
Gallic Acid Can Promote Low-Density Lipoprotein Uptake in HepG2 Cells via Increasing Low-Density Lipoprotein Receptor Accumulation

 ,
,
Abstract
:
1. Introduction
2. Results
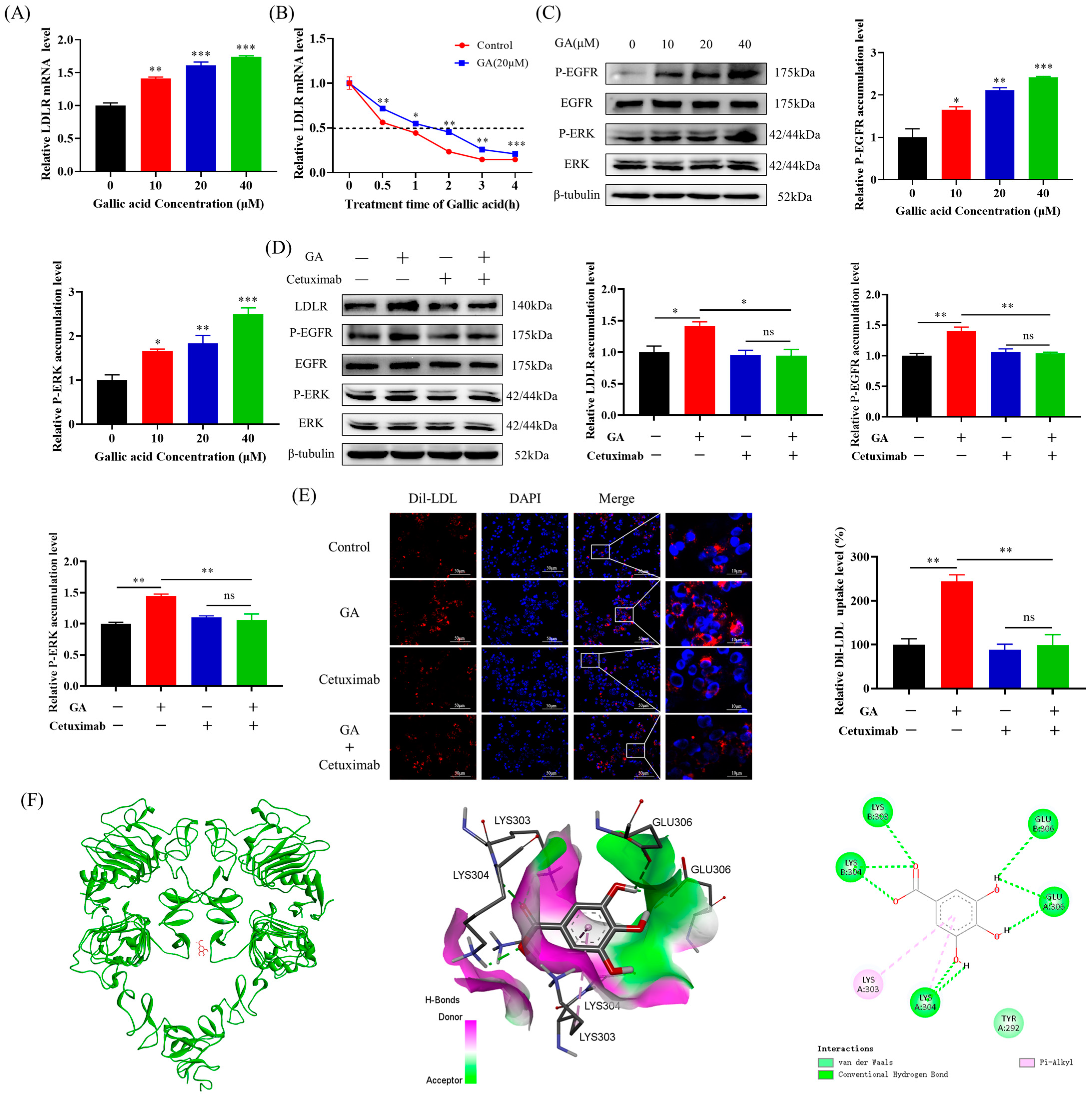
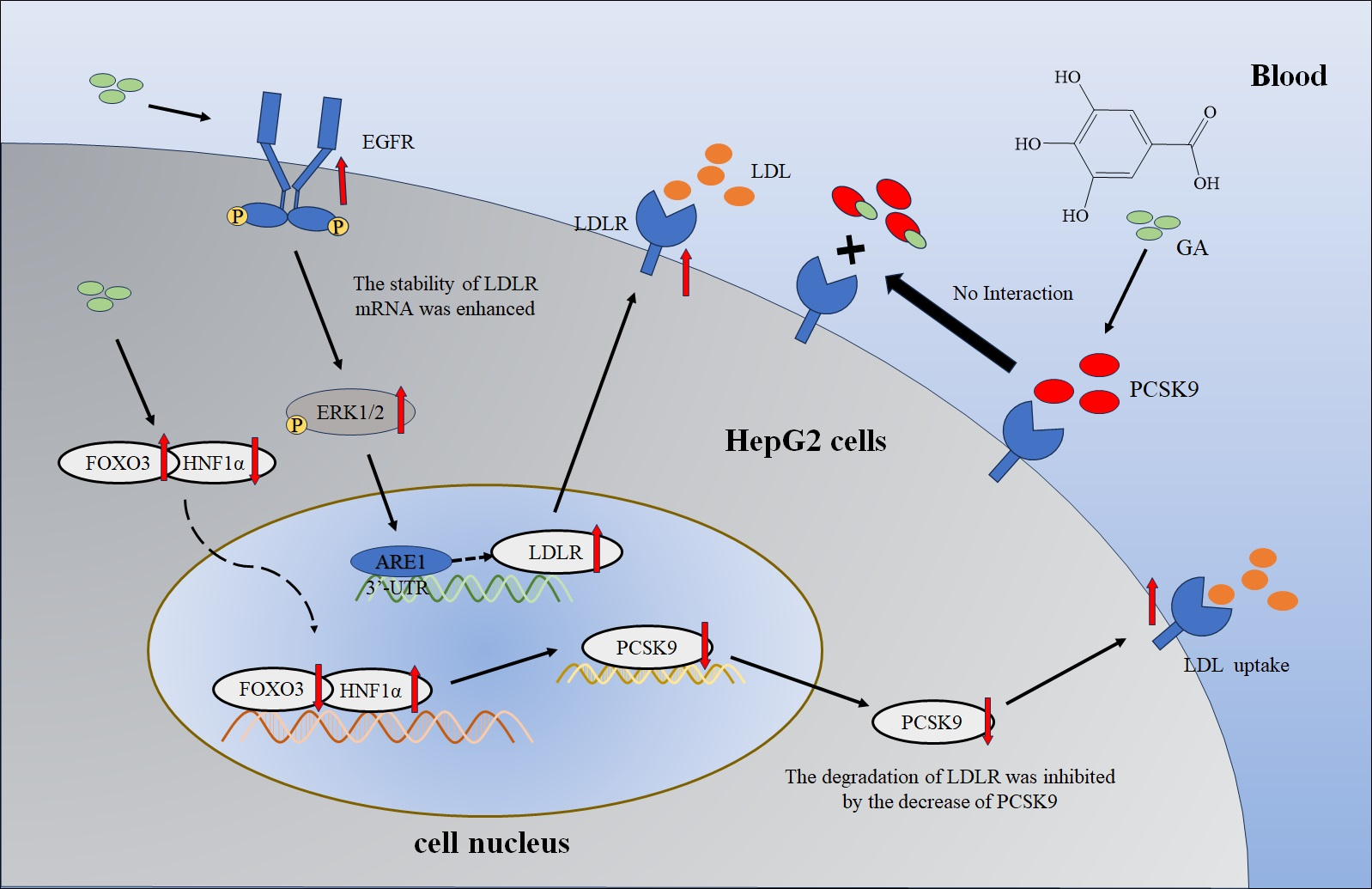
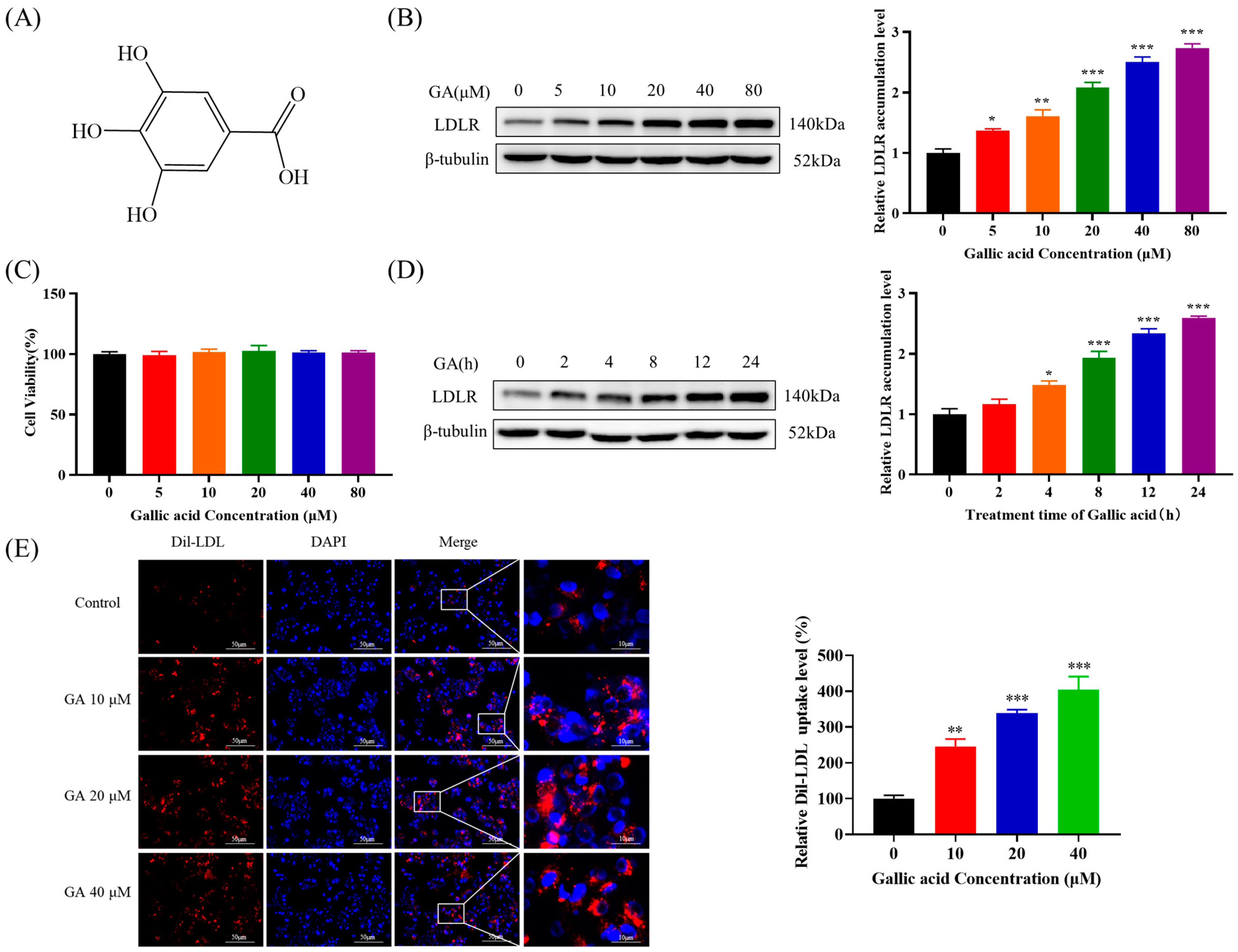
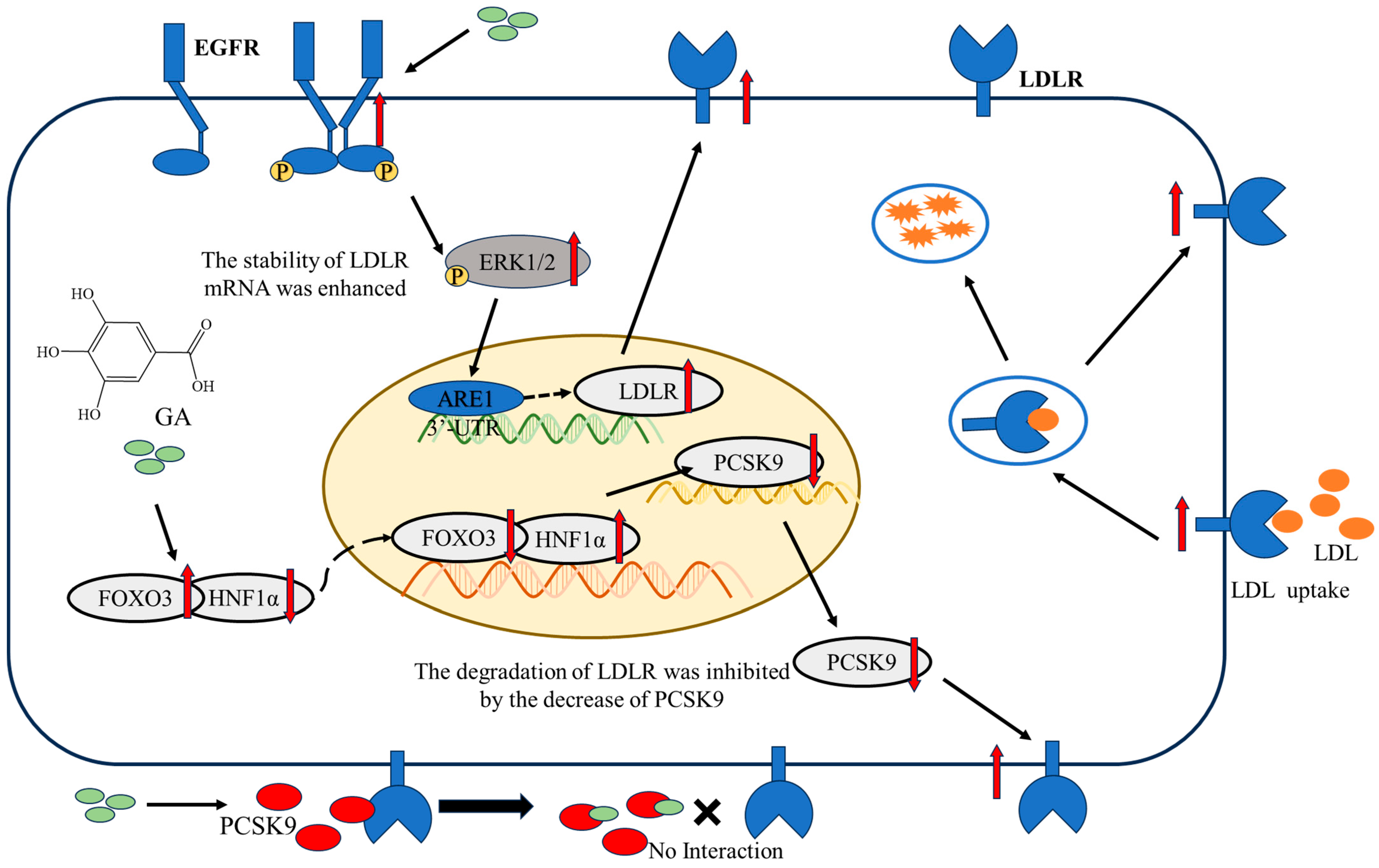
2.1. The Accumulation of LDLR Was Increased by GA, Leading to an Enhancement in the Uptake of LDL in HepG2 Cells
2.2. The EGFR-ERK1/2 Signaling Pathway Was Activated by GA, Leading to an Enhancement of LDLR Accumulation in HepG2 Cells
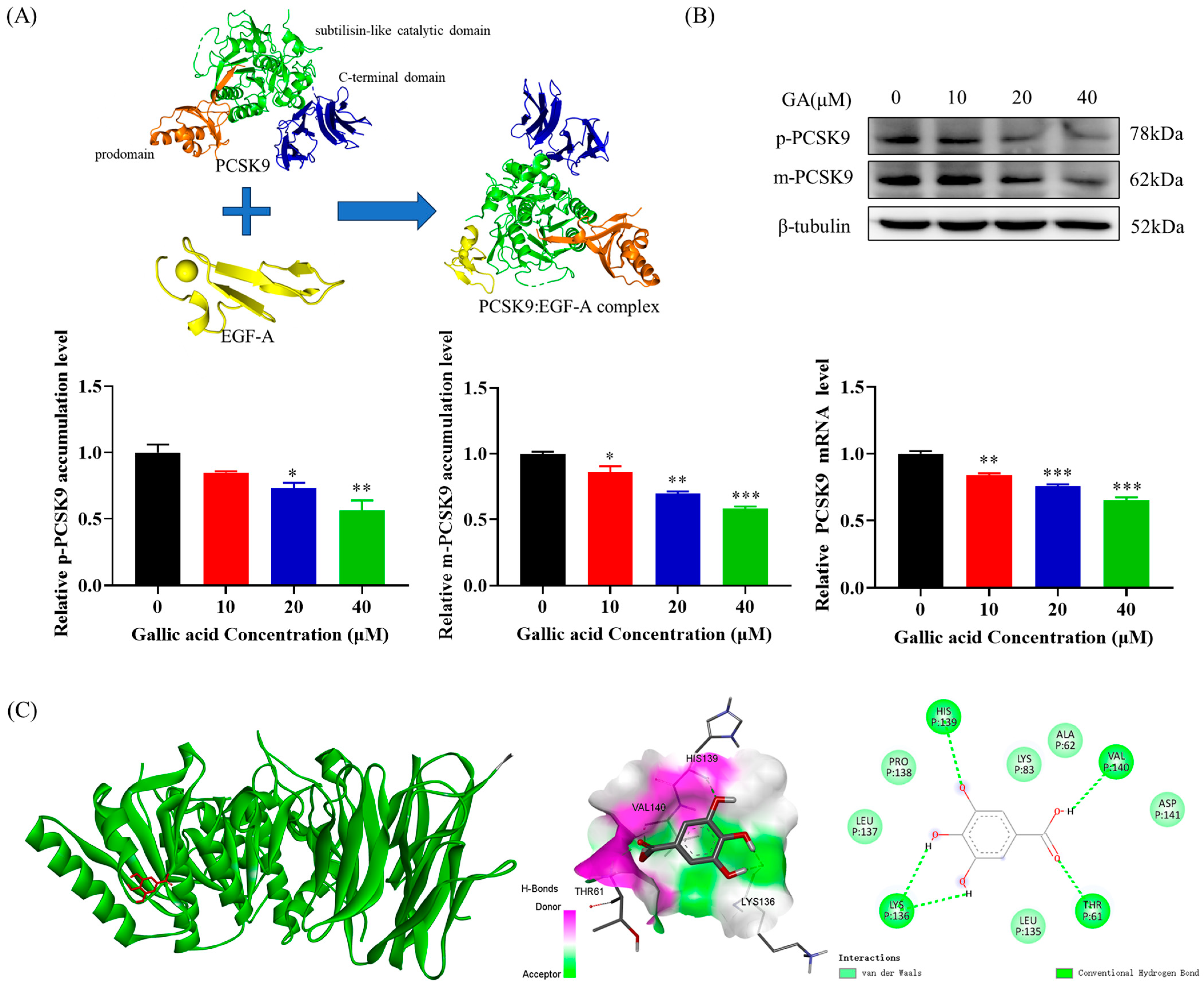
2.3. The Accumulation of PCSK9 Was Inhibited by GA, Leading to an Enhancement in Accumulation of LDLR in HepG2 Cells
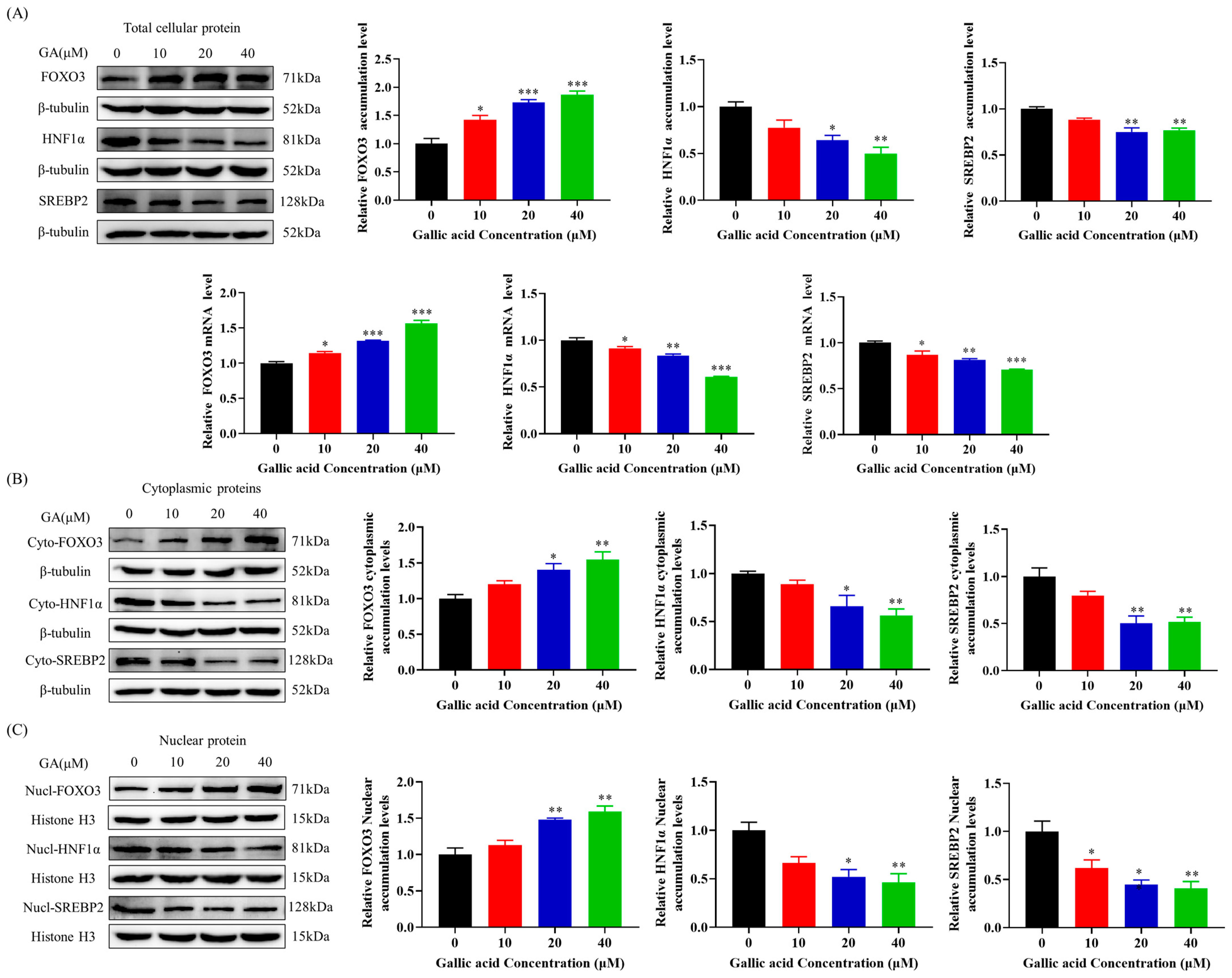
2.4. GA Activated FOXO3 Accumulation and Inhibited HNF1α and SREBP2 Accumulation in HepG2 Cells
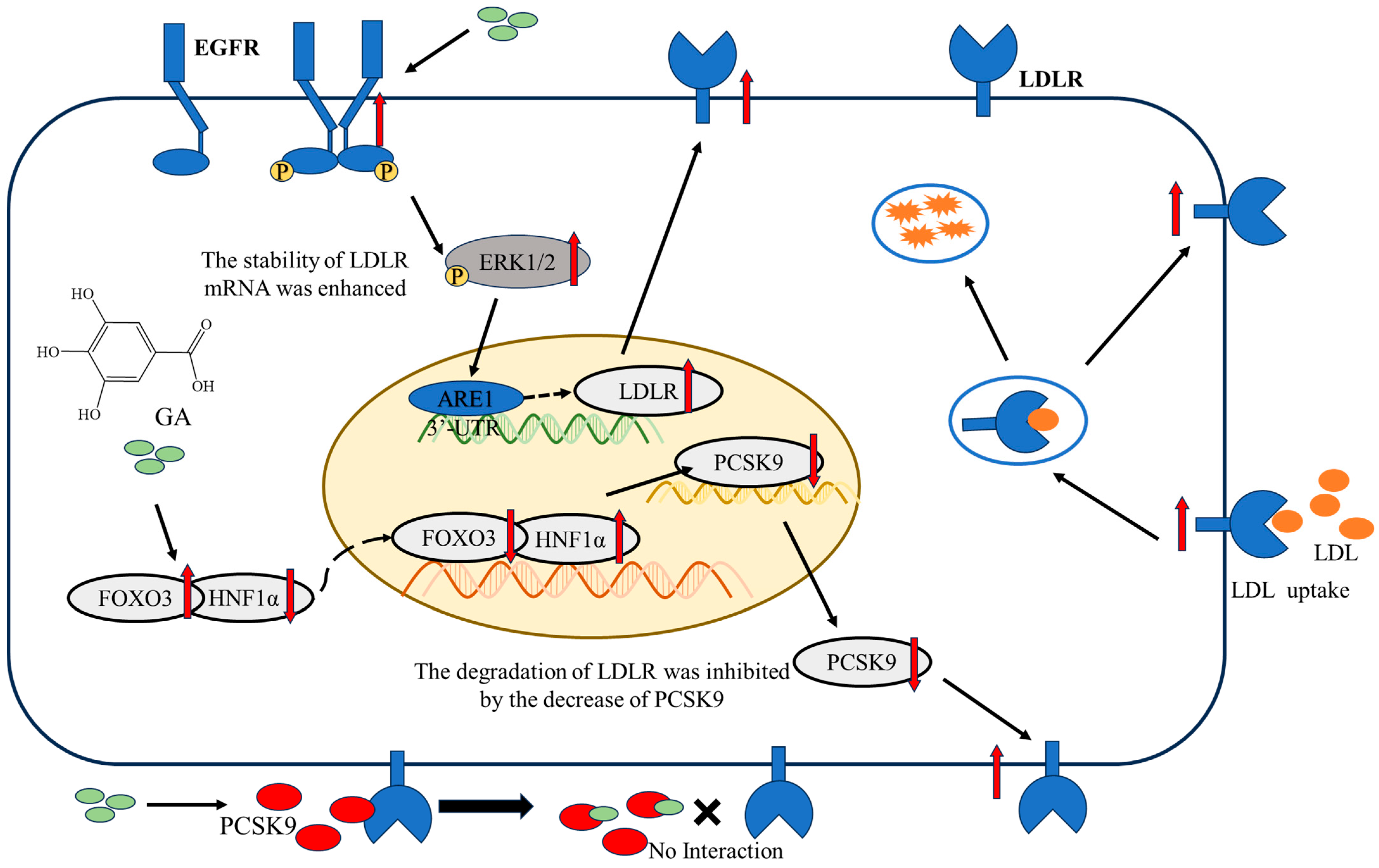
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Culture
4.3. MTT Assay
4.4. Western Blot Analysis
4.5. Dil-LDL Uptake Test
4.6. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
4.7. Molecular Docking
4.8. Data Analysis and Mapping
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, Y.; Rodrigues, R.M.; Wang, X.; Seo, W.; Ma, J.; Hwang, S.; Fu, Y.; Trojnar, E.; Matyas, C.; Zhao, S.; et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J. Clin. Investig. 2021, 131, e141513. [Google Scholar] [CrossRef]
- Li, H.; Yu, X.H.; Ou, X.; Ouyang, X.P.; Tang, C.K. Hepatic cholesterol transport and its role in non-alcoholic fatty liver disease and atherosclerosis. Prog. Lipid Res. 2021, 83, 101109. [Google Scholar] [CrossRef]
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahawar, K.; Ramnarain, D. Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef]
