
Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds




{kind=link}
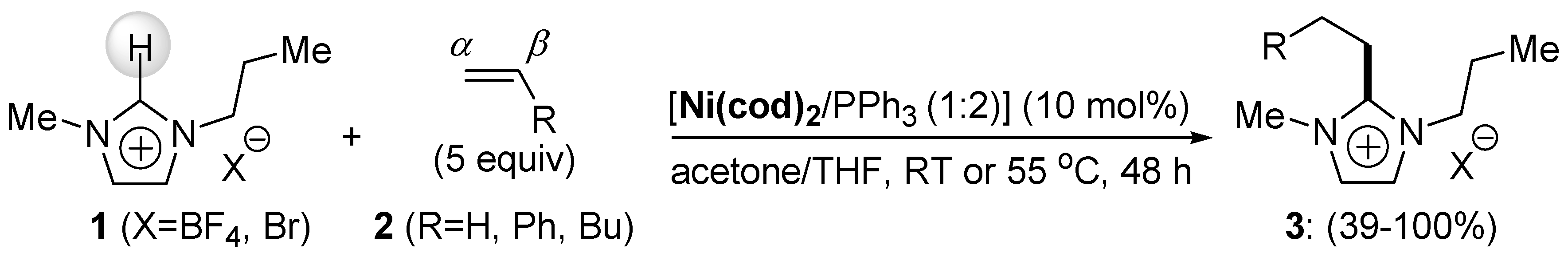{kind=link}
{kind=link}
{kind=link}
{kind=link}
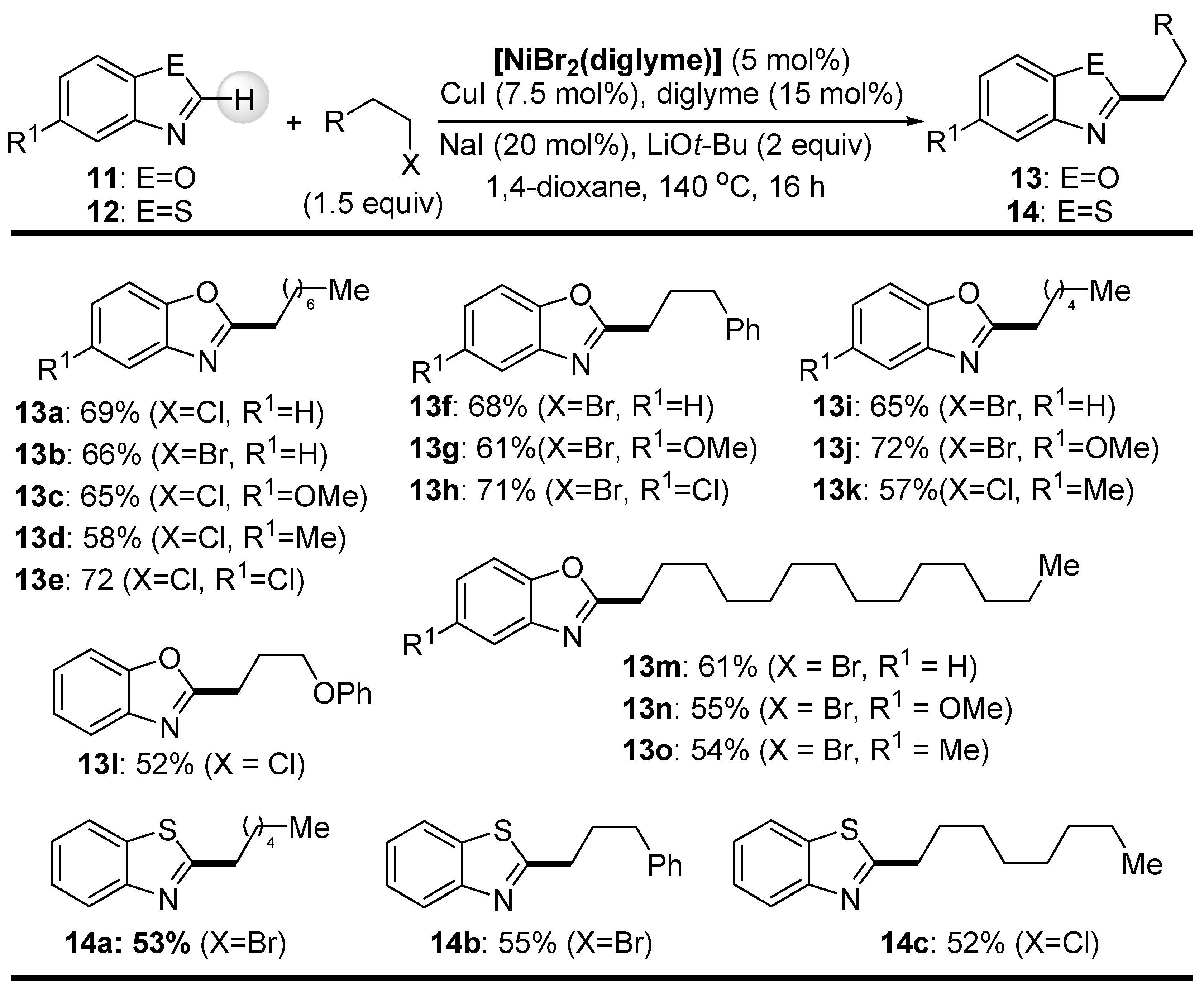{kind=link}
{kind=link}
{kind=link}
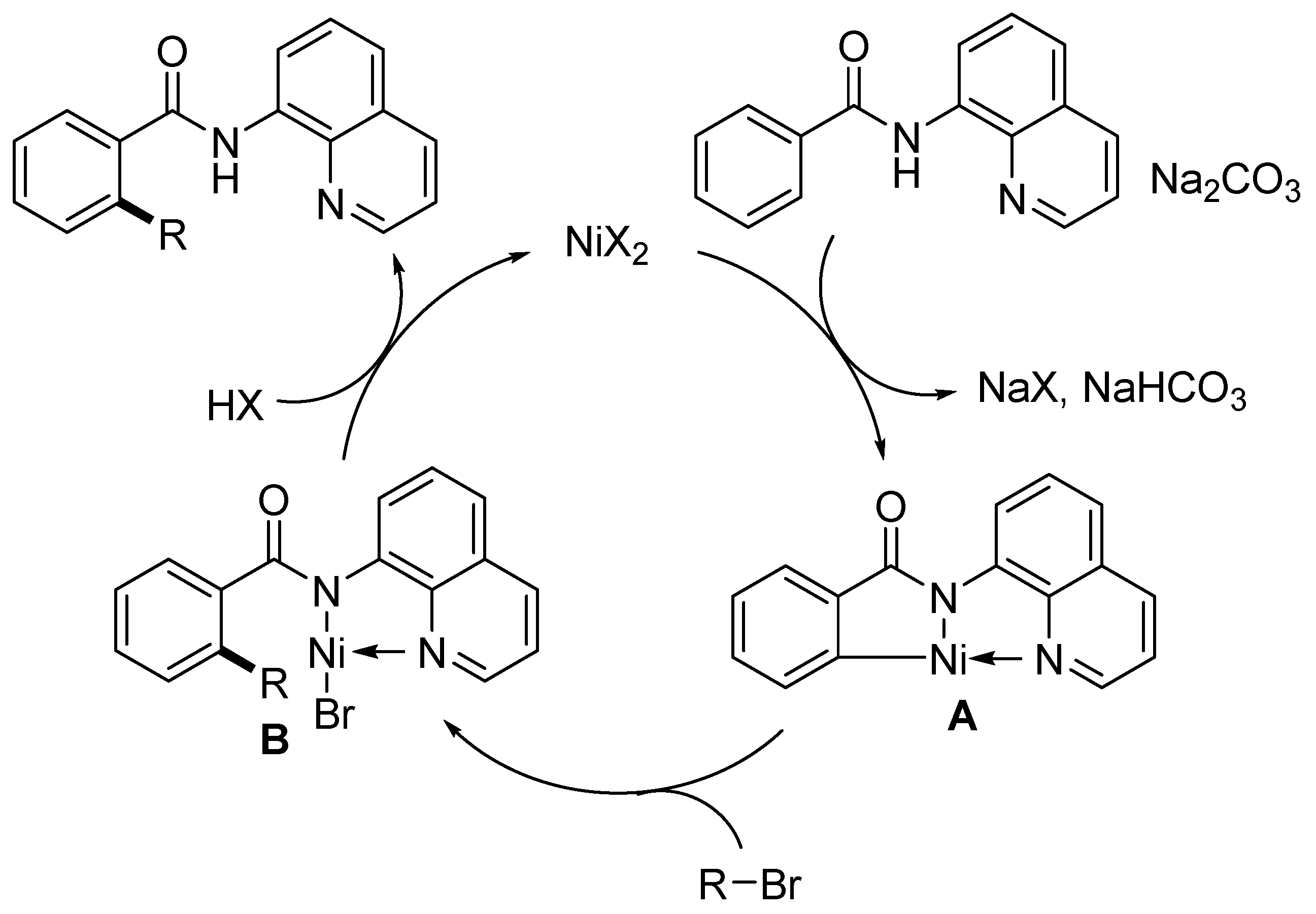{kind=link}
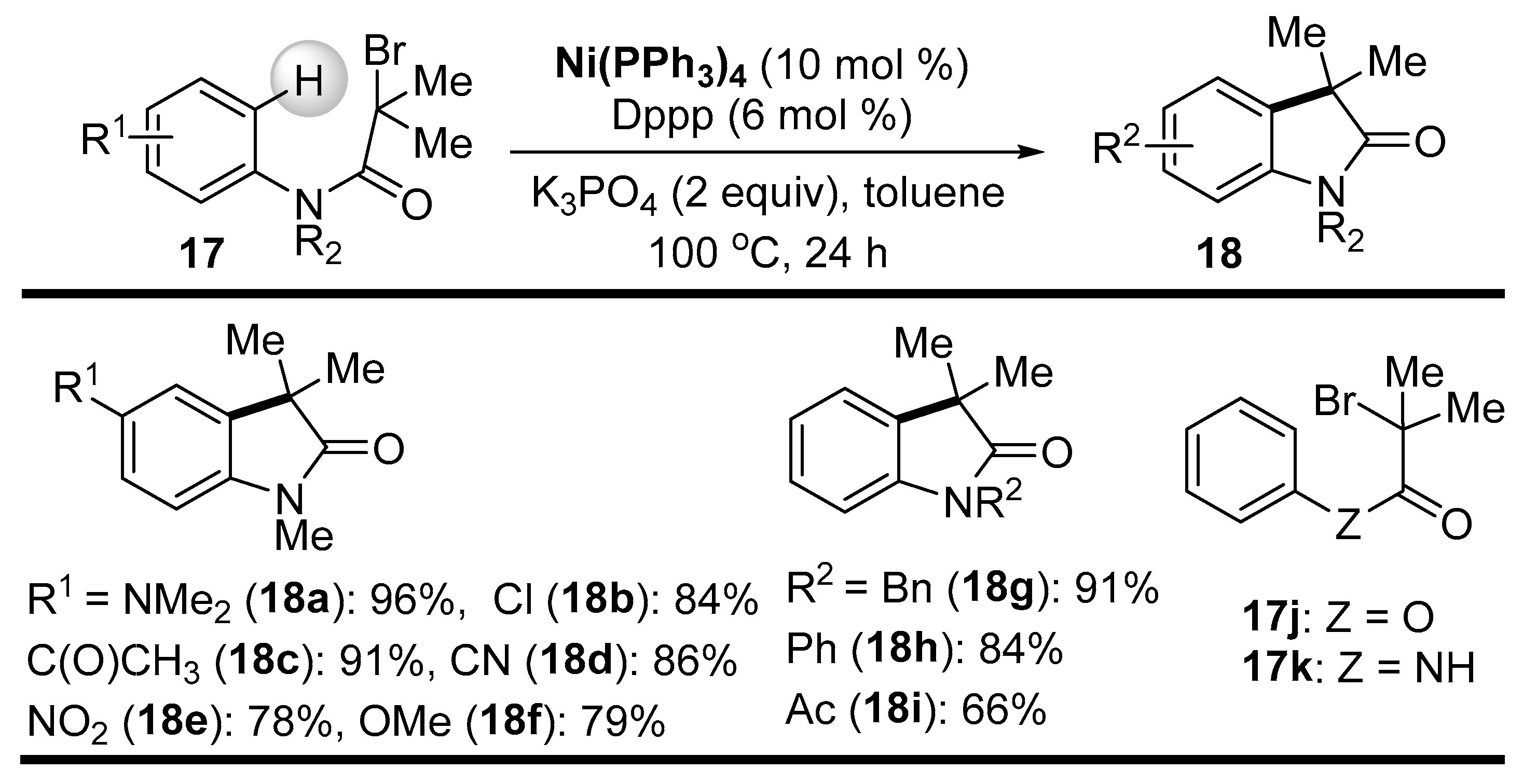{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
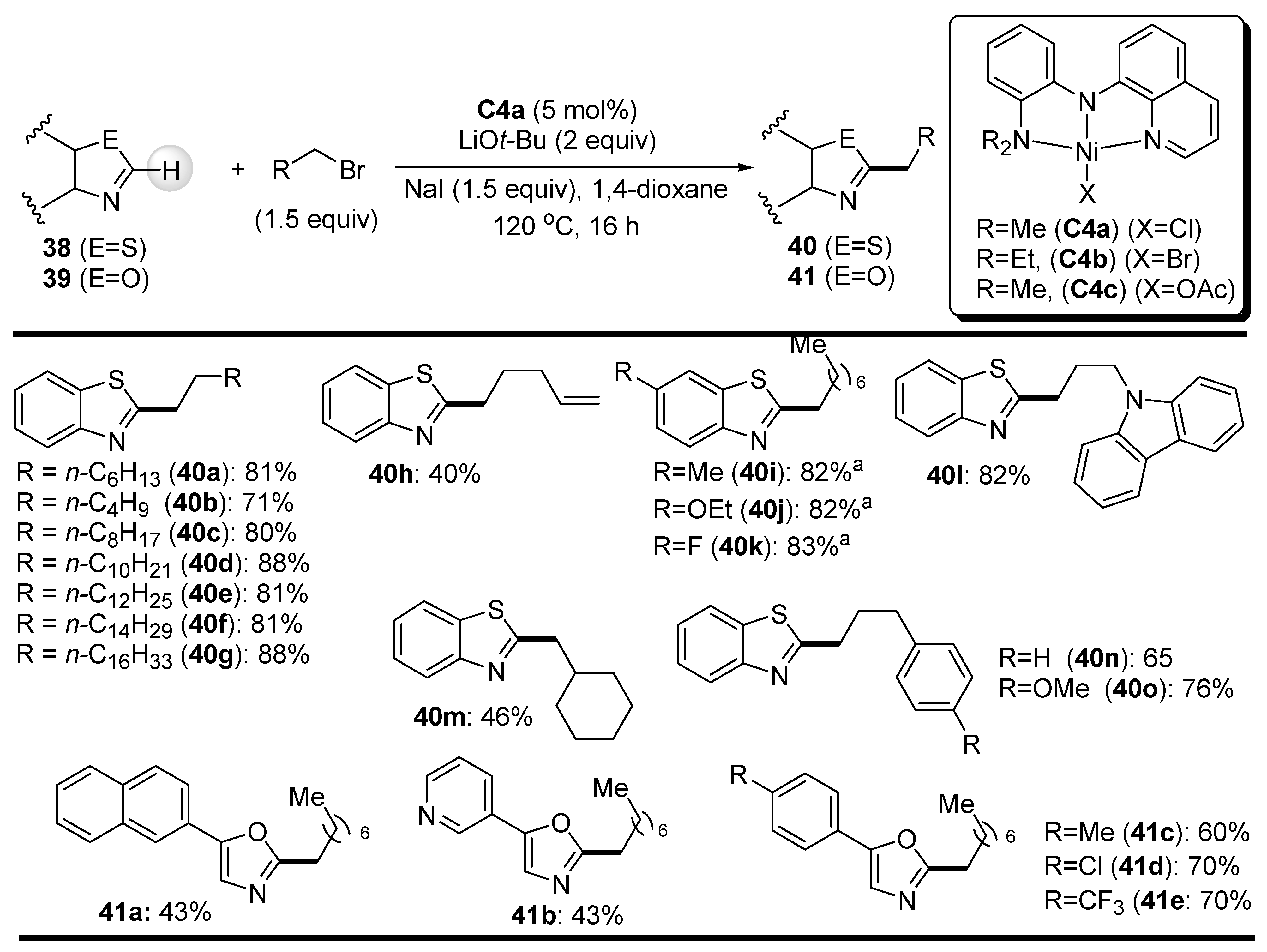{kind=link}
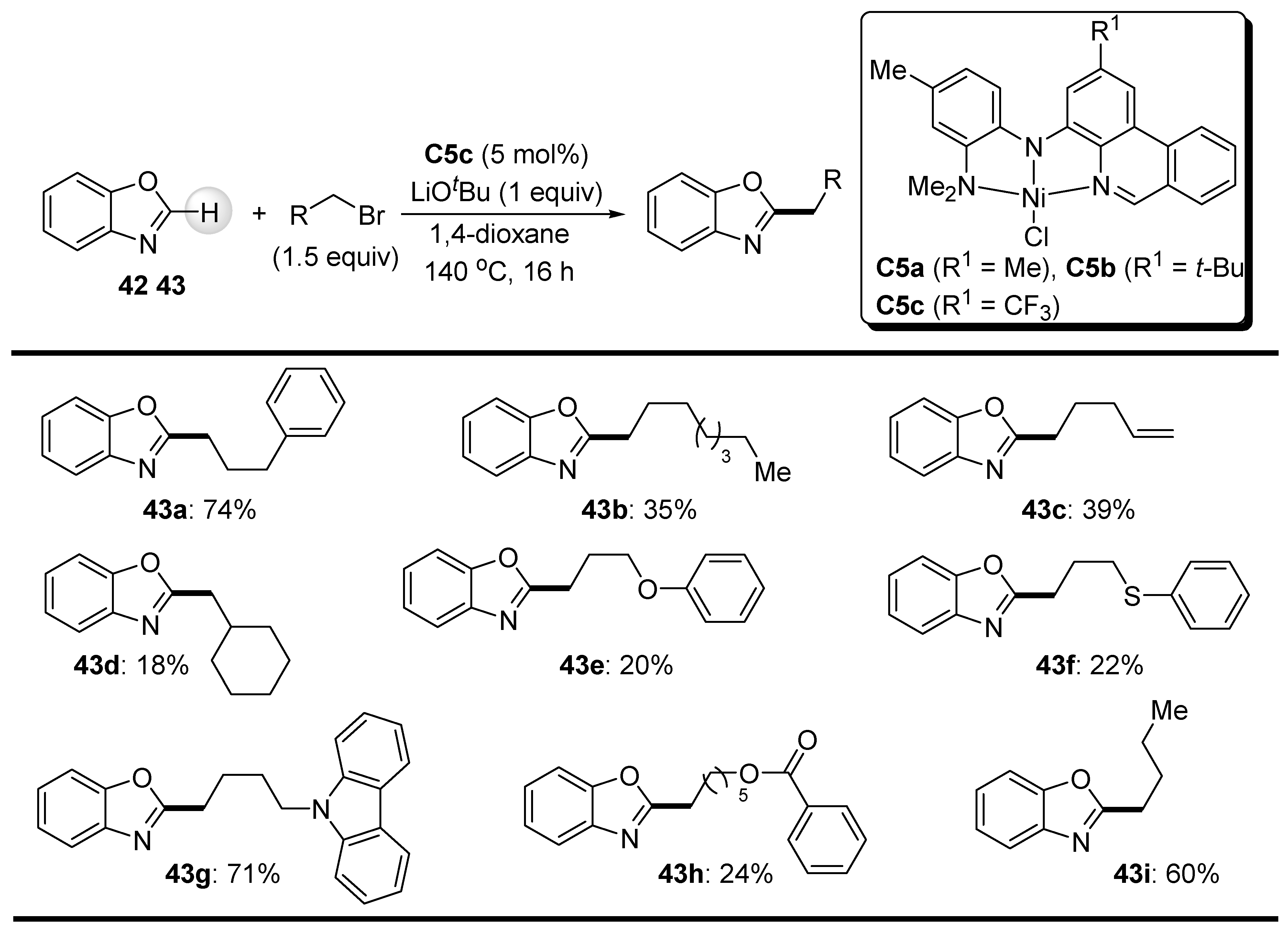{kind=link}
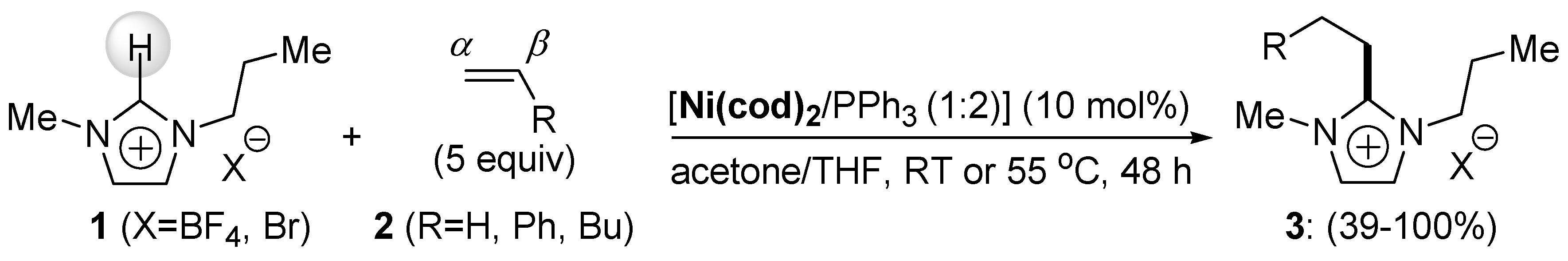
{kind=link}
{kind=link}
{kind=link}
{kind=link}
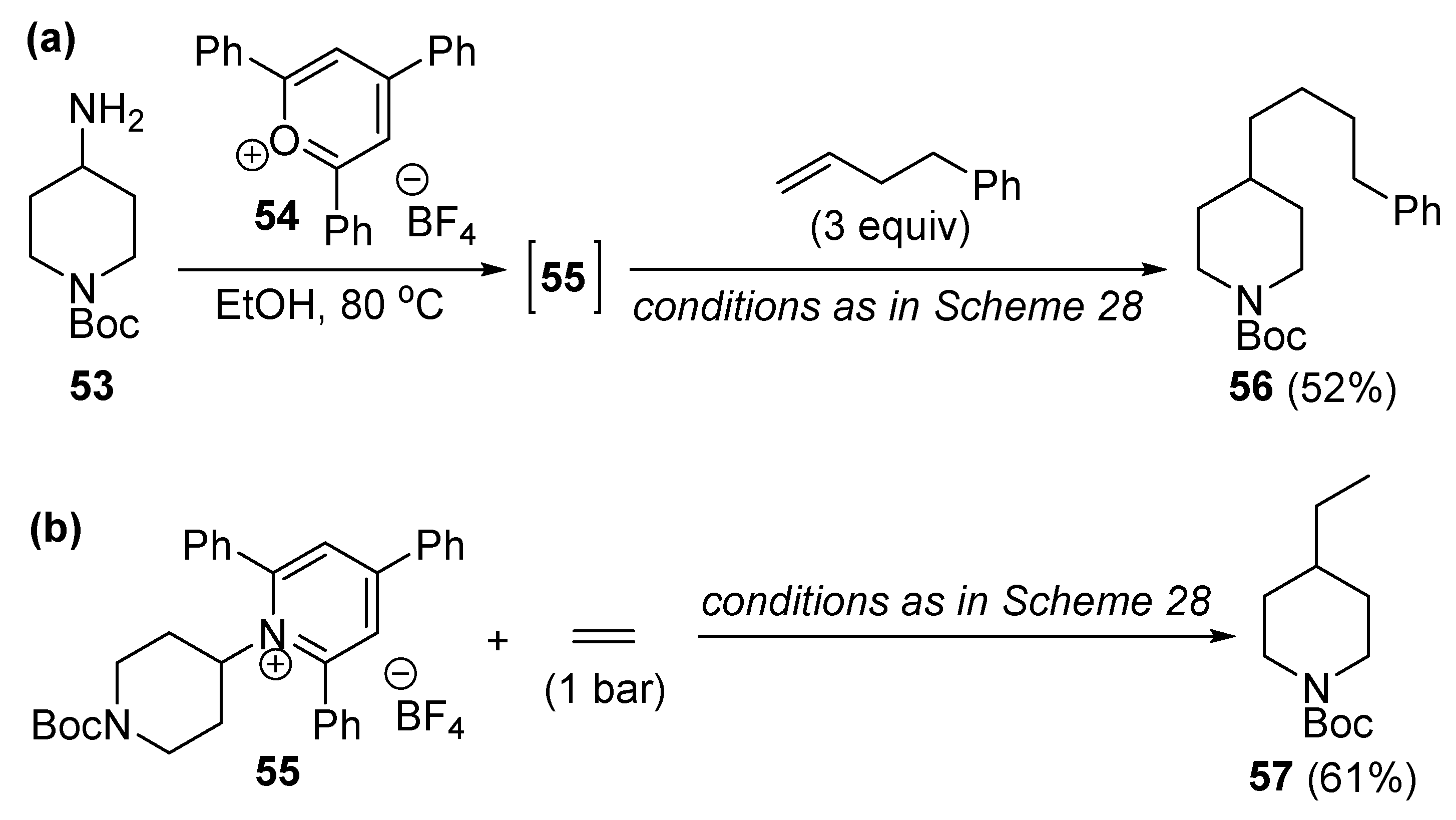{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discussion
2.1. C-H Bond Alkylation with Alkenes
2.2. C-H Bond Alkylation with Alkyl Halides
2.3. Miscellaneous C-H Bond Alkylations
2.4. Asymmetric C-H Bond Alkylation
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ac | Acyl |
| Ar | Aryl |
| Bn | Benzyl |
| Boc | tert-Butyloxycarbonyl |
| Bpy | 2,2-Bipyridine |
| Bu | Butyl |
| Cbz | Benzyloxycarbonyl |
| Cod | 1,5-Cyclooctadiene |
| Cp | Cyclopentadienyl |
| Cy | Cyclohexyl |
| Cyp | Cyclopentyl |
| DCM | Dichloromethane |
| diglyme | bis(2-methoxyethyl) ether |
| DMA | Dimethylacetamide |
| DME | Dimethoxyethane |
| DMSO | Dimethylsulfoxide |
| dppp | 1,3-Bis(diphenylphosphino)propane |
| DtBEDA | N1,N2-di-tert-butylethane-1,2-diamine |
| ee | enantiomeric excess |
| equiv | Equivalent |
| Et | Ethyl |
| glyme | Dimethoxyethane |
| Hept | Heptyl |
| Hex | Hexyl |
| HMDS | Hexamethyldisilazide |
| iPr | iso-Propyl |
| L | Ligand |
| LED | Light-emitting diode |
| Me | Methyl |
| MTBE | Methyl tert-butyl ether |
| NHC | N-Heterocyclic carbine |
| NMP | N-Methyl-2-pyrrolidone |
| Ph | Phenyl |
| Pr | Propyl |
| RT | Room Temperature |
| TBME | tert-Butyl methyl ether |
| TBS | tert-Butyldimethylsilyl |
| Tf | Triflyl |
| THF | Tetrahydrofuran |
| TM | Transition meta |
| TMS | Trimethylsilyl |
| Xantphos | (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) |
| Xylyl | Dimethylphenyl |
| μW | Microwave |
References
