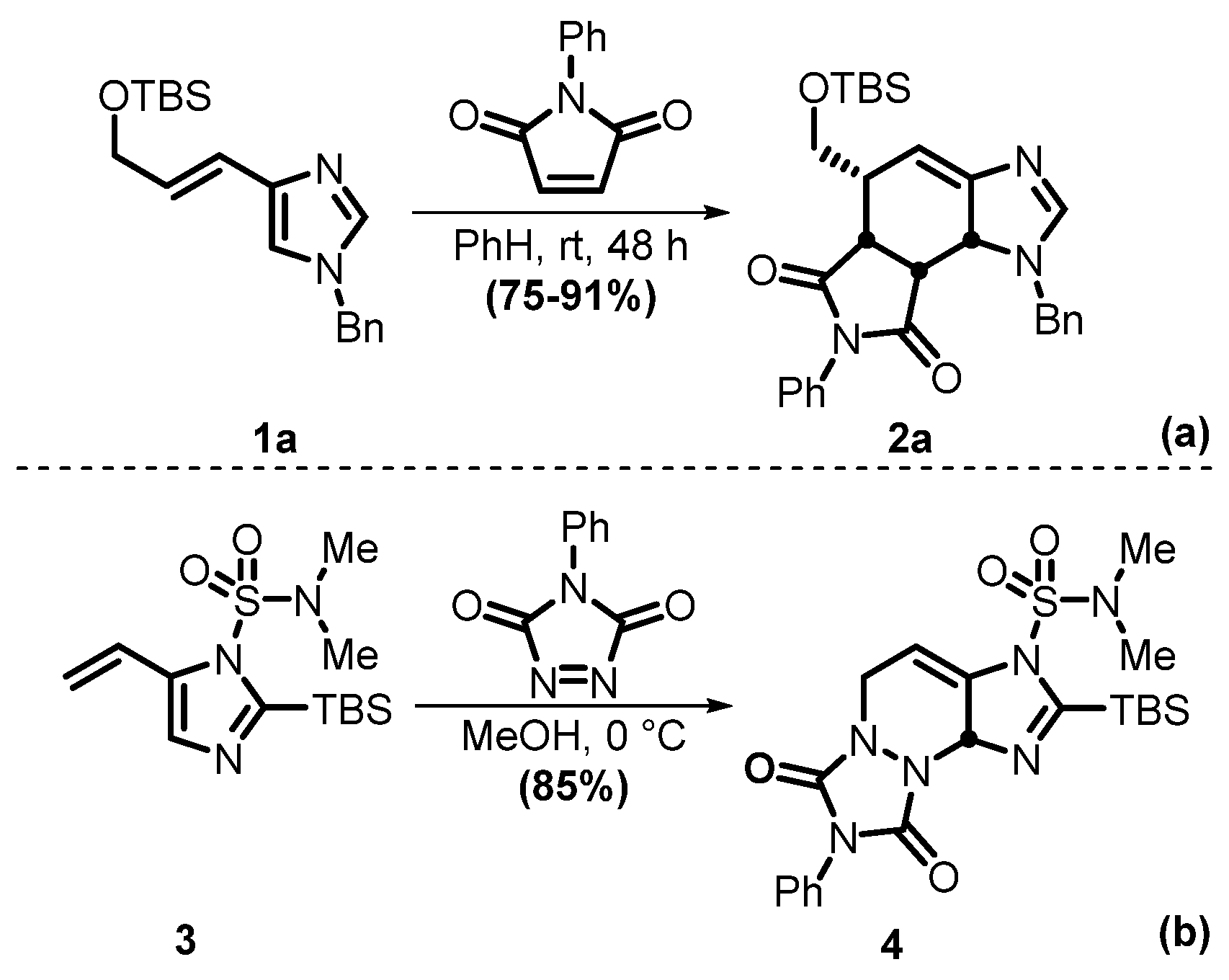
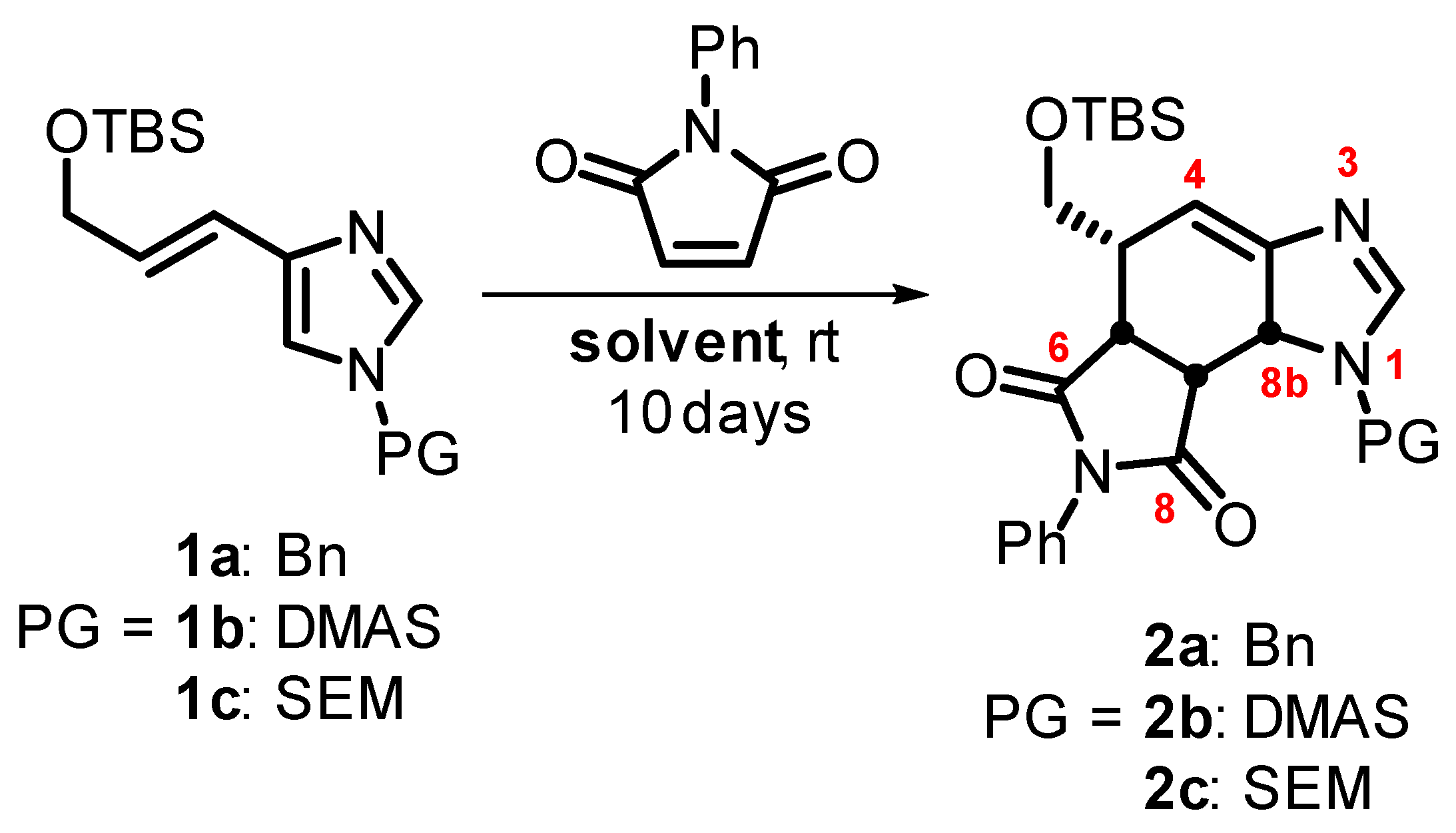
2.1. Chemistry
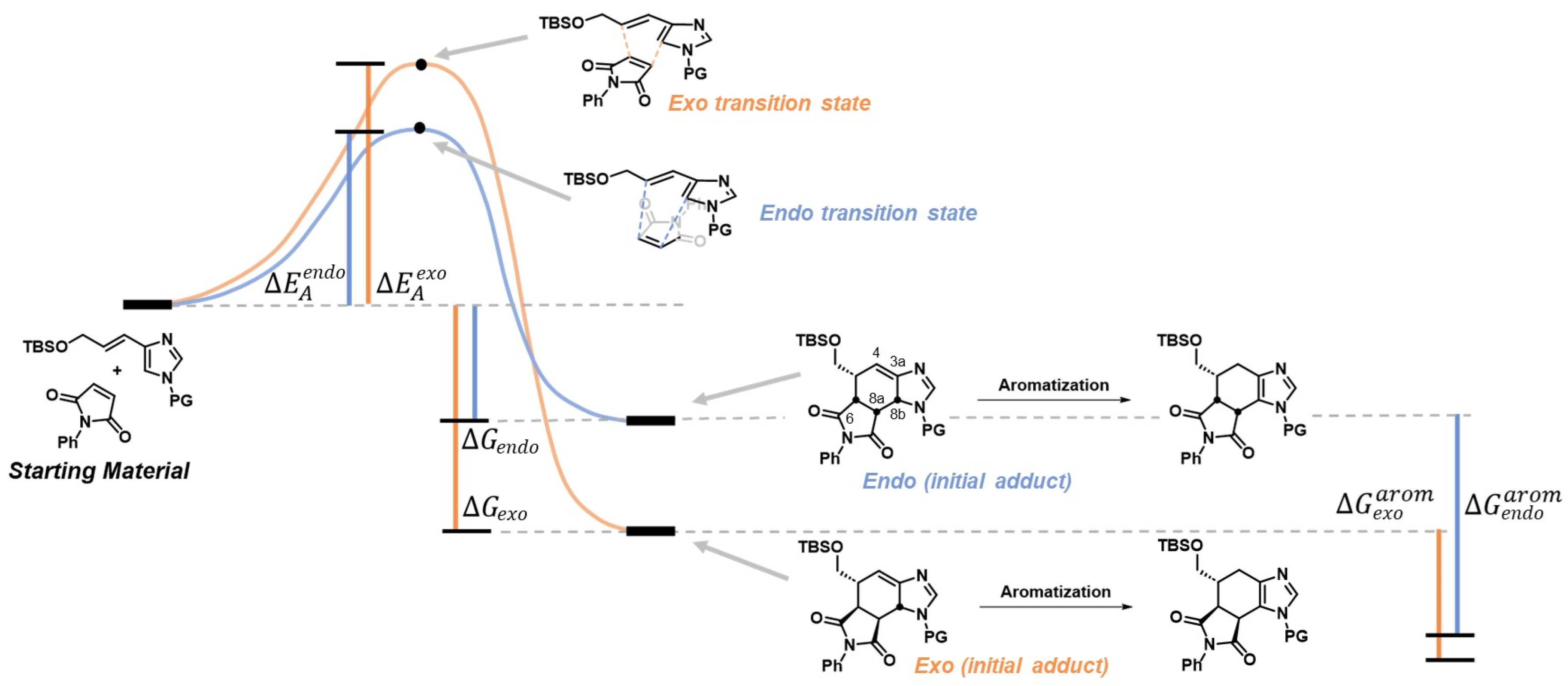
2.2. DFT Investigation
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sara, A.A.; Um-e-Farwa; Saeed, A.; Kalesse, M. Recent Applications of the Diels-Alder Reaction in the Synthesis of Natural Products (2017–2020). Synthesis 2022, 54, 975–988. [Google Scholar] [CrossRef]
- Krishna, G.; Grudinin, D.G.; Nikitina, E.V.; Zubkov, F.I. Intramolecular Diels-Alder Reactions of Vinylarenes and Alkynyl Arenes (the IMDAV reaction). Synthesis 2022, 54, 797–863. [Google Scholar]
- Laina-Martin, V.; Fernandez-Salas, J.A.; Aleman, J. Organocatalytic Strategies for the Development of Enantioselective Inverse Electron Demand Hetero-Diels-Alder Reaction. Chem. Eur. J. 2021, 27, 12509–12520. [Google Scholar] [CrossRef]
- Houk, K.N.; Liu, F.; Yong, Z.; Seeman, J.I. Evolution of the Diels-Alder Reaction Mechanism since the 1930s: Woodward, Houk with Woodward, and the Influence of Computational Chemistry on Understanding Cycloadditions. Angew. Chem. Int. Ed. 2021, 60, 12660–12681. [Google Scholar] [CrossRef] [PubMed]
- Flugel, L.L.; Hoye, T.R. Hexahydro-Diels-Alder Reaction: Benzyne Generation via Cycloisomerization of Tethered Triynes. Chem. Rev. 2021, 121, 2413–2444. [Google Scholar] [CrossRef]
- Min, L.; Hu, Y.-J.; Fan, J.-H.; Zhang, W.; Li, C.-C. Synthetic applications of type II intramolecular cycloadditions. Chem. Soc. Rev. 2020, 49, 7015–7043. [Google Scholar] [CrossRef] [PubMed]
- Andrus, M.B.; Saavedra, D.I. The Vinylarene Diels-Alder Reaction, Development and Potential. Tetrahedron 2019, 75, 2129–2142. [Google Scholar] [CrossRef]
