
o-Halogenation and -Alkoxylation of Phenylglycine Derivatives by Pd-Mediated C-H Functionalization: Scope and Limitations



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Halogenation Reactions of Phenylglycine
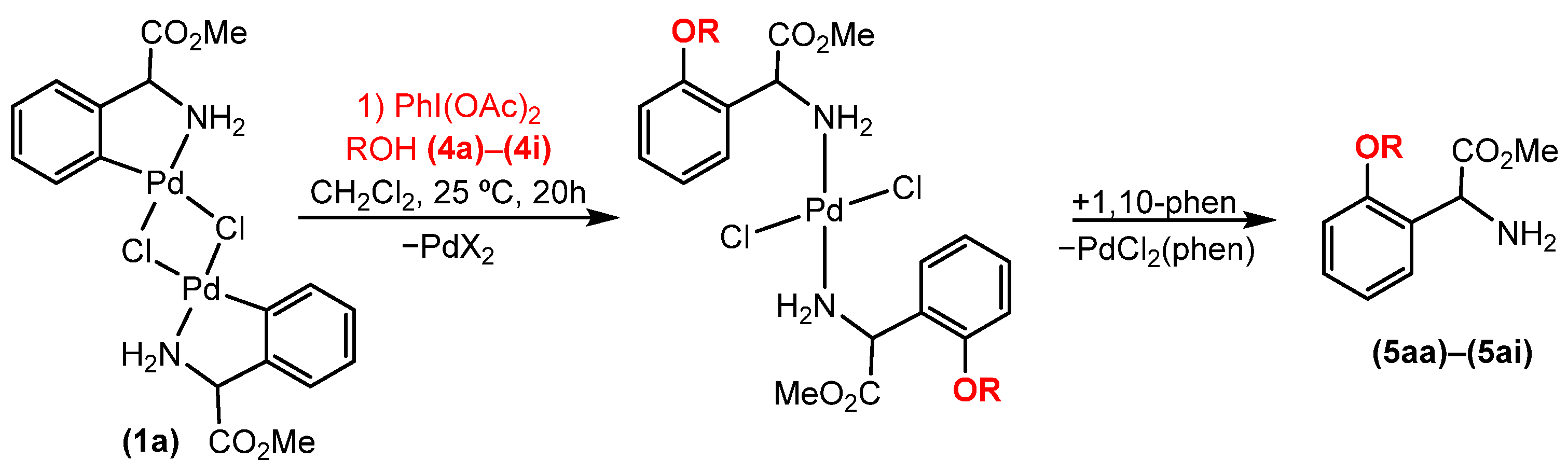
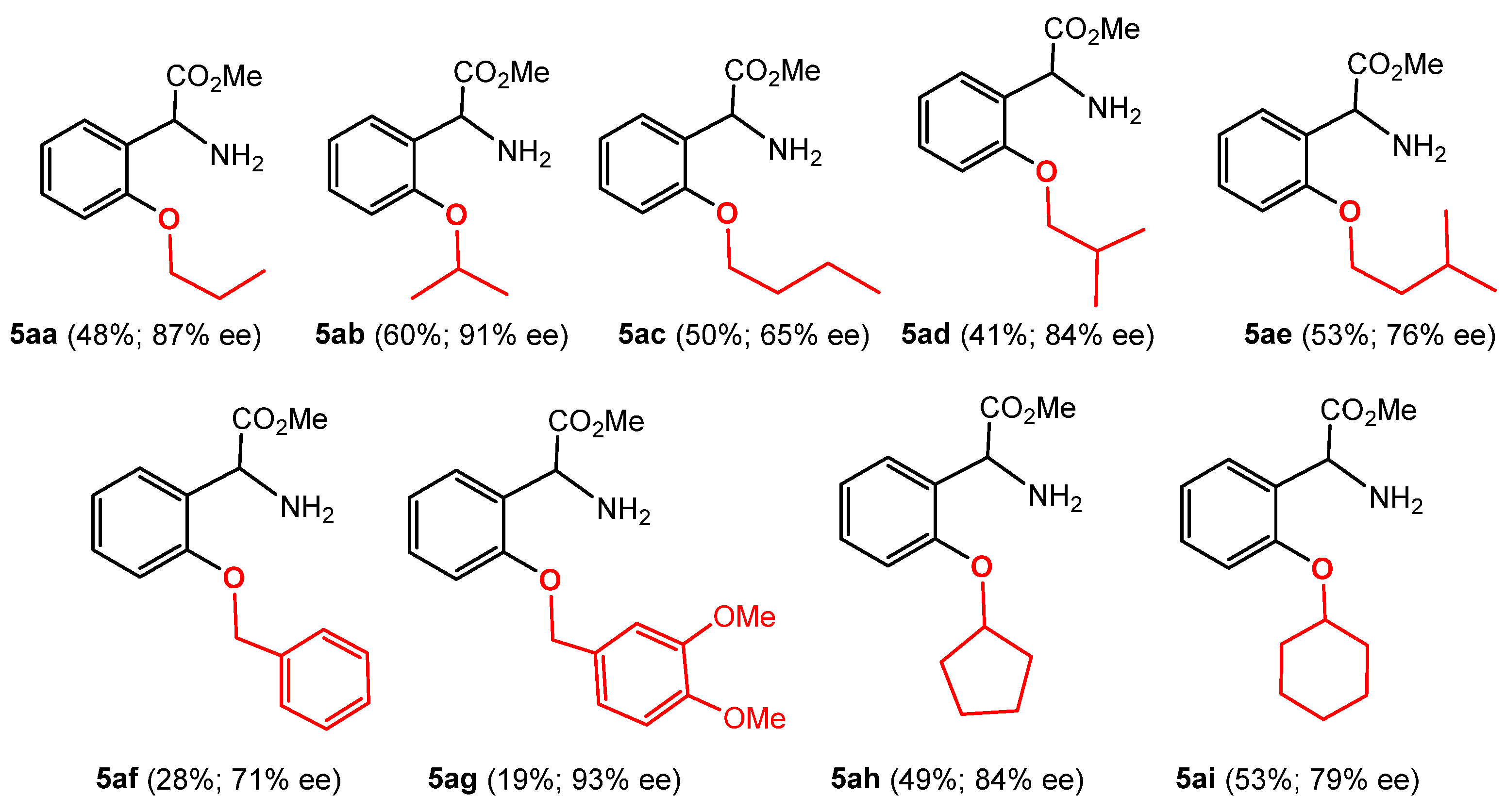
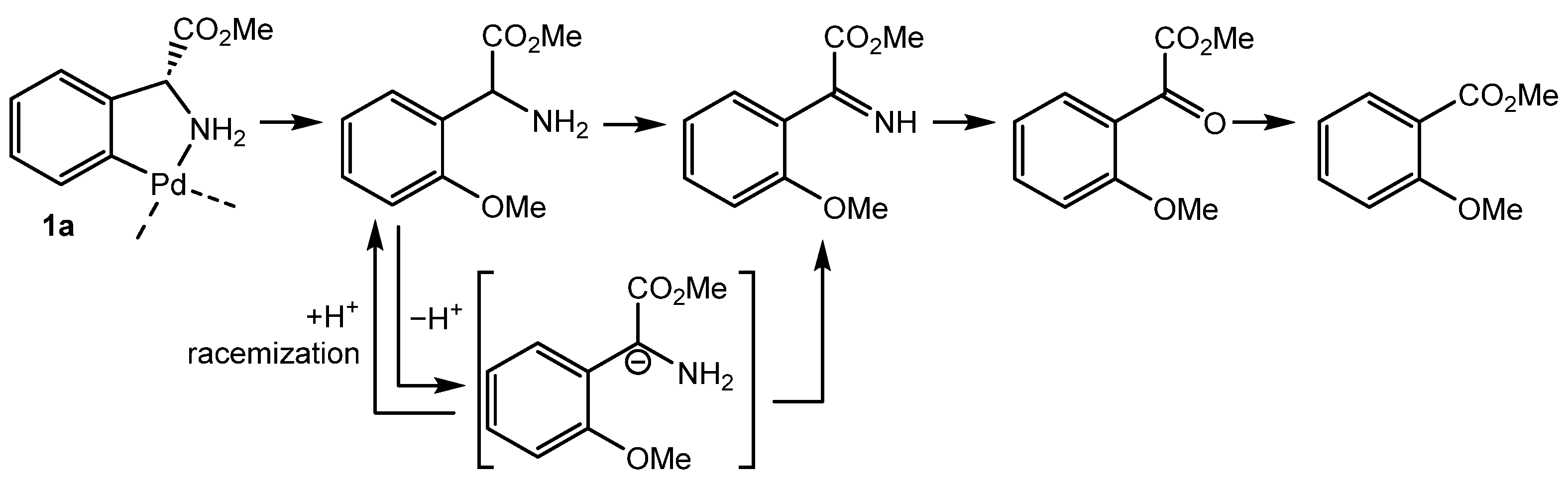
2.2. Alkoxylation Reactions of Phenylglycine
3. Materials and Methods
3.1. General Methods
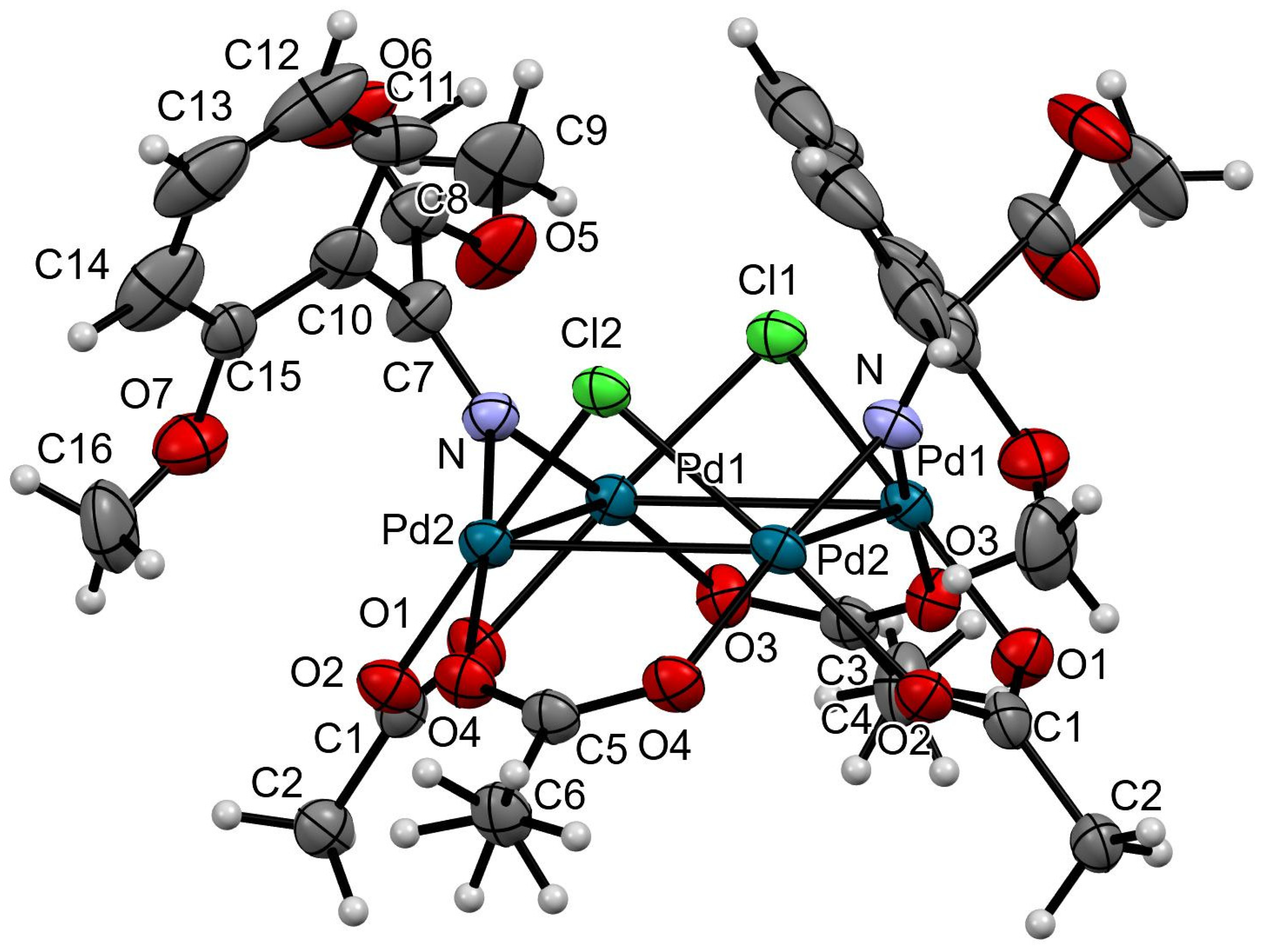
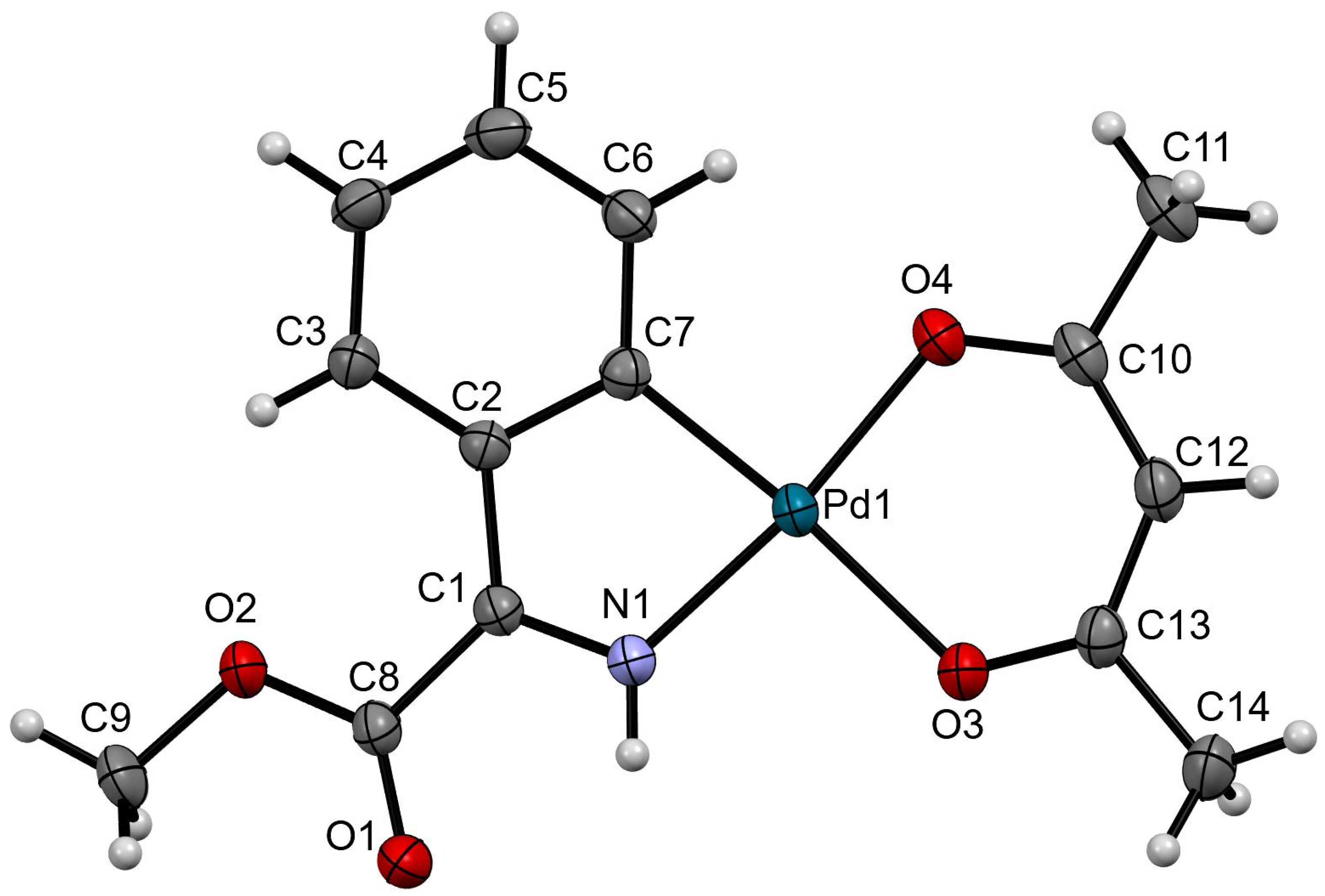
3.2. X-Ray Crystallography
3.2.1. Data Collection
3.2.2. Structure Solution and Refinement
3.3. General Halogenation Procedure
3.3.1. Synthesis of Methyl-2′-(chloro)phenylglycinate (3aa)
3.3.2. Synthesis of Methyl 2′-(Iodo)-6′-(bromo)phenylglycinate (3bc)
3.3.3. Synthesis of Methyl 5′-(Bromo)-2′-(iodo)phenylglycinate (3cc)
3.3.4. Synthesis of Methyl 4′-(Bromo)-2′-(iodo)-phenylglycinate (3dc)
3.3.5. Synthesis of Methyl N,N-Dimethyl-2′-(iodo)phenylglycinate (3ec)
3.3.6. Synthesis of Methyl α-Methyl-2′-(chloro)phenylglycinate (3fa)
3.3.7. Synthesis of Methyl α-Methyl-2′-(bromo)phenylglycinate (3fb)
3.3.8. Synthesis of Methyl α-Methyl-2′-(iodo)phenylglycinate (3fc)
3.3.9. Synthesis of Methyl α-Benzyl-2′-(chloro)phenylglycinate (3ga)
3.3.10. Synthesis of Methyl α-Benzyl-2′-(bromo)phenylglycinate (3gb)
3.3.11. Synthesis of Methyl α-Benzyl-2′-(iodo)phenylglycinate (3gc)
3.4. General Alkoxylation Procedure
3.4.1. Synthesis of (R)-Methyl 2′-(1-Propoxy)phenylglycinate (5aa)
3.4.2. Synthesis of (R)-Methyl 2′-(2-Propoxy)phenylglycinate (5ab)
3.4.3. Synthesis of (R)-Methyl 2′-(1-Butoxy)phenylglycinate (5ac)
3.4.4. Synthesis of (R)-Methyl 2′-(2-Methyl-1-propoxy)phenylglycinate (5ad)
3.4.5. Synthesis of (R)-Methyl 2′-(3-Methyl-1-butoxy)phenylglycinate (5ae)
3.4.6. Synthesis of (R)-Methyl 2′-(Benzyloxy)phenylglycinate (5af)
3.4.7. Synthesis of (R)-Methyl 2′-(3,4-Dimethoxybenzyloxy)phenylglycinate (5ag)
3.4.8. Synthesis of (R)-Methyl 2′-(Cyclopentyloxy)phenylglycinate (5ah)
3.4.9. Synthesis of (R)-Methyl 2′-(Cyclohexyloxy)phenylglycinate (5ai)
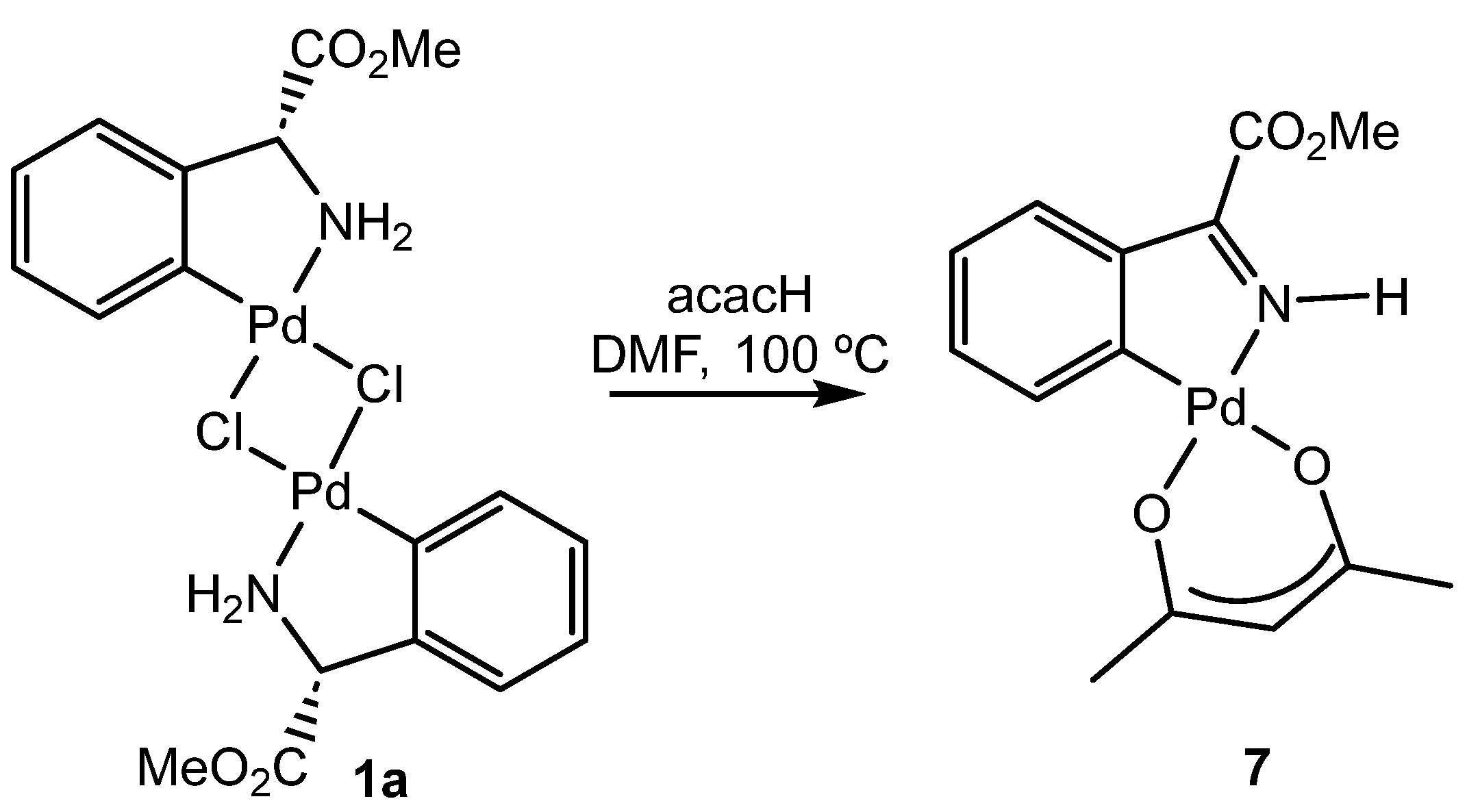
3.4.10. Synthesis of Compound (7)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guillemard, L.; Kaplaneris, N.; Ackermann, L.; Johansson, M.J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem. 2021, 5, 522–545. [Google Scholar] [CrossRef] [PubMed]