- Shilov, A.E.; Shul’pin, G.B. Activation of C-H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, F.; Chatani, N. Catalytic Methods for C-H Bond Functionalization: Application in Organic Synthesis. Adv. Synth. Catal. 2003, 345, 1077–1101. [Google Scholar] [CrossRef]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C-H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Das, A.; Chatani, N. Strategic Evolution in Transition Metal-Catalyzed Directed C-H Bond Activation and Future Directions. Coord. Chem. Rev. 2021, 431, 213683–213719. [Google Scholar] [CrossRef]
- Al Mamari, H.H.; Štefane, B.; Žugelj, H.B. Metal-Catalyzed C-H Bond Functionalization of Phenol Derivatives. Tetrahedron 2020, 76, 130925–130936. [Google Scholar] [CrossRef]
- Docherty, J.H.; Lister, T.M.; Mcarthur, G.; Findlay, M.T.; Domingo-Legarda, P.; Jacob Kenyon, J.; Choudhary, S.; Igor Larrosa, I. Transition-Metal-Catalyzed C-H Bond Activation for the Formation of C-C Bonds in Complex Molecules. Chem. Rev. 2023, 123, 7692–7760. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.; Faber, T.; Glorius, F. C-H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 2, 245–261. [Google Scholar] [CrossRef]
- Omae, I. Intramolecular five-membered ring compounds and their applications. Coord. Chem. Rev. 2004, 248, 995–1023. [Google Scholar] [CrossRef]
- Rouquet, G.N.; Chatani, N. Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnio, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A Comprehensive Overview of Directing Groups Applied in Metal-Catalyzed C-H Functionalization Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef]
- de Meijere, A.; Bräse, S.; Oestreich, M. (Eds.) Metal Catalyzed Cross-Coupling Reactions and More; Wiley: Hoboken, NJ, USA, 2013; Volume 3. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Del Zotto, A.; Marco Zecca, M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef]
- Campeau, L.-C.; Hazari, N. Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 2019, 38, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium-Catalyzed Transformations of Alkyl C-H Bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. Controlling Site Selectivity in Palladium-Catalyzed C-H Bond Functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef]
