

High Reactivity of Dimethyl Ether Activated by Zeolite Ferrierite within a Fer Cage: A Prediction Study


Abstract
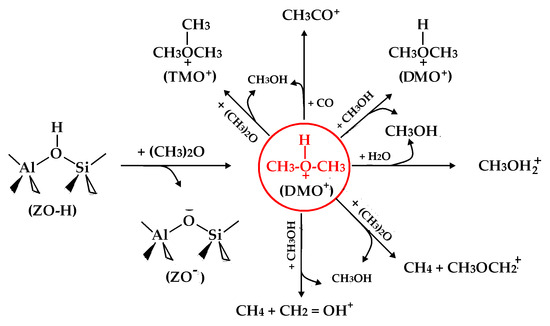
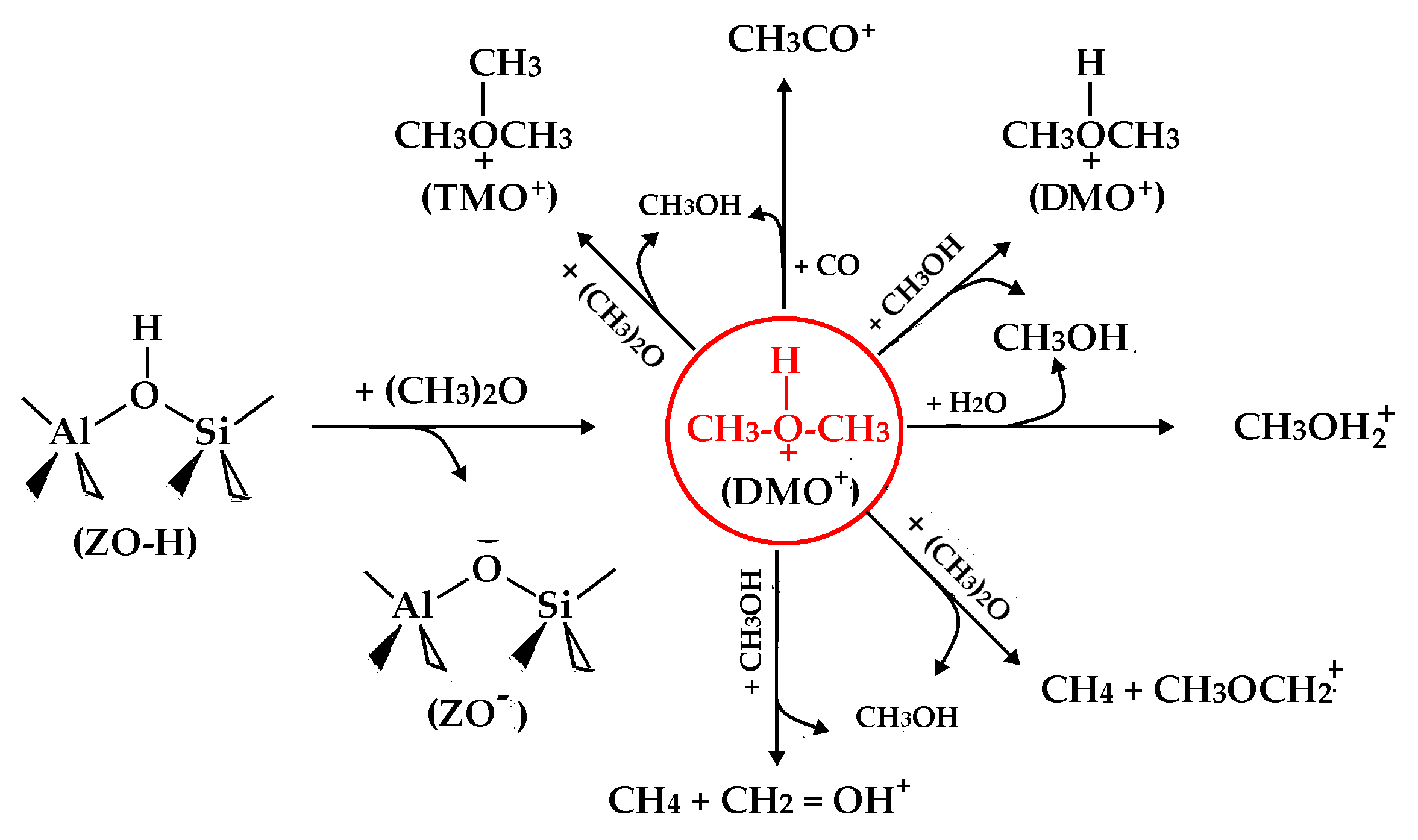
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- Methylation of hydrogen-bonded DME within the deprotonated zeolite (ZO−):(CH3)2OH+ + CH3OCH3 → (CH3)3O+ (i.e., TMO+) + CH3OH(CH3)2OH+ + CH3OH → CH3OH + (CH3)2OH+(CH3)2OH+ + H2O → CH3OH + CH3OH2+(CH3)2OH+ + CO → CH3CO+ + CH3OH
- H-abstraction of hydrogen-bonded DME within ZO-:(CH3)2OH+ + CH3OCH3 → CH3OH + CH4 + CH3OCH2+(CH3)2OH+ + CH3OH → CH3OH + CH4 + CH2OH+
2. Results and Discussion
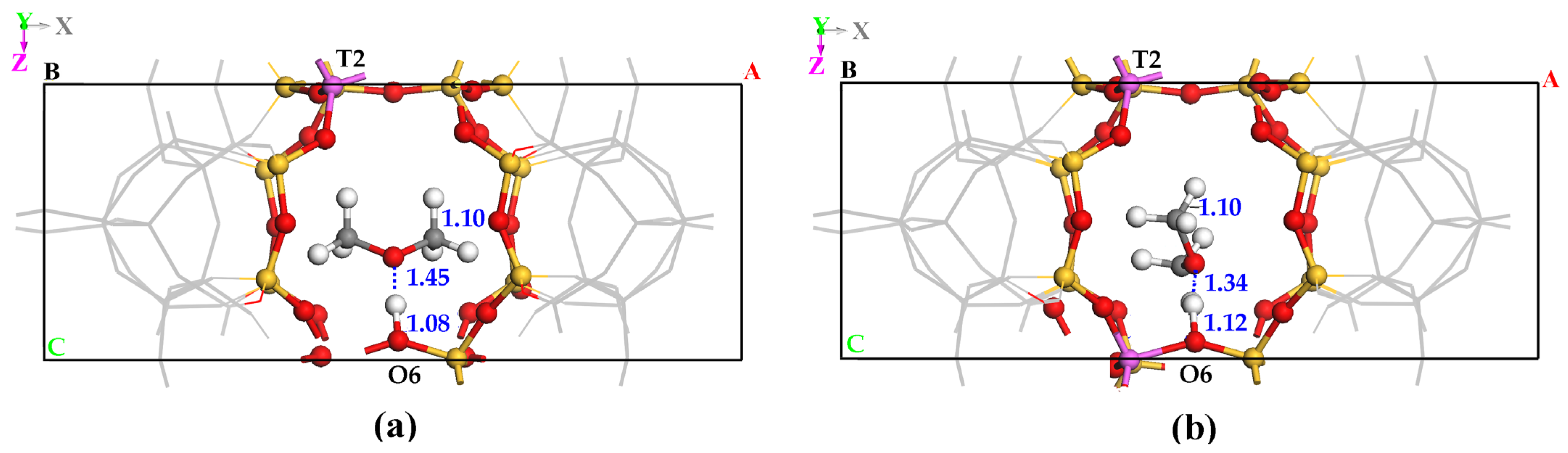
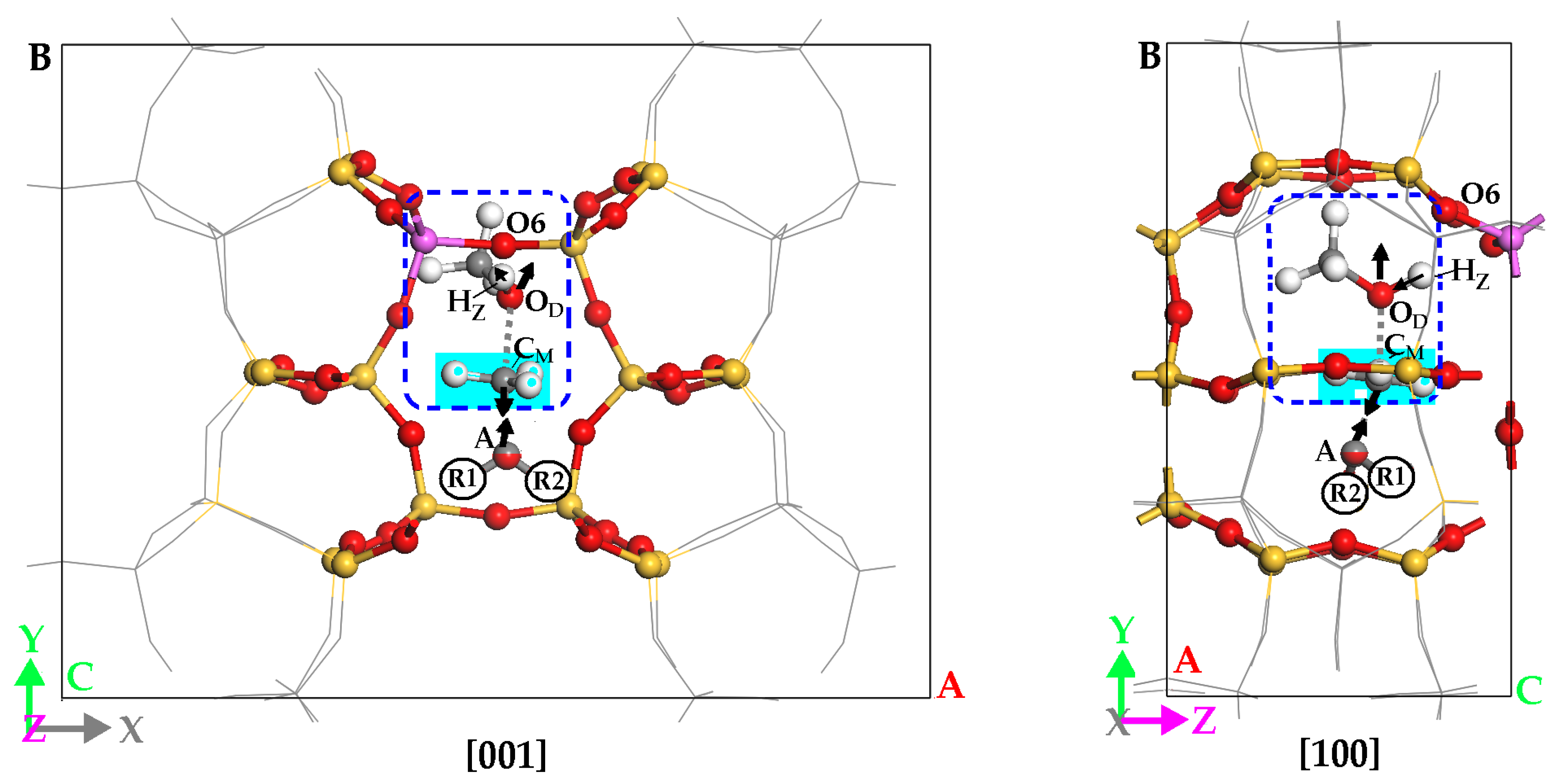
2.1. DME Adsorbed on the Zeolitic Surface
2.2. Attacking Molecule Serving as Electron Donor
2.3. Methylation Process of Hydrogen-Bonded DME (i.e., DMO+)
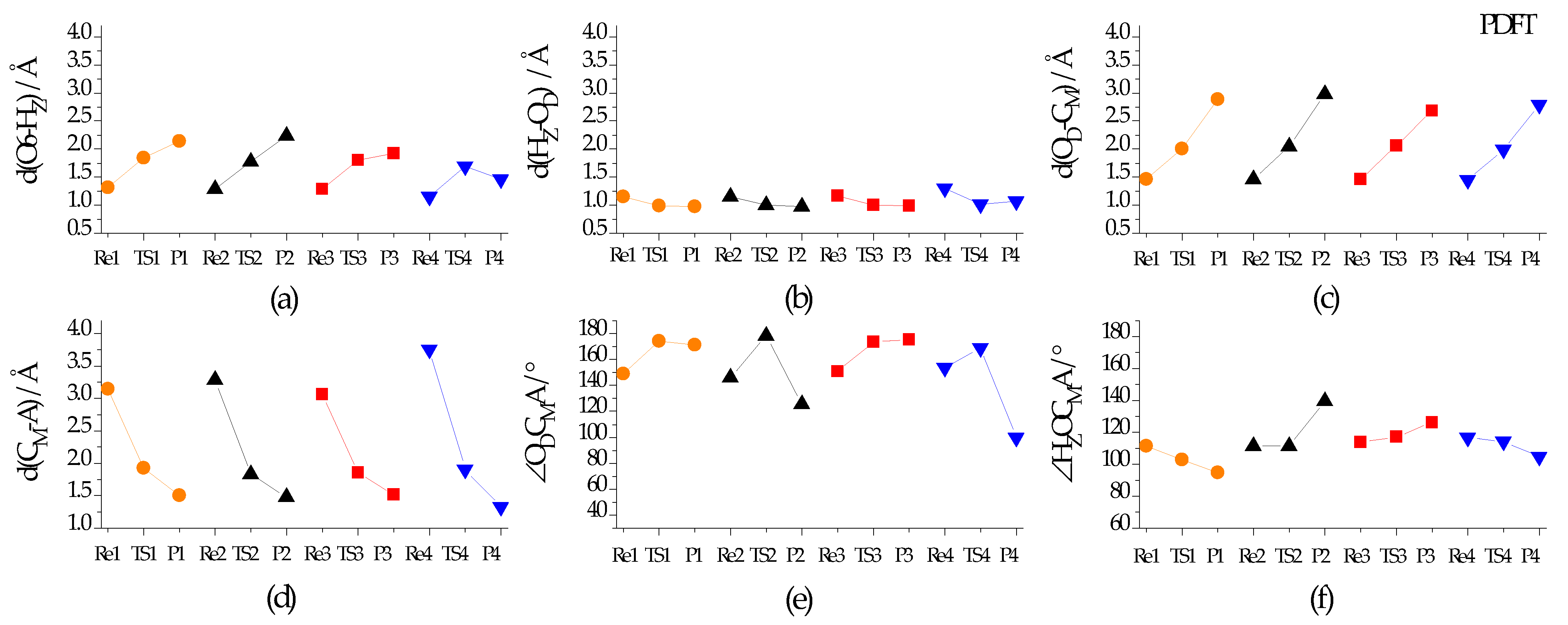
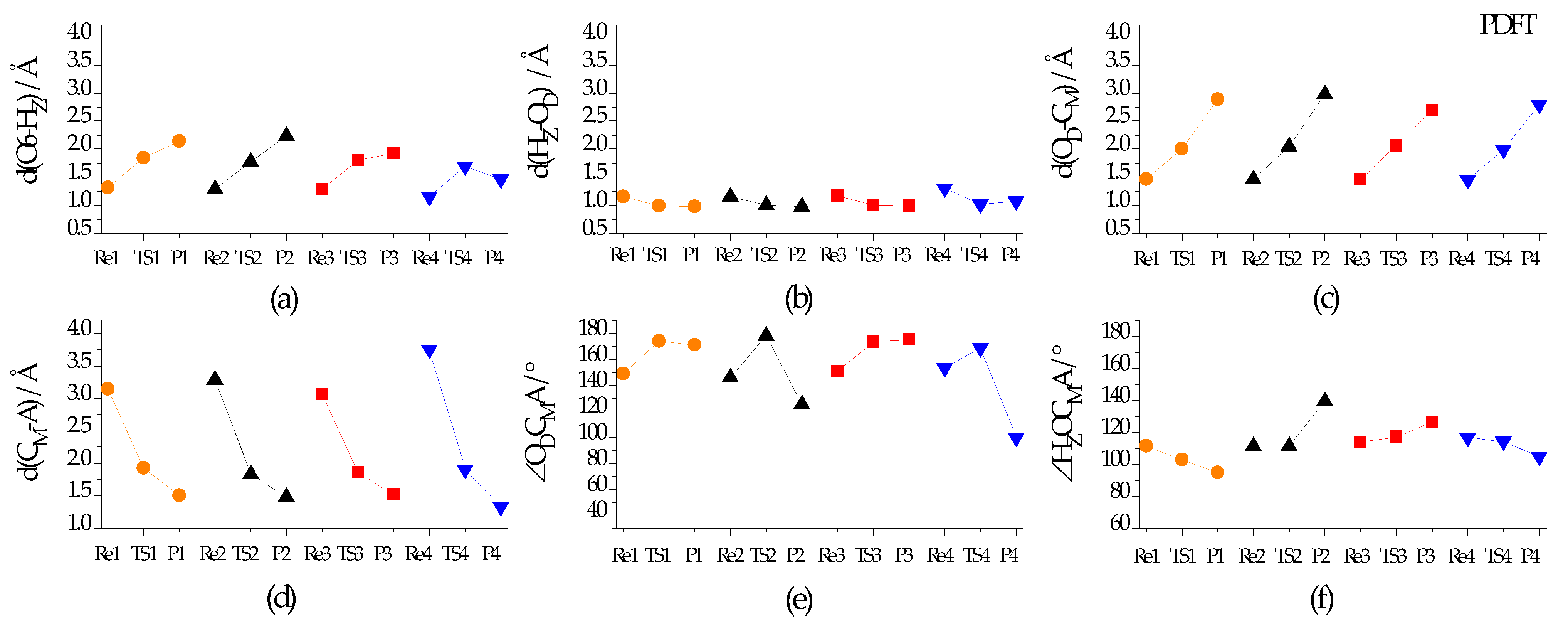
2.3.1. Reaction Course of DMO+ Methylation
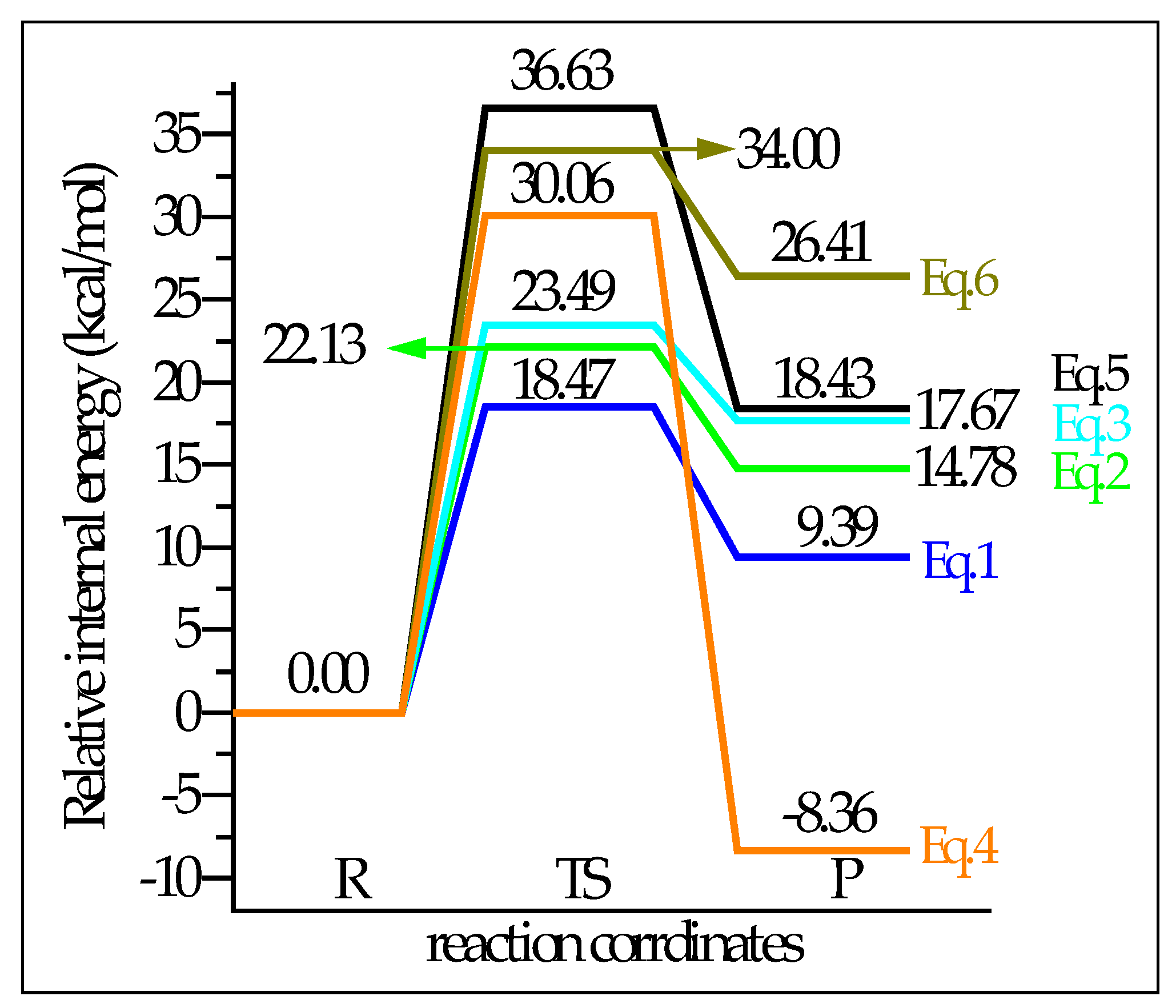
2.3.2. Energy Parameters and Products for DMO+ Methylation
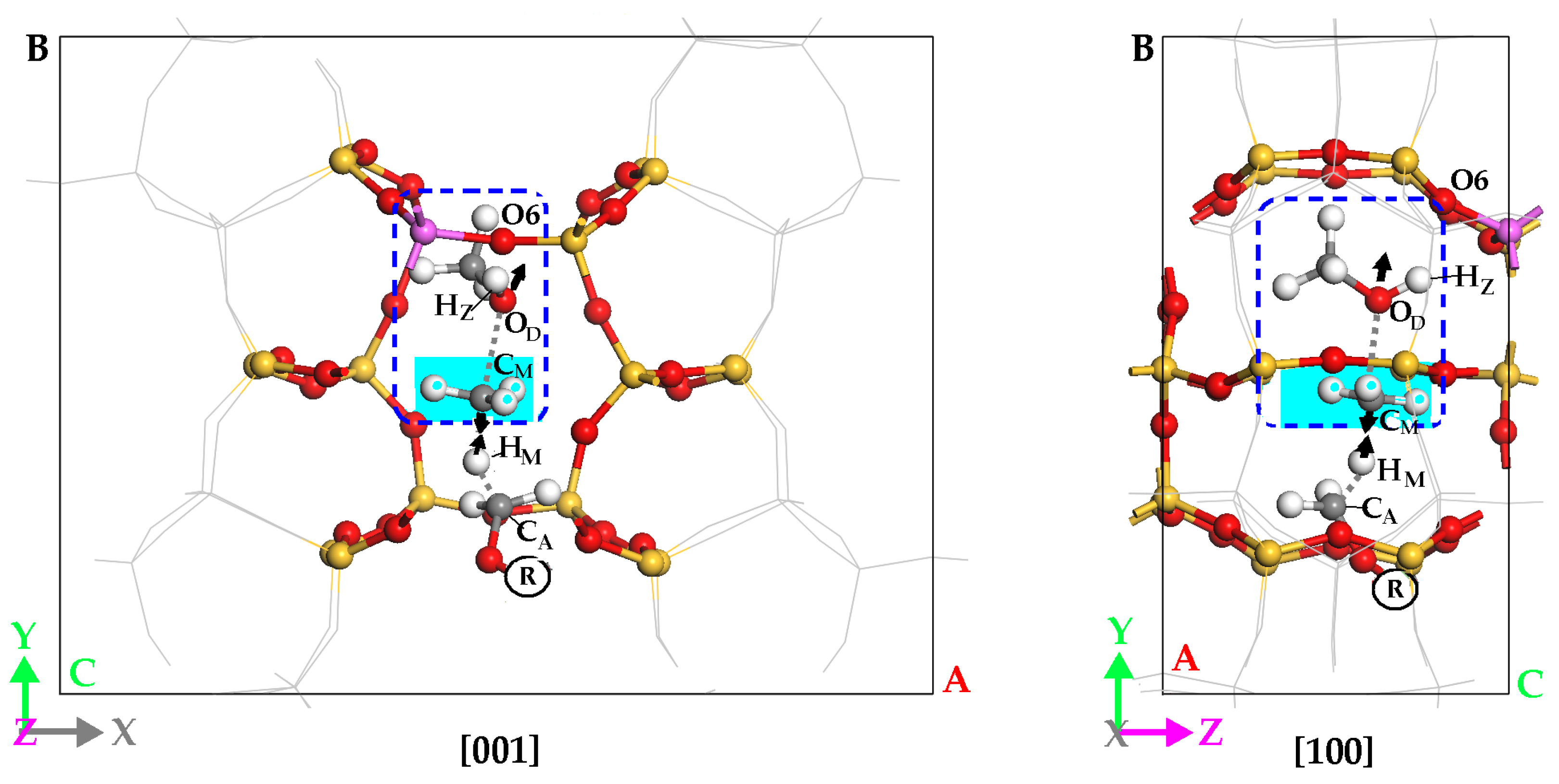
2.4. H-Abstract of Hydrogen-Bonded DME (i.e., DMO+)
2.4.1. Reaction Course of DMO+ H-Abstraction
2.4.2. Energy Parameters and Products for DMO+ H-Abstraction
2.5. Rate Constant
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BA | Brönsted acid; |
| MA | methyl acetate; |
| H-FER | the acidic zeolite ferrierite; |
| SMS or ZO-CH3 | surface methoxy species on zeolites; |
| DME | dimethyl ether; |
| DMO+ | dimethyl oxonium ion; |
| TMO+ | trimethyl oxonium ion; |
| CH3OH2+ | methoxonium ion; |
| ∆E≠ | the activation of internal energy. |
References
- Cheung, P.; Bhan, A.; Sunley, G.J.; Iglesia, E. Selective Carbonylation of Dimethyl Ether to Methyl Acetate Catalyzed by Acidic Zeolites. Angew. Chem. Int. Ed. 2006, 45, 1617–1620. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Law, D.J.; Iglesia, E. Site requirements and elementary steps in dimethyl ether carbonylation catalyzed by acidic zeolites. J. Catal. 2007, 245, 110–123. [Google Scholar] [CrossRef]