- Rogge, T.; Kaplaneris, N.; Chatani, N.; Kim, J.; Chang, S.; Punji, B.; Schafer, L.L.; Musaev, D.G.; Wencel-Delord, J.; Roberts, C.A.; et al. C–H activation. Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar] [CrossRef]
- Dutta, U.; Maiti, S.; Bhattacharya, T.; Maiti, D. Arene diversification through distal C(sp2)–H functionalization. Science 2021, 372, eabd5992. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.Y.S.; Wu, K.; Yu, J.Q. Advancing the Logic of Chemical Synthesis: C–H Activation as Strategic and Tactical Disconnections for C–C Bond Construction. Angew. Chem. Int. Ed. 2021, 60, 15767–15790. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Das, A.; Chatani, N. Strategic evolution in transition metal-catalyzed directed C–H bond activation and future directions. Coord. Chem. Rev. 2021, 431, 213683. [Google Scholar] [CrossRef]
- Das, J.; Mal, D.K.; Maji, S.; Maiti, D. Recent Advances in External-Directing-Group-Free C–H Functionalization of Carboxylic Acids without Decarboxylation. ACS Catal. 2021, 11, 4205–4229. [Google Scholar] [CrossRef]
- Zhang, L.; Ritter, T. A Perspective on Late-Stage Aromatic C–H Bond Functionalization. J. Am. Chem. Soc. 2022, 144, 2399–2414. [Google Scholar] [CrossRef]
- Liu, B.; Romine, A.M.; Rubel, C.Z.; Engle, K.M.; Shi, B.-F. Transition-Metal-Catalyzed, Coordination-Assisted Functionalization of Nonactivated C(sp3)–H Bonds. Chem. Rev. 2021, 121, 14957–15074. [Google Scholar] [CrossRef]
- Thombal, R.S.; Rubio, P.Y.M.; Lee, D.; Maiti, D.; Lee, Y.R. Modern Palladium-Catalyzed Transformations Involving C–H Activation and Subsequent Annulation. ACS Catal. 2022, 12, 5217–5230. [Google Scholar] [CrossRef]
- Monsigny, L.; Doche, F.; Besset, T. Transition-metal-catalyzed C–H bond activation as a sustainable strategy for the synthesis of fluorinated molecules: An overview. Beilstein J. Org. Chem. 2023, 19, 448–473. [Google Scholar] [CrossRef]
- Wu, K.; Lam, N.; Strassfeld, D.A.; Fan, Z.; Qiao, J.X.; Liu, T.; Stamos, D.; Yu, J.-Q. Palladium (II)-Catalyzed C-H Activation with Bifunctional Ligands: From Curiosity to Industrialization. Angew. Chem. Int. Ed. 2024, 63, e20240050929. [Google Scholar]