- Marcantonio, E.; Guazzetti, D.; Bugatti, K.; Battistini, L.; Sartori, A.; Pelosi, G.; Curti, C.; Zanardi, F. Turning the Imidazole Core into Three-Dimensional Ring Systems: Mild Organocatalytic Entry to Enantiopure 6,7-Dihydrobenzimidazoles. Chem. Eur. J. 2023, 43, e202301200. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Wipf, P. Intramolecular Diels-Alder Reactions of Oxazoles, Imidazoles and Thiazoles. Synthesis 2021, 53, 1181–1199. [Google Scholar]
- Sepulveda-Arques, J.; Abarca-Gonzalez, B.; Medio-Simon, M. Cycloaddition Reactions with Vinyl Heterocycles. Adv. Heterocyc. Chem. 1995, 63, 339–401. [Google Scholar]
- Elshaier, Y.A.A.M.; Nemr, M.T.M.; Refaey, M.S.; Fadaly, W.A.A.; Barakat, A. Chemistry of 2-Vinylindoles: Synthesis and Applications. Molecules 2022, 46, 13400. [Google Scholar] [CrossRef]
- Tu, M.-S.; Chen, K.-W.; Wu, P.; Zhang, Y.-C.; Liu, X.-Q.; Shi, F. Advances in Organocatalytic Asymmetric Reactions of Vinylindoles: Powerful Access to Enantioenriched Indole Derivatives. Org. Chem. Front. 2021, 8, 2643–2672. [Google Scholar] [CrossRef]
- Rossi, E.; Abbiati, G.; Pirovano, V. 2- and 3-Vinylimidazoles as 4p-Components in Cycloaddition Reactions. Eur. J. Org. Chem. 2017, 2017, 4512–4529. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Zaytsev, V.P.; Mertsalov, D.F.; Nikitina, E.V.; Horak, Y.I.; Lytvyn, R.Z.; Homza, Y.V.; Obushak, M.D.; Dorovatovskii, P.V.; Khrustalev, V.N.; et al. Easy construction of furo[2,3-f]isoindole core by the IMDAV reaction between 3-(furyl)allylamines and α,β-unsaturated acid anhydrides. Tetrahedron 2016, 72, 2239–2253. [Google Scholar] [CrossRef]
- Kotsuki, H.; Kawamura, A.; Ochi, M.; Tokoroyama, T. Intramolecular Diels-Alder Reactions of Vinylfuran Derivatives. A Novel Approach to Benzofurans. Chem. Lett. 1981, 7, 917–920. [Google Scholar] [CrossRef]