- Li, Y.H.; Hou, J. Research progress in the regulation of cholesterol metabolism and its involvement in the occurrence and development of liver cancer. Chin. J. Cancer Biother. 2021, 28, 1215–1218. [Google Scholar]
- Zhang, Q.; Xu, C.; Tang, Z.K. Regulation Mechanism of Intracellular Cholesterol Level. Prog. Biochem. BioPhys. 2022, 49, 292–302. [Google Scholar]
- Vourakis, M.; Mayer, G.; Rousseau, G. The Role of Gut Microbiota on Cholesterol Metabolism in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8074. [Google Scholar] [CrossRef]
- Sun, D.Q.; Liu, W.Y.; Wu, S.J.; Zhu, G.Q.; Braddock, M.; Zhang, D.C.; Shi, K.Q.; Song, D.; Zheng, M.H. Increased levels of low-density lipoprotein cholesterol within the normal range as a risk factor for nonalcoholic fatty liver disease. Oncotarget 2016, 7, 5728–5737. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Kong, W.; Wei, J.; Abidi, P.; Lin, M.; Inaba, S.; Li, C.; Wang, Y.; Wang, Z.; Si, S.; Pan, H.; et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat. Med. 2004, 10, 1344–1351. [Google Scholar] [CrossRef]
- Wilson, G.M.; Roberts, E.A.; Deeley, R.G. Modulation of LDL receptor mRNA stability by phorbol esters in human liver cell culture models. J. Lipid Res. 1997, 38, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Lagace, T.A. PCSK9 and LDLR degradation: Regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef]
- Wilson, G.M.; Vasa, M.Z.; Deeley, R.G. Stabilization and cytoskeletal-association of LDL receptor mRNA are mediated by distinct domains in its 3′ untranslated region. J. Lipid Res. 1998, 39, 1025–1032. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, Y.; Peng, P.; Zhan, J.; Wang, Z.; Zhu, Z.; Zhang, Z.; Liu, L.; Fang, W.; Zhang, L. Cholesterol synthesis disruption combined with a molecule-targeted drug is a promising metabolic therapy for EGFR mutant non-small cell lung cancer. Transl. Cancer Res. 2021, 10, 128–142. [Google Scholar] [CrossRef]