- Bay, K.L.; Yang, Y.-F.; Houk, K.N. Multiple Roles of Silver Salts in Palladium-Catalyzed C-H Activations. J. Organomet. Chem. 2018, 864, 19–25. [Google Scholar] [CrossRef]
- Colby, D.A.; Tsai, A.S.; Bergman, R.G.; Ellman, J.A. Rhodium Catalyzed Chelation-Assisted C-H Bond Functionalization Reactions. Acc. Chem. Res. 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Chatani, N. Rhodium-Catalyzed C(sp2)- or C(sp3)–H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 8304–8329, Erratum in Angew. Chem. 2019, 131, 8390–8416. [Google Scholar] [CrossRef]
- Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Direct Functionalization of Nitrogen Heterocycles via Rh-Catalyzed C-H Bond Activation. Acc. Chem. Res. 2008, 41, 1013–1025. [Google Scholar] [CrossRef]
- Vasquez-Cespedes, S.; Wang, X.; Glorius, F. Plausible Rh(V) Intermediates in Catalytic C-H Activation Reactions. ACS Catal. 2018, 8, 242–257. [Google Scholar] [CrossRef]
- Song, G.; Li, X. Substrate Activation Strategies in Rhodium(III)-Catalyzed Selective Functionalization of Arenes. Acc. Chem. Res. 2015, 48, 1007–1020. [Google Scholar] [CrossRef]
- Singh, K.S. Recent Advances in C-H Bond Functionalization with Ruthenium-Based Catalysts. Catalysts 2019, 9, 173. [Google Scholar] [CrossRef]
- Dana, S.; Yadav, M.R.; Sahon, A.K. Ruthenium-Catalyzed C-N and C–O Bond-Forming Processes from C-H Bond Functionalization. Top. Organomet. Chem. 2016, 55, 189–215. [Google Scholar] [CrossRef]
- Ruiz, S.; Villuendas, P.; Urriolabeitia, E.P. Ru-catalyzed C-H functionalizations as a tool for selective organic synthesis. Tetrahedron Lett. 2016, 57, 3413–3432. [Google Scholar] [CrossRef]
- Li, B.; Dixneuf, P.H. Ruthenium(II)-Catalyzed sp2 C-H Bond Functionalization by C-C Bond Formation. Top. Organomet. Chem. 2015, 48, 119–193. [Google Scholar] [CrossRef]
- Ackermann, L.; Vicente, R. Ruthenium-Catalyzed Direct Arylations through C-H Bond Cleavages. Top. Curr. Chem. 2010, 292, 211–229. [Google Scholar] [CrossRef]