- Cooper, J.A.; Cornwall, P.; Dell, C.P.; Knight, D.W. Furanodecalin synthesis using intramolecular Diels-Alder reactions of vinylfurans. Tetrahedron Lett. 1988, 29, 2107–2110. [Google Scholar] [CrossRef]
- Najera, C.; Sansano, J.M.; Yus, M. Diels-Alder Reactions of 1-Amino-1,3-dienes and Related Systems. Tetrahedron 2021, 94, 132316. [Google Scholar] [CrossRef]
- Yuan, J.; Wu, X.-Y.; Sha, F. Direct Assembly of Benzo[e]indoles by Diels-Alder Reactionof Arynes and 2-Vinylpyrroles. Eur. J. Org. Chem. 2016, 17, 2929–2932. [Google Scholar] [CrossRef]
- Cotterill, L.J.; Harrington, R.W.; Clegg, W.; Hall, M.J. Thermal 1,3-Trityl Migrations in Diels-Alder Domino Reactions of 1-Trityl-4-vinyl-1H-imidazoles. J. Org. Chem. 2010, 75, 4604–4607. [Google Scholar] [CrossRef]
- Watson, L.J.; Harrington, R.W.; Clegg, W.; Hall, M.J. Diastereoselective intermolecular ene reactions: Synthesis of 4,5,6,7-tetrahydro-1H-benzo[d]imidazoles. Org. Biomol. Chem. 2012, 10, 6649–6655. [Google Scholar] [CrossRef]
- Deghati, P.Y.F.; Wanner, M.J.; Koomen, G.-J. An Efficient Hetero Diels-Alder Approach to Imidazo[4,5-c]pyridazines as Purine Analogs. Tetrahedron Lett. 1998, 39, 4561–4564. [Google Scholar] [CrossRef]
- Walters, M.A.; Lee, M.D. The Use of Vinyl Imidazoles as Diels-Alder Dienes. Tetrahedron Lett. 1994, 35, 8307–8310. [Google Scholar] [CrossRef]
- Feldman, K.S.; Nuriye, A.Y. Extending Pummerer Reaction Chemistry: Application to the Assembly of the Pentacyclic Core of Dibromopalau’amine. Org. Lett. 2010, 12, 4532–4535. [Google Scholar] [CrossRef] [PubMed]