- Higham, J.I.; Ma, T.-K.; Bull, J.A. When is an Imine Directing Group a Transient Imine Directing Group in C–H Functionalization? Chem. Eur. J. 2024, 30, e202400345. [Google Scholar] [CrossRef] [PubMed]
- Holmberg-Douglas, N.; Nicewicz, D.A. Photoredox-Catalyzed C–H Functionalization. Chem. Rev. 2022, 122, 1925–2016. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Su, C.J.; Jiang, H.; Chiang, C.W.; Saha, P.S.; Gopinath, P. Dual Palladium-Photoredox Catalyzed C–H functionalization. Eur. J. Org. Chem. 2022, 35, e202200733. [Google Scholar]
- Guillemard, L.; Wencel-Delord, J. When metal-catalyzed C–H functionalization meets visible-light photocatalysis. Beilstein J. Org. Chem. 2020, 16, 1754–1804. [Google Scholar] [CrossRef]
- Bellotti, P.; Huang, H.-M.; Faber, T.; Glorius, F. Photocatalytic Late-Stage C–H Functionalization. Chem. Rev. 2023, 123, 4237–4352. [Google Scholar] [CrossRef]
- Liu, J.; Lu, L.; Wood, D.; Lin, S. New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry. ACS Cent. Sci. 2020, 6, 1317–1340. [Google Scholar] [CrossRef]
- Ma, C.; Fang, P.; Mei, T.-S. Recent Advances in C–H Functionalization Using Electrochemical Transition Metal Catalysis. ACS Catal. 2018, 8, 7179–7189. [Google Scholar] [CrossRef]
- Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis. ACC Chem. Res. 2020, 53, 300–310. [Google Scholar] [CrossRef]
- Wang, Y.; Dana, S.; Long, H.; Xu, Y.; Li, Y.; Kaplaneris, N.; Ackermann, L. Electrochemical Late-Stage Functionalization. Chem. Rev. 2023, 123, 11269–11335. [Google Scholar] [CrossRef]
- Zhu, C.; Ang, N.W.J.; Meyer, T.H.; Qiu, Y.; Ackermann, L. Organic Electrochemistry: Molecular Syntheses with Potential. ACS Cent. Sci. 2021, 7, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, C.; Su, W.; Yu, J. Mechanochemical C–X/C–H Functionalization: An Alternative Strategic Access to Pharmaceuticals. Eur. J. Org. Chem. 2022, 2022, e202101440. [Google Scholar] [CrossRef]
- Grover, J.; Prakash, G.; Goswami, N.; Maiti, D. Traditional and sustainable approaches for the construction of C–C bonds by harnessing C–H arylation. Nat. Commun. 2022, 13, 1085. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.G. C–H Bond Functionalization by Mechanochemistry. Chem. Eur. J. 2017, 23, 17157–17165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, Y.; Liu, C.; Zhao, Y. Recent advances in mechanochemical C–H functionalization reactions. Tetrahedron Lett. 2018, 59, 317–324. [Google Scholar] [CrossRef]