- Cao, S.; Xu, P.; Yan, J.; Liu, H.; Liu, L.; Cheng, L.; Qiu, F.; Kang, N. Berberrubine and its analog, hydroxypropyl-berberrubine, regulate LDLR and PCSK9 expression via the ERK signal pathway to exert cholesterol-lowering effects in human hepatoma HepG2 cells. J. Cell. Biochem. 2019, 120, 1340–1349. [Google Scholar] [CrossRef]
- Mnasri, N.; Mamarbachi, M.; Allen, B.G.; Mayer, G. 5-Azacytidine engages an IRE1alpha-EGFR-ERK1/2 signaling pathway that stabilizes the LDL receptor mRNA. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 29–40. [Google Scholar] [CrossRef]
- Zhu, X.; Bian, H.; Gao, X. The Potential Mechanisms of Berberine in the Treatment of Nonalcoholic Fatty Liver Disease. Molecules 2016, 21, 1336. [Google Scholar] [CrossRef]
- Huang, Y.W.; Wang, L.T.; Zhang, M.; Nie, Y.; Yang, J.B.; Meng, W.L.; Wang, X.J.; Sheng, J. Caffeine can alleviate non-alcoholic fatty liver disease by augmenting LDLR expression via targeting EGFR. Food Funct. 2023, 14, 3269–3278. [Google Scholar] [CrossRef]
- Bergeron, N.; Phan, B.A.; Ding, Y.; Fong, A.; Krauss, R.M. Proprotein convertase subtilisin/kexin type 9 inhibition: A new therapeutic mechanism for reducing cardiovascular disease risk. Circulation 2015, 132, 1648–1666. [Google Scholar] [CrossRef]
- Momtazi, A.A.; Banach, M.; Pirro, M.; Stein, E.A.; Sahebkar, A. PCSK9 and diabetes: Is there a link? Drug Discov. Today 2017, 22, 883–895. [Google Scholar] [CrossRef]
- Bottomley, M.J.; Cirillo, A.; Orsatti, L.; Ruggeri, L.; Fisher, T.S.; Santoro, J.C.; Cummings, R.T.; Cubbon, R.M.; Lo Surdo, P.; Calzetta, A.; et al. Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 2009, 284, 1313–1323. [Google Scholar] [CrossRef]