- Pöverlein, C.; Breckle, G.; Lindel, T. Diels−Alder Reactions of Oroidin and Model Compounds. Org. Lett. 2006, 8, 819–821. [Google Scholar] [CrossRef]
- Lovely, C.J.; Du, H.; Dias, H.V.R. Synthesis and Diels-Alder Reactivity of 4-Vinylimidazoles. Org. Lett. 2001, 3, 1319–1322. [Google Scholar] [CrossRef]
- He, Y.; Chen, Y.; Wu, H.; Lovely, C.J. Intramolecular Diels-Alder Reactions of 4-Vinylimidazoles. Org. Lett. 2003, 5, 3623–3626. [Google Scholar] [CrossRef] [PubMed]
- Lovely, C.J.; Du, H.; Dias, H.V.R. The Regioselective Synthesis of 1-Benzyl- and 1-Methyl-4-vinylimidazole and their Reactions with N-Phenylmaleimide. Heterocycles 2003, 60, 1–7. [Google Scholar] [CrossRef]
- Lovely, C.J.; Du, H.; Sivappa, R.; Bhandari, M.R.; He, Y.; Dias, H.V.R. Preparation and Diels−Alder Chemistry of 4-Vinylimidazoles. J. Org. Chem. 2007, 72, 3741–3749. [Google Scholar] [CrossRef] [PubMed]
- Sivappa, R.; Hernandez, N.M.; He, Y.; Lovely, C.J. Studies toward the Total Synthesis of Axinellamine and Massadine. Org. Lett. 2007, 9, 3861–3864. [Google Scholar] [CrossRef]
- Sivappa, R.; Mukherjee, S.; Dias, H.V.R.; Lovely, C.J. Studies Toward the Total Synthesis of Oroidin Dimers. Org. Biomol. Chem. 2009, 7, 3215–3218. [Google Scholar] [CrossRef]
- He, Y.; Krishnamoorthy, P.; Lima, H.M.; Chen, Y.; Wu, H.; Sivappa, R.; Dias, H.V.R.; Lovely, C.J. Intramolecular Diels-Alder Reactions of 4-Vinylimidazoles. Org. Biomol. Chem. 2011, 9, 2685–2701. [Google Scholar] [CrossRef] [PubMed]
- Lima, H.M.; Sivappa, R.; Yousufuddin, M.; Lovely, C.J. Total synthesis of 7′-desmethylkealiiquinone. Org. Lett. 2012, 14, 2274–2277. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Mukherjee, S.; Das, J.; Bhandari, M.R.; Du, H.; Yousufuddin, M.; Lovely, C.J. Preparation and Diels-Alder reactions of 1′-heterosubstituted vinylimidazoles. Tetrahedron Lett. 2015, 56, 3518–3522. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Yousufuddin, M.; Gout, D.; Lovely, C.J. Intramolecular Diels–Alder Reaction of a Silyl-Substituted Vinylimidazole en Route to the Fully Substituted Cyclopentane Core of Oroidin Dimers. Org. Lett. 2018, 20, 5964–5968. [Google Scholar] [CrossRef] [PubMed]
- Lovely, C.J.; Du, H.; He, Y.; Dias, H.V.R. Oxidative Rearrangement of Imidazoles with Dimethyldioxirane. Org. Lett. 2004, 6, 735–738. [Google Scholar] [CrossRef]
- Lima, H.M.; Sivappa, R.; Yousufuddin, M.; Lovely, C.J. Total Synthesis of 7′-Desmethylkealiiquinone, 4′-Desmethoxykealiiquinone, and 2-Deoxykealiiquinone. J. Org. Chem. 2014, 79, 2481–2490. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; He, Y.; Rasapalli, S.; Lovely, C.J. New Methods of Imidazole Functionalization—From Imidazole to Marine Alkaloids. Synlett 2006, 2006, 965–992. [Google Scholar] [CrossRef]
- Chu, M.-J.; Li, M.; Zhao, Y. Dimeric pyrrole-imidazole alkaloids: Sources, structures, bioactivities and biosynthesis. Bioorg. Chem. 2023, 133, 106332. [Google Scholar] [CrossRef]
- Gschwend, H.W.; Lee, A.O.; Meier, H.P. Rates of Intramolecular Diels-Alder Reactions of Pentadienylacrylamides. J. Org. Chem. 1973, 38, 2169. [Google Scholar] [CrossRef]
- Jung, M.E. Substituent and Solvent Effects in Intramolecular Diels-Alder Reactions. Synlett 1990, 1990, 186–190. [Google Scholar] [CrossRef]
- Jung, M.E.; Gervay, J. gem-Dialkyl Effect in the Intramolecular Diels-Alder Reaction of 2-Furfuryl Methyl Fumarates: The Reactive Rotamer Effect, the Enthalpic Basis for Acceleration, and Evidence for a Polar Transition State. J. Am. Chem. Soc. 1991, 113, 224–232. [Google Scholar] [CrossRef]