- Laskar, R.; Pal, T.; Bhattacharya, T.; Maiti, S.; Akita, M.; Maiti, D. Sustainable C–H functionalization under ball-milling, microwave-irradiation and aqueous media. Green Chem. 2022, 24, 2296–2320. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.M.; Cuchillo, C.; Lehninger, A.L. Principios de Bioquímica; Omega: Barcelona, Spain, 2009. [Google Scholar]
- Noisier, A.F.M.; Brimble, M.A. C–H Functionalization in the Synthesis of Amino Acids and Peptides. Chem. Rev. 2014, 114, 8775–8806. [Google Scholar] [CrossRef]
- He, G.; Wang, B.; Nack, W.A.; Chen, G. Syntheses and Transformations of α-Amino Acids via Palladium-Catalyzed Auxiliary-Directed sp3 C–H Functionalization. ACC Chem. Res. 2016, 49, 635–645. [Google Scholar] [CrossRef]
- Brandhofer, T.; García Macheño, O. Site-selective C–H Bond Activation/Functionalization on Alpha-Amino Acids and Peptide-like Derivatives. Eur. J. Org. Chem. 2018, 2018, 6050–6067. [Google Scholar] [CrossRef]
- Mondal, S.; Chowdhury, S. Recent Advances on Amino Acid Modifications via C–H Functionalization and Decarboxylative functionalization Strategies. Adv. Synth. Catal. 2018, 360, 1884–1912. [Google Scholar] [CrossRef]
- Correa, A. Metal-Catalyzed C(sp2)–H Functionalization Processes of Phenylalanine- and Tyrosine-Containing Peptides. Eur. J. Inorg. Chem. 2021, 2021, 2928–2941. [Google Scholar] [CrossRef]
- Böhm, A.; Polborn, K.; Sünkel, K.; Beck, W. Metal Complexes of Biologically Important Ligands, CIV [1]. ortho-Palladated Complexes of N,N-Dimethyl-C-phenylglycine-methylester. Synthesis of α-Amino Acid Derivatives by Insertion of Isocyanides, CO, Alkenes, and Alkynes into the Pd–C Bond. Z. Naturforsch. 1998, 53, 448–458. [Google Scholar] [CrossRef]
- García-López, J.A.; Saura-Llamas, I.; McGrady, J.E.; Bautista, D.; Vicente, J. Insertion of Allenes into the Pd–C Bond of Ortho-Palladated Primary Arylamines of Biological Relevance: Phenethylamine, Phentermine, (l)-Phenylalanine Methyl Ester, and (l)-Tryptophan Methyl Ester. Synthesis of Tetrahydro-3-benzazepines and Their Salts. Organometallics 2012, 31, 8333–8347. [Google Scholar] [CrossRef]