- Dixneuf, P.H.; Cadierno, V. (Eds.) Metal-Catalyzed C-H Bond Activation and C-C Bond Formation in Water; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Li, B.; Dixneuf, P.H. sp2 C-H bond activation in water and catalytic crosscoupling reactions. Chem. Soc. Rev. 2013, 42, 5744–5767. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Chelation-Assisted Nickel-Catalyzed C-H Functionalizations. Trends Chem. 2019, 1, 524–539. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Nickel-Catalyzed C-H Functionalization Using A Non-directed Strategy. Chem 2020, 6, 1056–1081. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Xia, Y.-N.; Shi, B.-F. Ni-Catalyzed Chelation-Assisted Direct Functionalization of Inert C-H Bonds. Chin. J. Chem. 2020, 38, 635–662. [Google Scholar] [CrossRef]
- Cano, R.; Mackey, K.; McGlacken, G.P. Recent Advances in Manganese-Catalyzed C-H Activation: Scope and Mechanism. Catal. Sci. Technol. 2018, 8, 1251–1266. [Google Scholar] [CrossRef]
- Liu, W.; Ackermann, L. Manganese-Catalyzed C-H Activation. ACS Catal. 2016, 6, 3743–3752. [Google Scholar] [CrossRef]
- Lanzi, M.; Cera, G. Iron-Catalyzed C-H Functionalizations under Triazole-Assistance. Molecules 2020, 25, 1806. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C-H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Prakash, S.; Kuppusamy, R.; Cheng, C.H. Cobalt-Catalyzed Annulation Reactions via C-H Bond Activation. ChemCatChem 2018, 10, 683–705. [Google Scholar] [CrossRef]
- Wang, S.; Chen, S.-Y.; Yu, X.-Q. C-H Functionalization by High-Valent Cp*Co(iii) Catalysis. Chem. Commun. 2017, 53, 3165–3180. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Matsunaga, S. High-Valent Cobalt-Catalyzed C-H Bond Functionalization. Adv. Organomet. Chem. 2019, 68, 197–247. [Google Scholar] [CrossRef]
- Baccalini, A.; Vergura, S.; Dolui, P.; Zanoni, G.; Maiti, D. Recent Advances in Cobalt-Catalyzed C-H Functionalizations. Org. Biomol. Chem. 2019, 17, 10119–10141. [Google Scholar] [CrossRef]