- Bhan, A.; Allian, A.D.; Sunley, G.J.; Law, D.J.; Iglesia, E. Specificity of sites within eight-membered ring zeolite channels for carbonylation of methyls to acetyls. J. Am. Chem. Soc. 2007, 129, 4919–4924. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xue, H.; Huang, X.; Li, Y.; Shen, W. Dimethyl Ether Carbonylation to Methyl Acetate over HZSM-35. Catal. Lett. 2010, 139, 33–37. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Liu, S.; Xie, S.; Zhu, X.; Chen, F.; Xu, L. Activity Enhancement of ZSM-35 in Dimethyl Ether Carbonylation Reaction through Alkaline Modifications. RSC Adv. 2013, 3, 16549–16557. [Google Scholar] [CrossRef]
- Boronat, M.; Martinez-Sanchez, C.; Law, D.; Corma, A. Enzyme-like Specificity in Zeolites: A Unique Site Position in Mordenite for Selective Carbonylation of Methanol and Dimethyl Ether with CO. J. Am. Chem. Soc. 2008, 130, 16316–16323. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.; Martínez, C.; Corma, A. Mechanistic differences between methanol and dimethyl ether carbonylation in side pockets and large channels of mordenite. Phys. Chem. Chem. Phys. 2011, 13, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, W.; Zhu, L.; Wang, H.; Qiu, K.; Cen, K. Methyl Acetate Synthesis from Dimethyl Ether Carbonylation over Mordenite Modified by Cation Exchange. J. Phys. Chem. C 2015, 119, 524–533. [Google Scholar] [CrossRef]
- He, T.; Liu, X.; Xu, S.; Han, X.; Pan, X.; Hou, G.; Bao, X. Role of 12-Ring Channels of Mordenite in DME Carbonylation Investigated by Solid-State NMR. J. Phys. Chem. C 2016, 120, 22526–22531. [Google Scholar] [CrossRef]
- Park, S.Y.; Shin, C.-H.; Bae, J.W. Selective carbonylation of dimethyl ether to methyl acetate on Ferrierite. Catal. Commun. 2016, 75, 28–31. [Google Scholar] [CrossRef]
- Feng, P.; Chen, X.-F.; Li, X.-J.; Zhao, D.; Xie, S.J.; Xu, L.-Y.; He, G.-Z. The distribution analysis on the proton siting and the acid strength of the zeolite ferrierite: A computational study. Microporous Mesoporous Mater. 2017, 239, 354–362. [Google Scholar] [CrossRef]
- Feng, P.; Zhang, G.; Zang, K.; Li, X.; Xu, L.; Chen, X. A theoretical study on the selective adsorption behavior of dimethyl ether and carbon monoxide on H-FER zeolites. Chem. Phys. Lett. 2017, 684, 279–284. [Google Scholar] [CrossRef]