- Lo Surdo, P.; Bottomley, M.J.; Calzetta, A.; Settembre, E.C.; Cirillo, A.; Pandit, S.; Ni, Y.G.; Hubbard, B.; Sitlani, A.; Carfí, A. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011, 12, 1300–1305. [Google Scholar] [CrossRef]
- Li, H.; Dong, B.; Park, S.W.; Lee, H.S.; Chen, W.; Liu, J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 2009, 284, 28885–28895. [Google Scholar] [CrossRef]
- Tao, R.; Xiong, X.; DePinho, R.A.; Deng, C.X.; Dong, X.C. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 2013, 288, 29252–29259. [Google Scholar] [CrossRef]
- Cariou, B.; Langhi, C.; Le Bras, M.; Bortolotti, M.; Lê, K.A.; Theytaz, F.; Le May, C.; Guyomarc’h-Delasalle, B.; Zaïr, Y.; Kreis, R.; et al. Plasma PCSK9 concentrations during an oral fat load and after short term high-fat, high-fat high-protein and high-fructose diets. Nutr. Metab. 2013, 10, 4. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zarrabi, A.; Mirzaei, S.; Hashemi, F.; Samarghandian, S.; Zabolian, A.; Hushmandi, K.; Ang, H.L.; Sethi, G.; Kumar, A.P.; et al. Gallic acid for cancer therapy: Molecular mechanisms and boosting efficacy by nanoscopical delivery. Food Chem. Toxicol. 2021, 157, 112576. [Google Scholar] [CrossRef]
- Chao, J.; Huo, T.I.; Cheng, H.Y.; Tsai, J.C.; Liao, J.W.; Lee, M.S.; Qin, X.M.; Hsieh, M.T.; Pao, L.H.; Peng, W.H. Gallic acid ameliorated impaired glucose and lipid homeostasis in high fat diet-induced NAFLD mice. PLoS ONE 2014, 9, e96969. [Google Scholar] [CrossRef]
- Kongpichitchoke, T.; Chiu, M.T.; Huang, T.C.; Hsu, J.L. Gallic Acid Content in Taiwanese Teas at Different Degrees of Fermentation and Its Antioxidant Activity by Inhibiting PKCdelta Activation: In Vitro and in Silico Studies. Molecules 2016, 21, 1346. [Google Scholar] [CrossRef]