- Moselage, M.; Lie, J.; Ackermann, L. Cobalt-Catalyzed C-H Activation. ACS Catal. 2016, 6, 498–525. [Google Scholar] [CrossRef]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Erratum: Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef]
- Ge, S.; Hartwig, J.F. Highly Reactive, Single-Component Nickel Catalyst Precursor for Suzuki–Miyuara Cross-Coupling of Heteroaryl Boronic Acids with Heteroaryl Halides. Angew. Chem. Int. Ed. 2012, 51, 12837–12841. [Google Scholar] [CrossRef]
- Ramgren, S.D.; Hie, L.; Ye, Y.; Garg, N.K. Nickel-Catalyzed Suzuki–Miyaura Couplings in Green Solvents. Org. Lett. 2013, 15, 3950–3953. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Kleiman, J.P.; Dubeck, M. The Preparation of Cyclopentadienyl [o-(Phenylazo)Phenyl]Nickel. J. Am. Chem. Soc. 1963, 85, 1544–1545. [Google Scholar] [CrossRef]
- Jagtap, R.A.; Punji, B. Nickel-Catalyzed C H Bond Functionalization of Azoles and Indoles. Chem. Rec. 2021, 21, 3573. [Google Scholar] [CrossRef] [PubMed]
- Harry, N.A.; Saranya, S.; Ujwaldev, S.M.; Anilkumar, G. Recent advances and prospects in nickel-catalyzed C-H activation. Catal. Sci. Technol. 2019, 9, 1726–1743. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Muto, K.; Itami, K. Recent Progress in Nickel-Catalyzed Biaryl Coupling. Eur. J. Org. Chem. 2013, 2013, 19–30. [Google Scholar] [CrossRef]
- Hu, F.; Shen, Y.-B.; Wang, L.; Li, S.-S. Merging dearomatization with redox-neutral C(sp3)–H functionalization via hydride transfer/cyclization: Recent advances and perspectives. Org. Chem. Front. 2022, 9, 5041–5052. [Google Scholar] [CrossRef]
- Dong, Y.; Hu, F.; Wu, H.; Guo, F.-W.; Wang, L.; Du, F.-Y.; Li, S.-S. Controllable Synthesis of N-Heterocycles via Hydride Transfer Strategy-Enabled Formal [5 + 1] and [5 + 2] Cyclizations. Org. Lett. 2024, 26, 332–337. [Google Scholar] [CrossRef]
- Guo, W.; Wang, Q.; Zhu, J. Visible light photoredox-catalysed remote C-H functionalisation enabled by 1,5-hydrogen atom transfer (1,5-HAT). Chem. Soc. Rev. 2021, 50, 7359–7377. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, Z.; Hu, Q.; Elsaid, M.; Liu, C.; Chen, J.; Ge, H. Recent Strategies in Nickel-Catalyzed C-H Bond Functionalization for Nitrogen-Containing Heterocycles. Catalysts 2022, 12, 1163. [Google Scholar] [CrossRef]