- Feng, P.; Zhang, G.; Chen, X.; Zang, K.; Li, X.; Xu, L. Specific zone within 8-membered ring channel as catalytic center for carbonylation of dimethyl ether and methanol over FER zeolite. Appl. Catal. A Gen. 2018, 557, 119–124. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Liu, H.; Sun, T.; Fan, D.; Yang, M.; Tian, P.; Liu, Z. Preparation of Spherical Mordenite Zeolite Assemblies with Excellent Catalytic Performance for Dimethyl Ether Carbonylation. ACS Appl. Mater. Interfaces 2018, 10, 32239–32246. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yao, J.; Li, H.; Fang, Y.; Yoneyama, Y.; Yang, G. A brand new zeolite catalyst for carbonylation reaction. Chem. Commun. 2019, 55, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Zhan, E.; Xiong, Z.; Shen, W. Dimethyl ether carbonylation over zeolites. J. Energy Chem. 2019, 36, 51–63. [Google Scholar] [CrossRef]
- Liua, S.; Fang, X.; Liu, Y.; Liu, H.; Ma, X.; Zhu, W.; Liu, Z. Dimethyl ether Carbonylation over Mordenite zeolite modified by Alkyimidazolium ions. Catal. Commun. 2020, 147, 106161. [Google Scholar] [CrossRef]
- Liu, S.; Liu, H.; Ma, X.; Yong, L.; Zhu, W.; Liu, Z. Identifying and controlling the acid site distributions in mordenite zeolite for dimethyl ether carbonylation reaction by means of selective ion-exchange. Catal. Sci. Technol. 2020, 10, 4663–4672. [Google Scholar] [CrossRef]
- Li, S.; Cai, K.; Li, Y.; Liu, S.; Yu, M.; Wang, Y.; Ma, X.; Huang, S. Identifying the Active Silver Species in Carbonylation of Dimethyl Ether over Ag-HMOR. ChemCatChem 2020, 12, 3290–3297. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhan, E.; Li, M.; Shen, W. DME carbonylation over a HSUZ-4 zeolite. Chem. Commun. 2020, 56, 3401–3404. [Google Scholar] [CrossRef]
- Kipnis, M.A.; Volnina, E.A. Methyl Acetate Synthesis by Dimethyl Ether Carbonylation in the Presence of Zeolites: A Review. Kinet. Catal. 2022, 63, 129–140. [Google Scholar] [CrossRef]