- Gandhi, G.R.; Jothi, G.; Antony, P.J.; Balakrishna, K.; Paulraj, M.G.; Ignacimuthu, S.; Stalin, A.; Al-Dhabi, N.A. Gallic acid attenuates high-fat diet fed-streptozotocin-induced insulin resistance via partial agonism of PPARgamma in experimental type 2 diabetic rats and enhances glucose uptake through translocation and activation of GLUT4 in PI3K/p-Akt signaling pathway. Eur. J. Pharmacol. 2014, 745, 201–216. [Google Scholar]
- Punithavathi, V.R.; Prince, P.S.; Kumar, R.; Selvakumari, J. Antihyperglycaemic, antilipid peroxidative and antioxidant effects of gallic acid on streptozotocin induced diabetic Wistar rats. Eur. J. Pharmacol. 2011, 650, 465–471. [Google Scholar] [CrossRef]
- Variya, B.C.; Bakrania, A.K.; Patel, S.S. Antidiabetic potential of gallic acid from Emblica officinalis: Improved glucose transporters and insulin sensitivity through PPAR-γ and Akt signaling. Phytomed. Int. J. Phytother. Phytopharm. 2020, 73, 152906. [Google Scholar] [CrossRef]
- Way, T.D.; Lin, H.Y.; Kuo, D.H.; Tsai, S.J.; Shieh, J.C.; Wu, J.C.; Lee, M.R.; Lin, J.K. Pu-erh tea attenuates hyperlipogenesis and induces hepatoma cells growth arrest through activating AMP-activated protein kinase (AMPK) in human HepG2 cells. J. Agric. Food Chem. 2009, 57, 5257–5264. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Cheng, H.Y.; Chang, M.L.; Huang, S.S.; Liao, J.W.; Cheng, Y.C.; Peng, W.H.; Pao, L.H. Gallic Acid Ameliorated Impaired Lipid Homeostasis in a Mouse Model of High-Fat Diet-and Streptozotocin-Induced NAFLD and Diabetes through Improvement of beta-oxidation and Ketogenesis. Front. Pharmacol. 2020, 11, 606759. [Google Scholar] [CrossRef]
- Kuan, Y.C.; Takahashi, Y.; Maruyama, T.; Shimizu, M.; Yamauchi, Y.; Sato, R. Ring finger protein 5 activates sterol regulatory element-binding protein 2 (SREBP2) to promote cholesterol biosynthesis via inducing polyubiquitination of SREBP chaperone SCAP. J. Biol. Chem. 2020, 295, 3918–3928. [Google Scholar] [CrossRef]