- Clement, N.D.; Cavell, K.J. Transition-Metal-Catalyzed Reactions Involving Imidazolium Salt/N-Heterocyclic Carbene Couples as Substrates. Angew. Chem. Int. Ed. 2004, 43, 3845–3847. [Google Scholar] [CrossRef] [PubMed]
- Nakao, Y.; Kashihara, N.; Kanyiva, K.S.; Hiyama, T. Nickel-catalyzed alkenylation and alkylation of fluoroarenes via activation of C-H bond over C-F bond. J. Am. Chem. Soc. 2008, 130, 16170–16171. [Google Scholar] [CrossRef] [PubMed]
- Keen, A.L.; Johnson, S.A. Nickel(0)-Catalyzed Isomerization of an Aryne Complex: Formation of a Dinuclear Ni(I) Complex via C-H Rather than C-F Bond Activation. J. Am. Chem. Soc. 2006, 128, 1806–1807. [Google Scholar] [CrossRef]
- Keen, A.L.; Doster, M.; Johnson, S.A. 1,4-Shifts in a Dinuclear Ni(I) Biarylyl Complex: A Mechanistic Study of C-H Bond Activation by Monovalent Nickel. J. Am. Chem. Soc. 2007, 129, 810–819. [Google Scholar] [CrossRef]
- Mukai, T.; Hirano, K.; Satoh, T.; Miura, M. Nickel-Catalyzed C-H Alkenylation and Alkylation of 1,3,4-Oxadiazoles with Alkynes and Styrenes. J. Org. Chem. 2009, 74, 6410–6413. [Google Scholar] [CrossRef] [PubMed]
- Tlili, A.; Schranck, J.; Pospech, J.; Neumann, H.; Beller, M. Ruthenium-Catalyzed Hydroaroylation of Styrenes in Water through Directed C-H Bond Activation. ChemCatChem 2014, 6, 1562–1566. [Google Scholar] [CrossRef]
- Thowfik, S.; Afsinab, M.A.; Anilkumar, G. Ruthenium-catalyzed hydroarylation reactions as the strategy towards the synthesis of alkylated arenes and substituted alkenes. RSC Adv. 2023, 13, 6246–6263. [Google Scholar] [CrossRef]
- Vechorkin, O.; Proust, V.; Hu, X. The Nickel/Copper-Catalyzed Direct Alkylation of Heterocyclic C-H Bonds. Angew. Chem. Int. Ed. 2010, 49, 3061–3064. [Google Scholar] [CrossRef]
- Ackermann, L.; Punji, B.; Song, W. User-Friendly [(Diglyme)NiBr2]-Catalyzed Direct Alkylations ofHeteroarenes with Unactivated Alkyl Halides through C-H Bond Cleavages. Adv. Synth. Catal. 2011, 353, 3325–3329. [Google Scholar] [CrossRef]