- Fan, B.; Zhang, W.; Gao, P.; Hou, G.; Liu, R.; Xu, S.; Wei, Y.; Liu, Z. Quantitatively Mapping the Distribution of Intrinsic Acid Sites in Mordenite Zeolite by High-Field 23Na Solid-State Nuclear Magnetic Resonance. J. Phys. Chem. Lett. 2022, 13, 5186–5194. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Feng, X.; Fan, J.; Komiyama, S.; Kugue, Y.; Guo, X.; He, Y.; Yang, G.; Tsubaki, N. Self-Assembled Nano-Filamentous Zeolite Catalyst to Realize Efficient One-Step Ethanol Synthesis. Chem. A Eur. J 2022, 28, e202201783. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-B.; Cao, J.-P.; Su, C.; He, Z.-M.; Zhao, X.-Y. Tailoring the acid distribution and identifying the active center of rod-shaped HSUZ-4 zeolite for enhancing dimethyl ether carbonylation performance. Fuel 2022, 315, 123267. [Google Scholar] [CrossRef]
- Xu, H.; Zhu, J.; Zhu, L.; Zhou, E.; Shen, C. Advances in the Synthesis of Ferrierite Zeolite. Molecules 2020, 25, 3722. [Google Scholar] [CrossRef]
- Vaughan, P.A. crystal structures of zeolite ferrierite. Am. Miner. 1965, 50, 293–294. [Google Scholar] [CrossRef]
- Kerr, I.S. Structure of Ferrierite. Nature 1966, 210, 294–295. [Google Scholar] [CrossRef]
- Vaughan, P.A. The Crystal Structure of the Zeolite Ferrierite. Acta. Crystallogr. 1966, 21, 983–990. [Google Scholar] [CrossRef]
- Morris, R.E.; Weigel, S.J.; Henson, N.J.; Bull, L.M.; Janicke, M.T.; Chmelka, B.F.; Cheetham, A.K. A Synchrotron X-ray Diffraction, Neutron Diffraction, 29Si MAS-NMR, and Computational Study of the Siliceous Form of Zeolite Ferrierite. J. Am. Chem. Soc. 1994, 116, 11849–11855. [Google Scholar] [CrossRef]
- Blanco, F.; Urbinavillalba, G.; Deagudelo, M.M.R. Theoretical calculations on zeolites—The aluminum substitution in mordenite, ferrierite and ZSM-5. Mol. Simul. 1995, 14, 165–176. [Google Scholar] [CrossRef]
- Dědeček, J.; Sobalík, Z.; Wichterlová, B. Siting and Distribution of Framework Aluminium Atoms in Silicon-Rich Zeolites and Impact on Catalysis. Cat. Rev. Sci. Eng. 2012, 54, 135–223. [Google Scholar] [CrossRef]
- Xiong, Z.; Qi, G.; Bai, L.; Zhan, E.; Chu, Y.; Xu, J.; Ta, N.; Hao, A.; Deng, F.; Shen, W. Preferential population of Al atoms at the T4 site of ZSM-35 for the carbonylation of dimethyl ether. Catal. Sci. Technol. 2022, 12, 4993–4997. [Google Scholar] [CrossRef]