- Li, J.-J.; Mei, T.-S.; Yu, J.-Q. Synthesis of Indolines and Tetrahydroisoquinolines from Arylethylamines by PdII-Catalyzed C–H Activation Reactions. Angew. Chem. Int. Ed. 2008, 47, 6452–6455. [Google Scholar] [CrossRef]
- Antermite, D.; Bull, J.A. Transition Metal-Catalyzed Directed C(sp3)–H Functionalization of Saturated Heterocycles. Synthesis 2019, 51, 3171–3204. [Google Scholar] [CrossRef]
- Piticari, A.-S.; Larionova, N.; Bull, J.A. C–H Functionalization of Saturated Heterocycles Beyond the C2 Position. In Transition-Metal-Catalyzed C–H Functionalization of Heterocycles; Punniyamurthy, T., Kumar, A., Eds.; Wiley: Hoboken, NJ, USA, 2023; pp. 567–607. [Google Scholar]
- Xia, G.; Zhuang, Z.; Liu, L.-Y.; Schreiber, S.L.; Melillo, B.; Yu, J.-Q. Ligand-Enabled b-methylene C(sp3)–H Arylation of Masked Aliphatic Alcohols. Angew. Chem. Int. Ed. 2020, 59, 7783–7787. [Google Scholar] [CrossRef]
- Shang, M.; Feu, K.S.; Vantourout, J.C.; Barton, L.M.; Osswald, H.L.; Kato, N.; Gagaring, K.; McNamara, C.W.; Chen, G.; Hu, L.; et al. Modular, stereocontrolled Cβ–H/Cα–C activation of alkyl carboxylic acids. Proc. Nat. Acad. Sci. USA 2019, 116, 8721–8727. [Google Scholar] [CrossRef]
- Hutskalova, V.; Mykhailiuk, P.K. Pd-Catalyzed directed CH-(hetero)arylation of cyclic α-amino acids: Effects of substituents and the ring size. Org. Biomol. Chem. 2019, 17, 4342–4349. [Google Scholar] [CrossRef]
- Affron, D.P.; Davis, O.A.; Bull, J.A. Regio- and Stereospecific Synthesis of C-3 Functionalized Proline Derivatives by Palladium Catalyzed Directed C(sp3)–H Arylation. Org. Lett. 2014, 16, 4956–4959. [Google Scholar] [CrossRef]
- Affron, D.P.; Bull, J.A. Palladium-Catalyzed Directed C(sp3)–H Arylation of Saturated Heterocycles at C-3 Using a Concise Optimization Approach. Eur. J. Org. Chem. 2016, 2016, 139–149. [Google Scholar] [CrossRef]
- Verho, O.; Maetani, M.; Melillo, B.; Zoller, J.; Schreiber, S.L. Stereospecific Palladium-Catalyzed C–H Arylation of Pyroglutamic Acid Derivatives at the C3 Position Enabled by 8-Aminoquinoline as a Directing Group. Org. Lett. 2017, 19, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.C.; Collingridge, G.L. Phenylglycine Derivatives as Antagonists of Metabotropic Glutamate Receptors. Trends Pharmacol. Sci. 1994, 15, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Stoica, B.; Hanscom, M.; Kabadi, S.; Faden, A. Positive Allosteric Modulators (PAMs) of Metabotropic Glutamate Receptor 5 (MGluR5) Attenuate Microglial Activation. CNS Neurol. Disord.