- Aihara, Y.; Chatani, N. Ruthenium-catalyzed direct arylation of C-H bonds in aromatic amides containing a bidentate directing group: Significant electronic effects on arylation. Chem. Sci. 2013, 4, 664–670. [Google Scholar] [CrossRef]
- Aihara, Y.; Chatani, N. Nickel-Catalyzed Direct Alkylation of C-H Bonds in Benzamides and Acrylamides with Functionalized Alkyl Halides via Bidentate-Chelation Assistance. J. Am. Chem. Soc. 2013, 135, 5308–5311. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L. Metal-catalyzed direct alkylations of (hetero)arenes via C-H bond cleavages with unactivated alkyl halides. Chem. Commun. 2010, 46, 4866–4877. [Google Scholar] [CrossRef] [PubMed]
- Chao Liu, C.; Dong Liu, D.; Wei Zhang, W.; Liangliang Zhou, L.; Lei, A. Nickel-Catalyzed Aromatic C-H Alkylation with Secondary or Tertiary Alkyl–Bromine Bonds for the Construction of Indolones. Org. Lett. 2013, 15, 6166–6169. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Lackner, S.; Ackermann, L. A General Strategy for the Nickel-Catalyzed C-H Alkylation of Anilines. Angew. Chem. Int. Ed. 2016, 55, 3153–3157. [Google Scholar] [CrossRef] [PubMed]
- Vijayarajan Devannah, V.; Watson, D.A. Nickel-Catalyzed C-Alkylation of Nitroalkanes with Unactivated Alkyl Iodides. J. Am. Chem. Soc. 2017, 139, 8110–8113. [Google Scholar] [CrossRef]
- Soni, V.; Jagtap, R.A.; Gonnade, R.G.; Punji, B. Unified Strategy for Nickel-Catalyzed C-2 Alkylation of Indoles through Chelation Assistance. ACS Catal. 2016, 6, 5666–5672. [Google Scholar] [CrossRef]
- Pandey, D.K.; Ankade, S.B.; Ali, A.; Vinod, C.B.; Punji, B. Nickel-catalyzed C-H alkylation of indoles with unactivated alkyl chlorides: Evidence of a Ni(I)/Ni(III) pathway. Chem. Sci. 2019, 10, 9493–9500. [Google Scholar] [CrossRef]
- Patel, U.N.; Pandey, D.K.; Rajesh, G.; Gonnade, R.G.; Punji, B. Synthesis of Quinoline-Based NNN-Pincer Nickel(II) Complexes: A Robust and Improved Catalyst System for C-H Bond Alkylation of Azoles with Alkyl Halides. Organometallics 2016, 35, 1785–1793. [Google Scholar] [CrossRef]