- Martucci, A.; Alberti, A.; Cruciani, G.; Radaelli, P.; Ciambelli, P.; Rapacciulo, M. Location of Brønsted sites in D-ferrierite by neutron powder diffraction. Microporous Mesoporous Mater. 1999, 30, 95–101. [Google Scholar] [CrossRef]
- van Santen, R.A.; Kramer, G.J. Reactivity Theory of Zeolitic Brarnsted Acidic Sites. Chem. Rev. 1995, 95, 637–660. [Google Scholar] [CrossRef]
- Petersen, H.; Weidenthaler, C. A review of recent developments for the in situ/operando characterization of nanoporous materials. Inorg. Chem. Front. 2022, 9, 4244–4271. [Google Scholar] [CrossRef]
- Bhan, A.; Iglesia, E. A Link between Reactivity and Local Structure in Acid Catalysis on Zeolites. Acc. Chem. Res. 2008, 41, 559–567. [Google Scholar] [CrossRef]
- Voleska, I.; Nachtigall, P.; Ivanova, E.; Hadjiivanov, K.; Bulanek, R. Theoretical and experimental study of CO adsorption on Ca-FER zeolite. Catal. Today 2015, 243, 53–61. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. The effect of FER zeolite acid sites in methanol-to-dimethyl-ether catalytic dehydration. J. Energy. Chem. 2017, 26, 406–415. [Google Scholar] [CrossRef]
- Xu, F.; Hong, Z.; Lv, J.; Chen, C.; Zhao, G.; Miao, L.; Yang, W.; Zhu, Z. Mg enhances the catalytic selectivity and stability of mordenite for carbonylation of dimethyl ether. Appl. Catal. A Gen. 2022, 648, 118928. [Google Scholar] [CrossRef]
- Nachtigall, P.; Bulánek, R. Theoretical investigation of site-specific characteristics of CO adsorption complexes in the Li+-FER zeolite. Appl. Catal. A Gen 2006, 307, 118–127. [Google Scholar] [CrossRef]
- Chu, Y.; Lo, A.-Y.; Wang, C.; Deng, F. Origin of High Selectivity of Dimethyl Ether Carbonylation in the 8-Membered Ring Channel of Mordenite Zeolite. J. Phys. Chem. C 2019, 123, 15503–15512. [Google Scholar] [CrossRef]
- Bordiga, S.; Palomino, G.T.; Paze, C.; Zecchina, A. Vibrational spectroscopy of H2, N2, CO and NO adsorbed on H, Li, Na, K-exchanged ferrierite. Microporous Mesoporous Mater. 2000, 34, 67–80. [Google Scholar] [CrossRef]
- Cairon, O. CO adsorption on the Na, K-Ferrierite zeolite at 77 K and 193 K: New IR insights and consequences on the previously endorsed assignments of adsorbed species. Microporous Mesoporous Mater. 2014, 183, 48–53. [Google Scholar] [CrossRef]