—Drug Targets 2014, 13, 558–566. [Google Scholar] [CrossRef]
- Mortell, K.H.; Anderson, D.J.; Lynch, J.J.; Nelson, S.L.; Sarris, K.; McDonald, H.; Sabet, R.; Baker, S.; Honore, P.; Lee, C.-H.; et al. Structure–Activity Relationships of α-Amino Acid Ligands for the A2δ Subunit of Voltage-Gated Calcium Channels. Bioorg. Med. Chem. Lett. 2006, 16, 1138–1141. [Google Scholar] [CrossRef]
- Ylikangas, H.; Malmioja, K.; Peura, L.; Gynther, M.; Nwachukwu, E.O.; Leppänen, J.; Laine, K.; Rautio, J.; Lahtela-Kakkonen, M.; Huttunen, K.M.; et al. Quantitative Insight into the Design of Compounds Recognized by the L-Type Amino Acid Transporter 1 (LAT1). ChemMedChem 2014, 9, 2699–2707. [Google Scholar] [CrossRef]
- Shiau, C.-Y.; Pai, S.-C.; Lin, W.-P.; Ji, D.-D.; Liu, Y.-T. Purification and Characterization of Inducible Cephalexin Synthesizing Enzyme in Gluconobacter Oxydans. Biosci. Biotechnol. Biochem. 2005, 69, 463–469. [Google Scholar] [CrossRef]
- McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar] [CrossRef]
- Top Pharmaceuticals Poster|Njarðarson. Available online: https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster (accessed on 11 November 2024).
- Sierra, M.A.; Gómez-Gallego, M.; Alcaraz, R.; Ramírez, P.; Moreno, J.; Lucena, J.J. Novel Method of Preparing Hydroxyarylglycines, Alkoxyarylglycines and the Glycinates Thereof. Patent WO 02/102762 A1, 27 December 2002. [Google Scholar]
- Wu, Y.; Qi, Z.; Wang, B.; Wang, J.; Liu, Q.; Wang, A.; Shi, C.; Zhou, B.; Liang, Q.; Wang, W.; et al. Discovery of IHMT-MST1-58 as a Novel, Potent, and Selective MST1 Inhibitor for the Treatment of Type 1/2 Diabetes. J. Med. Chem. 2022, 65, 11818–11839. [Google Scholar] [CrossRef]
- Gómez-Gallego, M.; Sierra, M.A.; Alcázar, R.; Ramírez, P.; Piñar, C.; Mancheño, M.J.; García-Marco, S.; Yunta, F.; Lucena, J.J. Synthesis of o,p-EDDHA and Its Detection as the Main Impurity in o,o-EDDHA Commercial Iron Chelates. J. Agric. Food Chem. 2002, 50, 6395–6399. [Google Scholar] [CrossRef]