- Mandapati, P.; Braun, J.D.; Sidhu, B.K.; Wilson, G.; Herbert, D.E. Catalytic C-H Bond Alkylation of Azoles with Alkyl Halides Mediated by Nickel(II) Complexes of Phenanthridine-Based N^N–^N Pincer Ligands. Organometallics 2020, 39, 1989–1997. [Google Scholar] [CrossRef]
- Buendia, M.B.; Higginson, B.; Kegnæs, S.; Kramer, S.; Martin, R. Redox-Neutral Ni-Catalyzed sp3 C-H Alkylation of α-Olefins with Unactivated Alkyl Bromides. ACS Catal. 2022, 12, 3815–3820. [Google Scholar] [CrossRef]
- Sun, S.-Z.; Romano, C.; Martin, R. Site-Selective Catalytic Deaminative Alkylation of Unactivated Olefins. J. Am. Chem. Soc. 2019, 141, 16197–16201. [Google Scholar] [CrossRef] [PubMed]
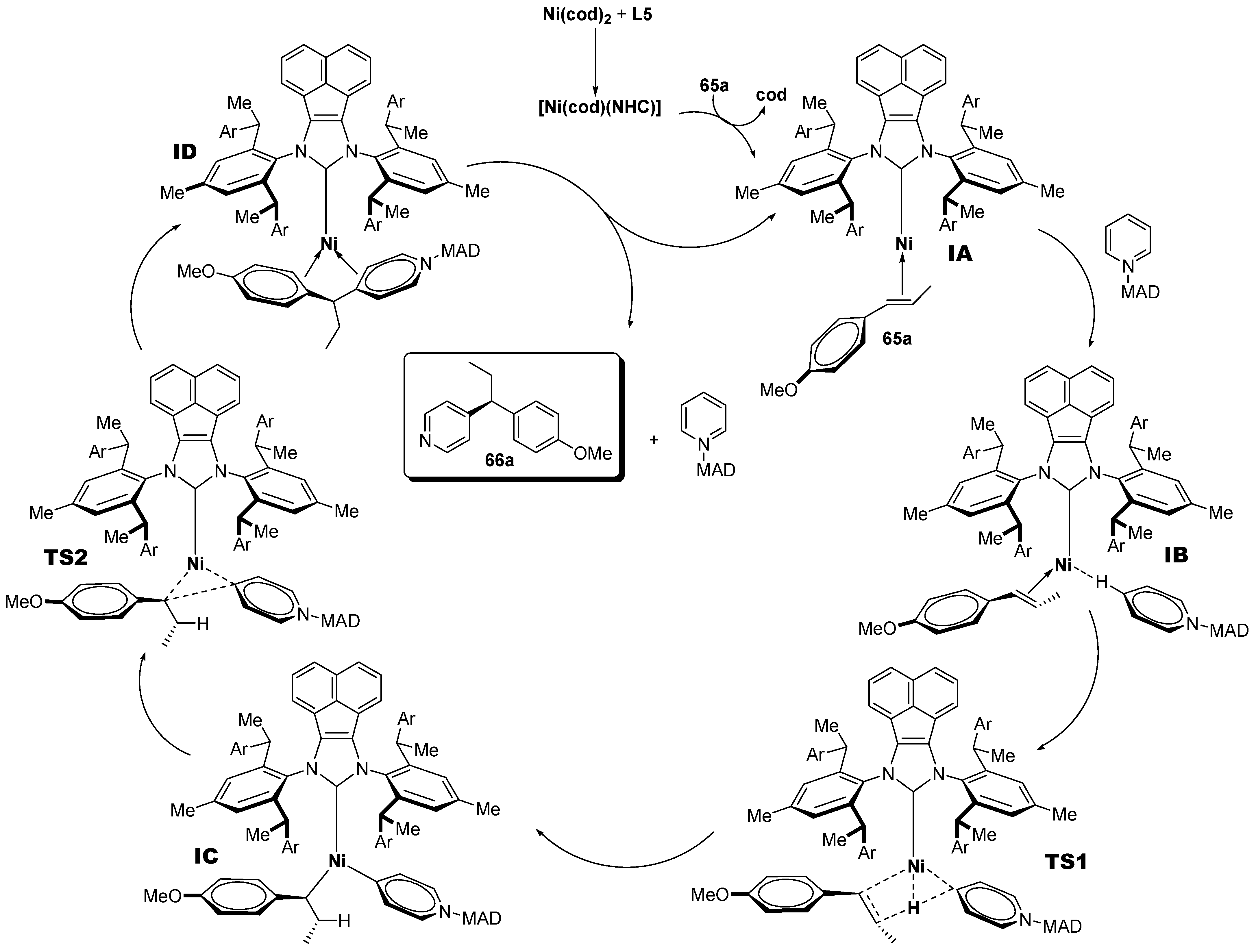
- Ma, J.-B.; Zhao, X.; Zhang, D.; Sh, S.-L. Enantio- and Regioselective Ni-Catalyzed para-C-H Alkylation of Pyridines with Styrenes via Intermolecular Hydroarylation. J. Am. Chem. Soc. 2022, 144, 13643–13651. [Google Scholar] [CrossRef] [PubMed]


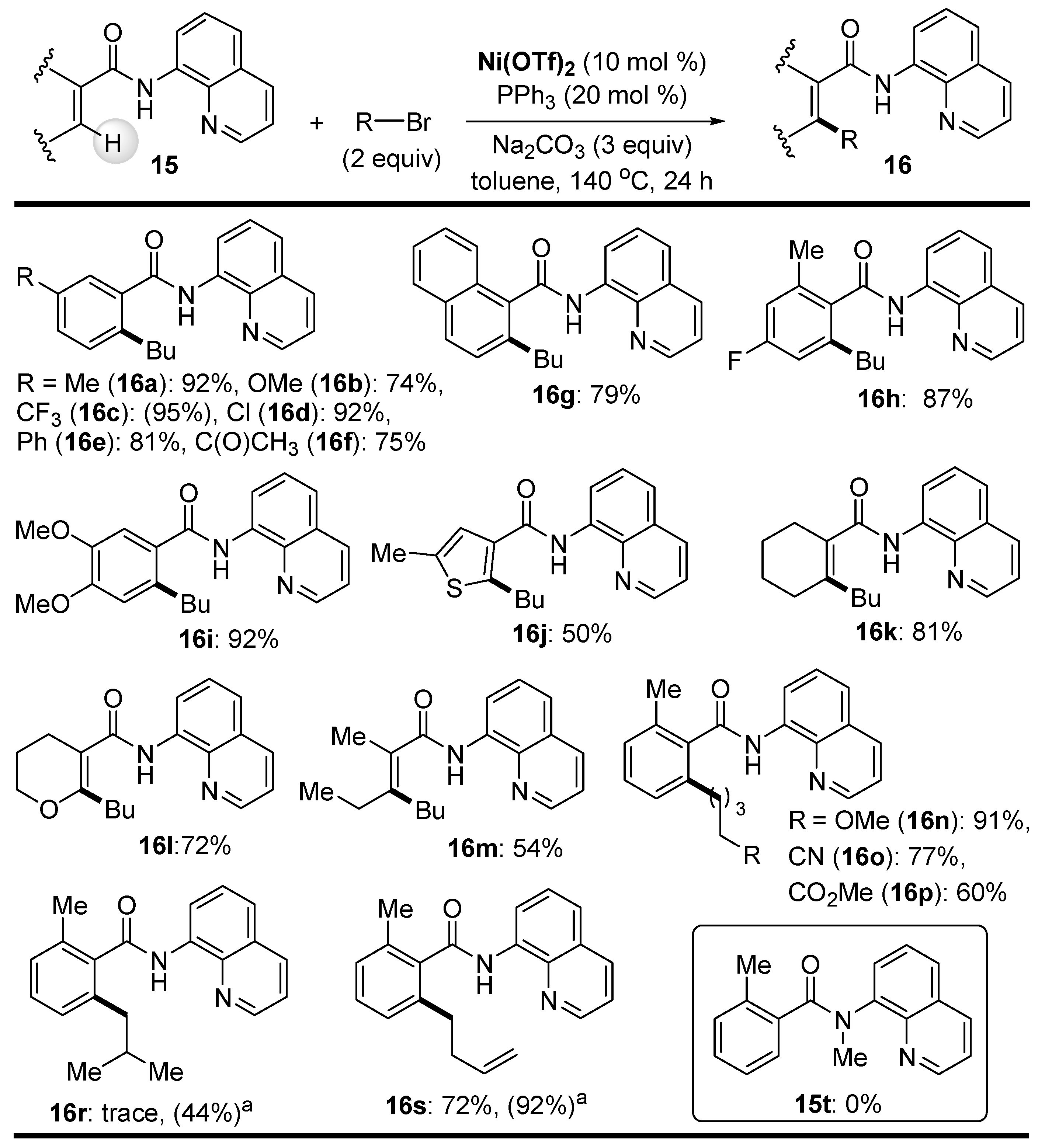
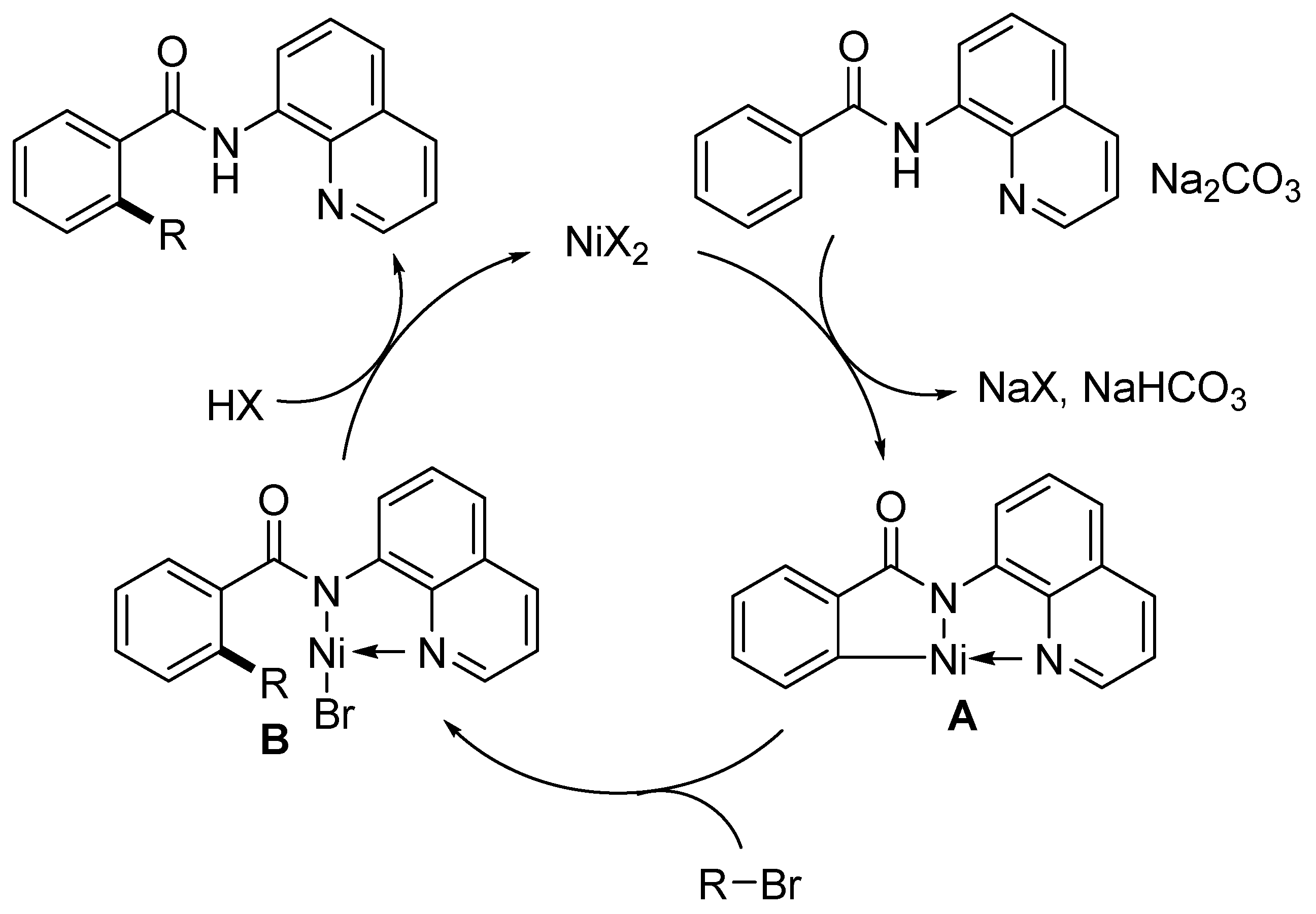
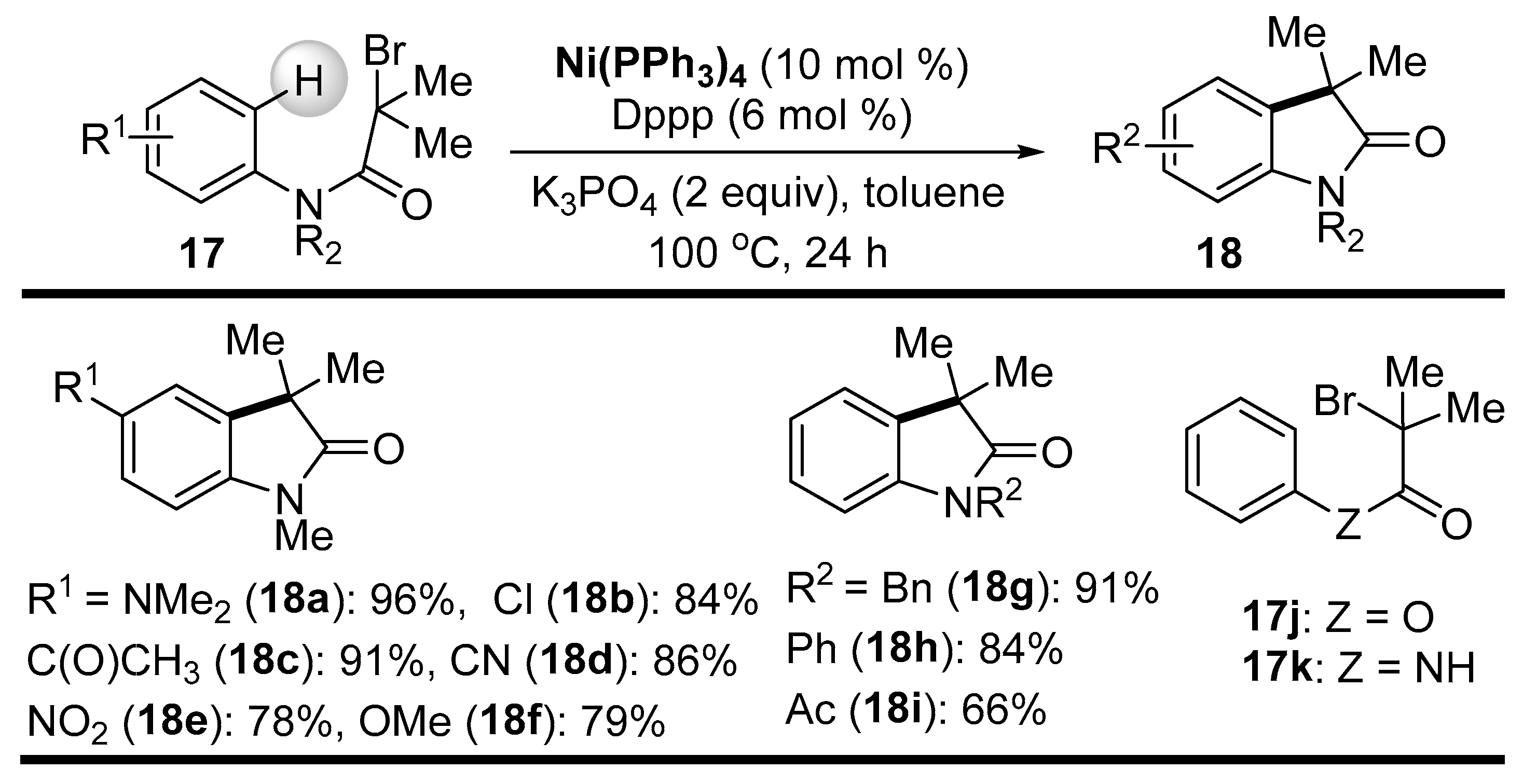
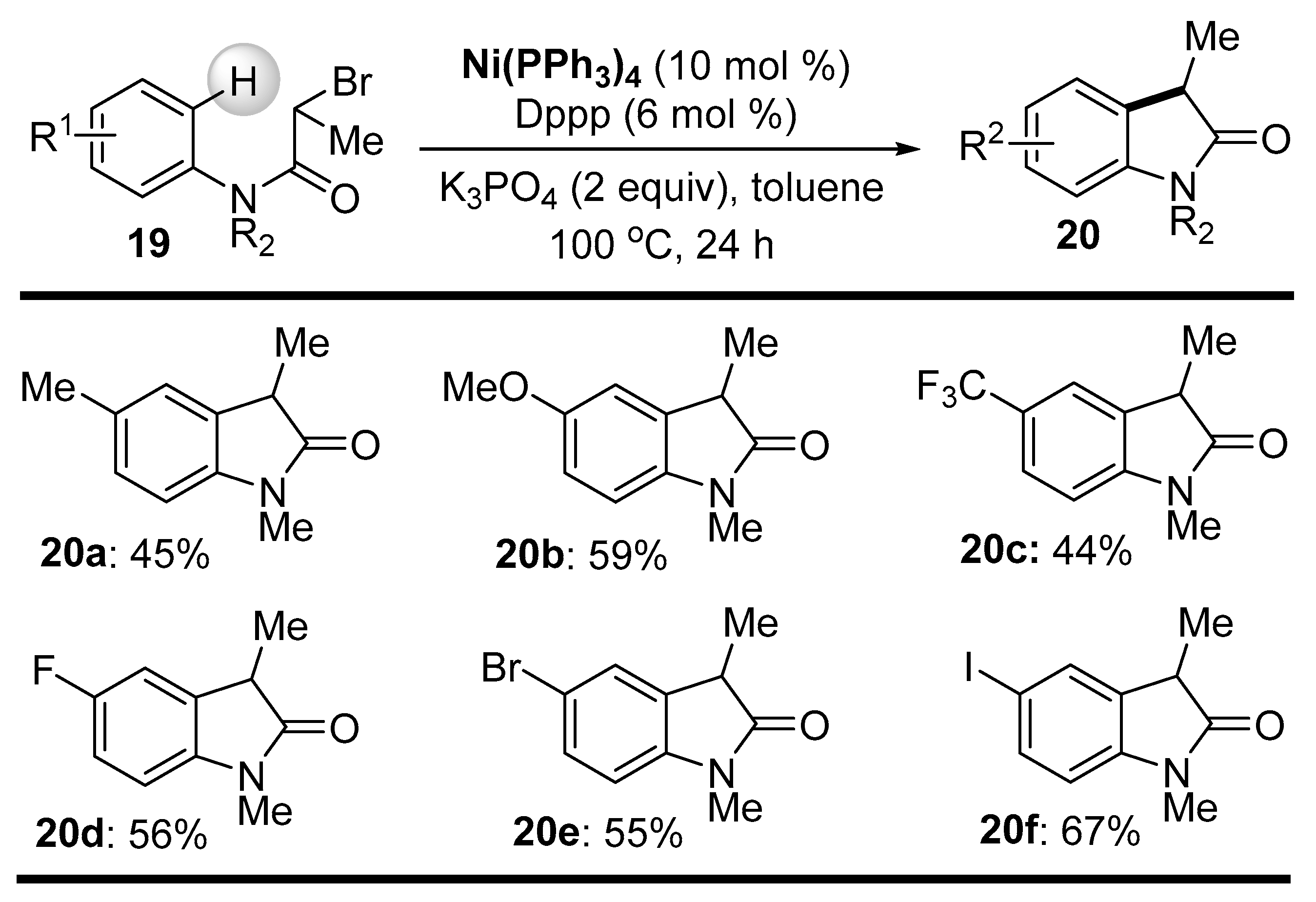









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Požgan, F.; Grošelj, U.; Svete, J.; Štefane, B.; Al Mamari, H.H. Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds. Molecules 2024, 29, 1917. https://doi.org/10.3390/molecules29091917
Požgan F, Grošelj U, Svete J, Štefane B, Al Mamari HH. Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds. Molecules. 2024; 29(9):1917. https://doi.org/10.3390/molecules29091917
Chicago/Turabian StylePožgan, Franc, Uroš Grošelj, Jurij Svete, Bogdan Štefane, and Hamad H. Al Mamari. 2024. "Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds" Molecules 29, no. 9: 1917. https://doi.org/10.3390/molecules29091917
APA StylePožgan, F., Grošelj, U., Svete, J., Štefane, B., & Al Mamari, H. H. (2024). Recent Advances in the Nickel-Catalyzed Alkylation of C-H Bonds. Molecules, 29(9), 1917. https://doi.org/10.3390/molecules29091917