- van Well, W.J.M.; Cottin, X.; Smit, B.; van Hooff, J.H.C.; van Santen, R.A. Chain Length Effects of Linear Alkanes in Zeolite Ferrierite. 2. Molecular Simulations1. J. Phys. Chem. B 1998, 102, 3952–3958. [Google Scholar] [CrossRef]
- Rasmussen, D.B.; Christensen, J.M.; Temel, B.; Studt, F.; Moses, P.G.; Rossmeisl, J.; Riisager, A.; Jensen, A.D. Ketene as a Reaction Intermediate in the Carbonylation of Dimethyl Ether to Methyl Acetate over Mordenite. Angew. Chem. 2015, 127, 7369–7372. [Google Scholar] [CrossRef]
- Forester, T.R.; Howe, R.F. In Situ FTIR Studies of Methanol and Dimethyl Ether in ZSM-5. J. Am. Chem. Soc. 1987, 109, 5076–5082. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Viruela, P.; Corma, A. Formation of Surface Methoxy Groups on H-Zeolites from Methanol. A Quantum Chemical Study. J. Phys. Chem. 1995, 99, 13224–13231. [Google Scholar] [CrossRef]
- Olah, G.A.; Burrichter, A.; Rasul, G.; Gnann, R.; Christe, K.O.; Prakash, G.K.S. 17O and 13C NMR/ab Initio/IGLO/GIAO-MP2 Study of Oxonium and Carboxonium Ions (Dications) and Comparison with Experimental Data. J. Am. Chem. Soc. 1997, 119, 8035–8042. [Google Scholar] [CrossRef]
- Fang, H.; Zheng, A.; Chu, Y.; Deng, F. 13C Chemical Shift of Adsorbed Acetone for Measuring the Acid Strength of Solid Acids: A Theoretical Calculation Study. J. Phys. Chem. C 2010, 114, 12711–12718. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, G.K.S.; Ellis, R.W.; Olah, J.A. Remarks on the Mechanism of Ethylene Formation from Methyl Alcohol. J. Chem. Soc. Chem. Commun. 1986, 9–10. [Google Scholar] [CrossRef]
- Olah, G.A.; Doggweiler, H.; Felberg, J.D. Onium Ylide chemistry. 2. Methylenedialkyloxonium Ylides. J. Org. Chem. 1984, 49, 2116–2122. [Google Scholar] [CrossRef]
- Poreddy, R.; Mossin, S.; Jensen, A.D.; Riisager, A. Promoting Effect of Copper Loading and Mesoporosity on Cu-MOR in the Carbonylation of Dimethyl Ether to Methyl Acetate. Catalysts 2021, 11, 696. [Google Scholar] [CrossRef]