- Nájera, C.; Sansano, J.M. Catalytic Asymmetric Synthesis of α-Amino Acids. Chem. Rev. 2007, 107, 4584–4671. [Google Scholar] [CrossRef]
- Olefination: Xiao, K.-J.; Chu, L.; Yu, J.-Q. Enantioselective C–H Olefination of α-Hydroxy and α-Amino Phenylacetic Acids by Kinetic Resolution. Angew. Chem. Int. Ed. 2016, 55, 2856–2860. [Google Scholar] [CrossRef]
- Zeng, W.; Nukeyeva, M.; Wang, Q.; Jiang, C. Synthesis of unnatural α-amino acid derivatives via selective o-C–H functionalization. Org. Biomol. Chem. 2018, 16, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Dastbaravardeh, N.; Toba, T.; Farmer, M.E.; Yu, J.Q. Monoselective o-C–H Functionalizations of Mandelic Acid and α-Phenylglycine. J. Am. Chem. Soc. 2015, 137, 9877–9884. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Chand-Thakuri, P.; Young, M.C. Carbon Dioxide-Mediated C(sp2)–H Arylation of Primary and Secondary Benzylamines. J. Am. Chem. Soc. 2019, 141, 7980–7989. [Google Scholar] [CrossRef] [PubMed]
- Kinsinger, T.; Kazmaier, U. C–H Functionalization of N-Methylated Amino Acids and Peptides as Tool in Natural Product Synthesis: Synthesis of Abyssenine A and Mucronine E. Org. Lett. 2018, 20, 7726–7730. [Google Scholar] [CrossRef]
- Terrey, M.J.; Holmes, A.; Perry, C.C.; Cross, W.B. C–H Olefination of Tryptophan Residues in Peptides: Control of Residue Selectivity and Peptide–Amino Acid Cross-linking. Org. Lett. 2019, 21, 7902–7907. [Google Scholar] [CrossRef]
- Martínez-Mingo, M.; García-Viada, A.; Alonso, I.; Rodríguez, N.; Gómez Arrayás, R.; Carretero, J.C. Overcoming the Necessity of γ-Substitution in δ-C(sp3)–H Arylation: Pd-Catalyzed Derivatization of α-Amino Acids. ACS Catal. 2021, 11, 5310–5317. [Google Scholar] [CrossRef]
- San Segundo, M.; Correa, A. Radical C–H Alkylation with Ethers and Unactivated Cycloalkanes toward the Assembly of Tetrasubstituted Amino Acid Derivatives. Adv. Synth. Catal. 2022, 364, 3161–3167. [Google Scholar] [CrossRef]
- Nieto, S.; Arnau, P.; Serrano, E.; Navarro, R.; Soler, T.; Cativiela, C.; Urriolabeitia, E.P. Functionalization of Methyl (R)-Phenylglycinate Through Orthopalladation: C–Hal, C–O, C–N, and C–C Bond Coupling. Inorg. Chem. 2009, 48, 11963–11975. [Google Scholar] [CrossRef]