- Ishikawa, T.; Senzaki, M.; Kadoya, R.; Morimoto, T.; Miyake, N.; Izawa, M.; Saito, S.; Kobayashi, H. Intramolecular Diels-Alder Reactions Employing Hydroxamate Tethers: The First Examples and Promising Prospects. J. Am. Chem. Soc. 2001, 123, 4607. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.A.; Cridland, A.P.; Rolla, G.A.; Iqbal, M.; Bainbridge, N.P.; Whitwood, A.C.; Wilson, C. Studies on the Synthesis of the ABC Rings of (±)-Hexacyclinic Acid. J. Org. Chem. 2009, 74, 7812–7821. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, P.; Sivappa, R.; Du, H.; Lovely, C.J. Palladium-Catalyzed Substitution Reactions of 4-Allylimidazole Derivatives. Tetrahedron 2006, 62, 10555. [Google Scholar] [CrossRef]
- Saavedra, D.I.; Rencher, B.D.; Kwon, D.-H.; Smith, S.J.; Ess, D.H.; Andrus, M.B. Synthesis and Computational Studies Demonstrate the Utility of an Intramolecular Styryl Diels-Alder Reaction and Di-t-butylhydroxytoluene Assisted [1,3]-Shift to Construct Anticancer dl-Deoxypodophyllotoxin. J. Org. Chem. 2018, 83, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Mukherjee, S.; Das, J.; Bhandari, M.; Herath, A.; Yousufuddin, M.; Lovely, C.J. Heterosubstituted 4-Vinylimidazoles—Preparation and Diels-Alder Reactions. Heterocycles 2019, 99, 324–349. [Google Scholar]
- Boeckman, R.K.; Ko, S.S. Stereochemical control in the intramolecular Diels-Alder reaction. 2. Structural and electronic effects on reactivity and selectivity. J. Am. Chem. Soc. 1982, 104, 1033–1041. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations. J. Chem. Theory Comput. 2019, 15, 2847–2862. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/citation/ (accessed on 23 October 2023).