- Olah, G.A.; Mathew, T.; Prakash, G.K.S.; Rasul, G. Chemical Aspects of Astrophysically Observed Extraterrestrial Methanol, Hydrocarbon Derivatives, and Ions. J. Am. Chem. Soc. 2016, 138, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wei, Z.; Chen, Y.; Jing, B.; He, Y.; Dong, M.; Jiao, H.; Li, X.; Qin, Z.; Wang, J.; et al. A route to form initial hydrocarbon pool species in methanol conversion to olefins over zeolites. J. Catal. 2014, 317, 277–283. [Google Scholar] [CrossRef]
- Wei, Z.; Chen, Y.-Y.; Li, J.; Guo, W.; Wang, S.; Dong, M.; Qin, Z.; Wang, J.; Jiao, H.; Fan, W. Stability and Reactivity of Intermediates of Methanol Related Reactions and C-C Bond Formation over H-ZSM-5 Acidic Catalyst: A Computational Analysis. J. Phys. Chem. C 2016, 120, 6075–6087. [Google Scholar] [CrossRef]
- Svelle, S.; Visur, M.; Olsbye, U.; Saepurahman; Bjørgen, M. Mechanistic Aspects of the Zeolite Catalyzed Methylation of Alkenes and Aromatics with Methanol: A Review. Top. Catal. 2011, 54, 897–906. [Google Scholar] [CrossRef]
- Olah, G.A.; Doggweiler, H.; Felberg, J.D.; Frohlich, S.; Grdina, M.J.; Karpeles, R.; Keumi, T.; Inaba, S.-i.; Lammertsma, W.M.I.K.; Salem, G.; et al. Onium Ylide Chemistry. 1. Bifunctional Acid-Base-Catalyzed Conversion of Heterosubstituted Methanes into Ethylenee and Derived Hydrocarbons. The Onium Ylide Mechanism of the C1→C2 conversion. J. Am. Chem. Soc. 1984, 106, 2143–2149. [Google Scholar] [CrossRef]
- Olah, G.A.; Doggweiler, H.; Felberg, J.D.; Frohlich, S. Onium Ions. 33. (Trimethylsily1)- and [(Trimethylsilyl)methyl]oxonium and -halonium Ions. J. Org. Chem. 1985, 50, 4847–4851. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Moleclar-Dynamics Simulation of the Liquid-Metal Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B Condens. Matter. Mater. Phys. 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mat. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-energy Calculations Using a Plane—Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases (accessed on 14 April 2024).
- Grajciar, L.; Areán, C.O.; Pulido, A.; Nachtigall, P. Periodic DFT investigation of the effect of aluminium content on the properties of the acid zeolite H-FER. Phys. Chem. Chem. Phys. 2010, 12, 1497–1506. [Google Scholar] [CrossRef]