- Yan, R.; Cao, P.; Song, W.; Qian, H.; Du, X.; Coates, H.W.; Zhao, X.; Li, Y.; Gao, S.; Gong, X.; et al. A structure of human Scap bound to Insig-2 suggests how their interaction is regulated by sterols. Science 2021, 371, eabb2224. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Huang, J.; Zhang, W.Y.; Qin, S.; Yang, Y.X.; Ren, H.; Yang, Q.B.; Hu, H. Effects of probiotics on nonalcoholic fatty liver disease: A systematic review and meta-analysis. Ther. Adv. Gastroenterol. 2019, 12, 1756284819878046. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Zhang, L.; She, Z.G.; Li, H.; Zhang, X.J. Non-alcoholic fatty liver disease: A metabolic burden promoting atherosclerosis. Clin. Sci. 2020, 134, 1775–1799. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 2016, 12, 569–579. [Google Scholar] [CrossRef]
- Averna, M. The effect of ezetimibe on NAFLD. Atheroscler. Suppl. 2015, 17, 27–34. [Google Scholar] [CrossRef]
- Nascimbeni, F.; Pellegrini, E.; Lugari, S.; Mondelli, A.; Bursi, S.; Onfiani, G.; Carubbi, F.; Lonardo, A. Statins and nonalcoholic fatty liver disease in the era of precision medicine: More friends than foes. Atherosclerosis 2019, 284, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Wang, D.; Huang, W.; Song, Y.; Ge, L.; Zhang, X.; Yang, L.; Lu, J.; Tu, X.; Chen, Q.; et al. Feedback regulation of coronary artery disease susceptibility gene ADTRP and LDL receptors LDLR/CD36/LOX-1 in endothelia cell functions involved in atherosclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166130. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Z.; Tung, Y.T.; Hsia, S.M.; Wu, C.H.; Yen, G.C. The hepatoprotective effect of Phyllanthus emblica L. fruit on high fat diet-induced non-alcoholic fatty liver disease (NAFLD) in SD rats. Food Funct. 