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Shelxt–Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with Shelxl. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | PG | Solvent | Time (Days) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||||
| Percent Conversion (%) a | ||||||||||||||
| 1 | 1a | Bn b | CDCl3 | 0 | 20 | 40 | 55 | 66 | 76 | 83 | 87 | 89 | 91 | 89 |
| 2 | 1b | DMAS b | CDCl3 | 0 | 0 | 0 | 1 | 4 | 7 | 10 | 12 | 12 | 20 | 21 |
| 3 | 1b | MeOH-d4 | 0 | 7 | 14 | 18 | 21 | 23 | 23 | 22 | 20 | 18 | 16 | |
| 4 | 1b | DMSO-d6 | 0 | 16 | 31 | 44 | 53 | 63 | 70 | 73 | 76 | 79 | 80 | |
| 5 | 1c | SEM c | C6D6 | 0 | 30 | 48 | 59 | 68 | 74 | 76 | 79 | 81 | 81 | 81 |
| 6 | 1c | CDCl3 | 0 | 17 | 30 | 42 | 48 | 54 | 62 | 68 | 72 | 77 | 79 | |
| Entry | Compound | Protecting Group | Endo vs. Exo TS Energy Comparison (kcal/mol) | ||||
|---|---|---|---|---|---|---|---|
| Gas Phase | DMSO | Methanol | Chloroform | Benzene | |||
| 1 | 1a | Bn | 4.10 | 3.91 | 4.80 | 4.59 | 4.33 |
| 2 | 1b | DMAS | 3.83 | 3.73 | 3.48 | 2.71 | 2.88 |
| 3 | 1c | SEM | 5.15 | 3.54 | 4.38 | 4.26 | 4.30 |
| 4 | 9 | Bn (amide) | 0.77 | −0.04 | 0.30 | 0.56 | 0.58 |
| 5 | 13a | Bn (ester) | 0.96 | −0.85 | −0.36 | −0.59 | −0.52 |
| 6 | 17 | Bn (hydroxamate) | 1.24 | −0.31 | −0.01 | 0.31 | 0.56 |
| Entry | Compound | Protecting Group | Rearomatization Stabilization Energy (kcal/mol) | ||||
|---|---|---|---|---|---|---|---|
| Gas Phase | DMSO | Methanol | Chloroform | Benzene | |||
| 1 | 2a | Bn | −22.88 | −21.68 | −20.93 | −20.90 | −21.34 |
| 2 | 2b | DMAS | −17.48 | −15.89 | −15.28 | −15.95 | −16.15 |
| 3 | 2c | SEM | −22.00 | −20.54 | −19.09 | −19.66 | −19.92 |
| 4 | 10 | Bn (amide) | −24.76 | −25.06 | −24.84 | −25.07 | −25.43 |
| 5 | Bn (ester) | −24.48 | −24.76 | −24.29 | −24.62 | −25.07 | |
| 6 | 18 | Bn (hydroxamate) | −24.76 | −23.56 | −23.43 | −23.59 | −23.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fulton, B.B.; Hartzell, A.J.; Dias, H.V.R.; Lovely, C.J. Room Temperature Diels–Alder Reactions of 4-Vinylimidazoles. Molecules 2024, 29, 1902. https://doi.org/10.3390/molecules29081902
Fulton BB, Hartzell AJ, Dias HVR, Lovely CJ. Room Temperature Diels–Alder Reactions of 4-Vinylimidazoles. Molecules. 2024; 29(8):1902. https://doi.org/10.3390/molecules29081902
Chicago/Turabian StyleFulton, Brandon B., Alexia J. Hartzell, H. V. Rasika Dias, and Carl J. Lovely. 2024. "Room Temperature Diels–Alder Reactions of 4-Vinylimidazoles" Molecules 29, no. 8: 1902. https://doi.org/10.3390/molecules29081902
APA StyleFulton, B. B., Hartzell, A. J., Dias, H. V. R., & Lovely, C. J. (2024). Room Temperature Diels–Alder Reactions of 4-Vinylimidazoles. Molecules, 29(8), 1902. https://doi.org/10.3390/molecules29081902