- Chen, X.; Yu, T. Simulating Crystal Structure, Acidity, Proton Distribution, and IR Spectra of Acid Zeolite HSAPO-34: A High Accuracy Study. Molecules 2023, 28, 8087. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-F. Periodic Density Functional Theory (PDFT) Simulating Crystal Structures with Microporous CHA Framework: An Accuracy and Efficiency Study. Inorganics 2023, 11, 215. [Google Scholar] [CrossRef]
- Brogaard, R.Y.; Weckhuysen, B.M.; Nørskov, J.K. Guest–host interactions of arenes in H-ZSM-5 and their impact on methanol-to-hydrocarbons deactivation processes. J. Catal. 2013, 300, 235–241. [Google Scholar] [CrossRef]
- Brogaard, R.Y.; Henry, R.; Schuurman, Y.; Medford, A.J.; Moses, P.G.; Beato, P.; Svelle, S.; Nørskov, J.K.; Olsbye, U. Methanol-to-hydrocarbons conversion: The alkene methylation pathway. J. Catal. 2014, 314, 159–169. [Google Scholar] [CrossRef]
- Ghorbanpour, A.; Rimer, J.D.; Grabow, L.C. Computational Assessment of the Dominant Factors Governing the Mechanism of Methanol Dehydration over H-ZSM-5 with Heterogeneous Aluminum Distribution. ACS Catal. 2016, 6, 2287–2298. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldson, A.; Jopsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 31, 354–360. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Feng, P.; Li, X. High Reactivity of Dimethyl Ether Activated by Zeolite Ferrierite within a Fer Cage: A Prediction Study. Molecules 2024, 29, 2000. https://doi.org/10.3390/molecules29092000
Chen X, Feng P, Li X. High Reactivity of Dimethyl Ether Activated by Zeolite Ferrierite within a Fer Cage: A Prediction Study. Molecules. 2024; 29(9):2000. https://doi.org/10.3390/molecules29092000
Chicago/Turabian StyleChen, Xiaofang, Pei Feng, and Xiujie Li. 2024. "High Reactivity of Dimethyl Ether Activated by Zeolite Ferrierite within a Fer Cage: A Prediction Study" Molecules 29, no. 9: 2000. https://doi.org/10.3390/molecules29092000
APA StyleChen, X., Feng, P., & Li, X. (2024). High Reactivity of Dimethyl Ether Activated by Zeolite Ferrierite within a Fer Cage: A Prediction Study. Molecules, 29(9), 2000. https://doi.org/10.3390/molecules29092000