2017, 8, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Fanaei, H.; Mard, S.A.; Sarkaki, A.; Goudarzi, G.; Khorsandi, L. Gallic acid treats dust-induced NAFLD in rats by improving the liver’s anti-oxidant capacity and inhibiting ROS/NFκβ/TNFα inflammatory pathway. Iran. J. Basic Med. Sci. 2021, 24, 240–247. [Google Scholar]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Homoto, M.; Tanaka, R.; Hioki, Y.; Murakami, H.; Suga, H.; Matsumoto, M.; Nakayama, K.I.; Hatta, T.; Iemura, S.; et al. ZFP36L1 and ZFP36L2 control LDLR mRNA stability via the ERK-RSK pathway. Nucleic Acids Res. 2014, 42, 10037–10049. [Google Scholar] [CrossRef] [PubMed]
- Choung, S.; Kim, J.M.; Joung, K.H.; Lee, E.S.; Kim, H.J.; Ku, B.J. Epidermal growth factor receptor inhibition attenuates non-alcoholic fatty liver disease in diet-induced obese mice. PLoS ONE 2019, 14, e0210828. [Google Scholar] [CrossRef] [PubMed]
- Kubota, S.; Tanaka, Y.; Nagaoka, S. Ellagic acid affects mRNA expression levels of genes that regulate cholesterol metabolism in HepG2 cells. Biosci. Biotechnol. Biochem. 2019, 83, 952–959. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Bittner, V.A.; Giugliano, R.P.; Brinton, E.A.; Guyton, J.R. PCSK9 inhibitors for prevention of atherosclerotic cardiovascular disease. J. Clin. Lipidol. 2018, 12, 835–843. [Google Scholar] [CrossRef]
- Li, H.H.; Li, J.; Zhang, X.J.; Li, J.M.; Xi, C.; Wang, W.Q.; Lu, Y.L.; Xuan, L.J. 23,24-Dihydrocucurbitacin B promotes lipid clearance by dual transcriptional regulation of LDLR and PCSK9. Acta Pharmacol. Sin. 2020, 41, 327–335. [Google Scholar] [CrossRef]
- Cui, C.J.; Jin, J.L.; Guo, L.N.; Sun, J.; Wu, N.Q.; Guo, Y.L.; Liu, G.; Dong, Q.; Li, J.J. Beneficial impact of epigallocatechingallate on LDL-C through PCSK9/LDLR pathway by blocking HNF1α and activating FoxO3a. J. Transl. Med. 2020, 18, 195. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequences (5′-3′) | |
|---|---|---|
| LDLR | F: GAACCCATCAAAGAGTGCG | R: TCTTCCTGACCTCGTGCC |
| PCSK9 | F: CCAAGCCTCTTCTTACTTCACC | R: GCATCGTTCTGCCATCACT |
| SREBP2 | F: CCCTGGGAGACATCGACGA | R: CGTTGCACTGAAGGGTCCA |
| HNF1α | F: GTGGCGAAGATGGTCAAGTCC | R: CCCTTGTTGAGGTGTTGGG |
| FOXO3 | F: ACATGGGCTTGAGTGAGT | R: GCCTGAGAGAGAGTCCGAGA |
| β-actin | F: ACAGAGCCTCGCCTTTGCCG | R: ACATGCCGGAGCCGTTGTCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Zhou, Q.; Yang, X.; Zhang, Z.; Wang, D.; Hu, D.; Huang, Y.; Sheng, J.; Wang, X. Gallic Acid Can Promote Low-Density Lipoprotein Uptake in HepG2 Cells via Increasing Low-Density Lipoprotein Receptor Accumulation. Molecules 2024, 29, 1999. https://doi.org/10.3390/molecules29091999
Zhang D, Zhou Q, Yang X, Zhang Z, Wang D, Hu D, Huang Y, Sheng J, Wang X. Gallic Acid Can Promote Low-Density Lipoprotein Uptake in HepG2 Cells via Increasing Low-Density Lipoprotein Receptor Accumulation. Molecules. 2024; 29(9):1999. https://doi.org/10.3390/molecules29091999
Chicago/Turabian StyleZhang, Dongying, Qixing Zhou, Xiangxuan Yang, Zhen Zhang, Dongxue Wang, Dandan Hu, Yewei Huang, Jun Sheng, and Xuanjun Wang. 2024. "Gallic Acid Can Promote Low-Density Lipoprotein Uptake in HepG2 Cells via Increasing Low-Density Lipoprotein Receptor Accumulation" Molecules 29, no. 9: 1999. https://doi.org/10.3390/molecules29091999
APA StyleZhang, D., Zhou, Q., Yang, X., Zhang, Z., Wang, D., Hu, D., Huang, Y., Sheng, J., & Wang, X. (2024). Gallic Acid Can Promote Low-Density Lipoprotein Uptake in HepG2 Cells via Increasing Low-Density Lipoprotein Receptor Accumulation. Molecules, 29(9), 1999. https://doi.org/10.3390/molecules29091999