- Urriolabeitia, E.P.; Laga, E.; Cativiela, C. Synthesis of Conformationally Restricted Glutamate and Glutamine Derivatives from Carbonylation of Orthopalladated Phenylglycine Derivatives. Beilstein J. Org. Chem. 2012, 8, 1569–1575. [Google Scholar] [CrossRef]
- Laga, E.; García-Montero, A.; Sayago, F.J.; Soler, T.; Moncho, S.; Cativiela, C.; Martínez, M.; Urriolabeitia, E.P. Cyclopalladation and Reactivity of Amino Esters through C-H Bond Activation: Experimental, Kinetic, and Density Functional Theory Mechanistic Studies. Chem.—Eur. J. 2013, 19, 17398–17412. [Google Scholar] [CrossRef] [PubMed]
- Laga, E.; Cativiela, C.; Urriolabeitia, E.P. Alkenylation of ortho-palladated phenylglycine: Synthesis of stilbene derivatives and 3-aryl-isoquinoline-1-carboxylates. J. Organomet. Chem. 2020, 925, 121482. [Google Scholar] [CrossRef]
- Ruiz, S.; Sayago, F.J.; Cativiela, C.; Urriolabeitia, E.P. Ru-Catalyzed C H Functionalization of Phenylglycine Derivatives: Synthesis of Isoquinoline-1-Carboxylates and Isoindoline-1-Carboxylates. J. Mol. Catal. A Chem. 2017, 426, 407–418. [Google Scholar] [CrossRef]
- Fuchita, Y.; Yoshinaga, K.; Ikeda, Y.; Kinoshita-Kawashima, J. Synthesis of Optically Active Cyclopalladated Complexes of Primary Benzylamine Derivatives, (R)-(−)-2-Phenylglycine Methyl Ester and (±)-1-Phenylethylamine. J. Chem. Soc. Dalton Trans. 1997, 2495–2500. [Google Scholar] [CrossRef]
- Breveglieri, F.; Mazzotti, M. Role of Racemization Kinetics in the Deracemization Process via Temperature Cycles. Cryst. Growth Des. 2019, 19, 3551–3558. [Google Scholar] [CrossRef]
- Zhao, X.-F.; Zhang, C. Iodobenzene Dichloride as a Stoichiometric Oxidant for the Conversion of Alcohols into Carbonyl Compounds; Two Facile Methods for Its Preparation. Synthesis 2007, 4, 551–557. [Google Scholar] [CrossRef]
- Rathore, P.S.; Advani, J.; Rathore, S.; Thakore, S. Metal nanoparticles assisted amine catalyzed transesterification under ambient conditions. J. Mol. Catal. A Chem. 2013, 377, 129–136. [Google Scholar] [CrossRef]
- Sun, X.; Sun, Y.; Zhang, C.; Rao, Y. Room-temperature Pd-catalyzed C–H chlorination by weak coordination: One-pot synthesis of 2-chlorophenols with excellent regioselectivity. Chem. Commun. 2014, 50, 1262–1264. [Google Scholar] [CrossRef]
- Lu, C.; Zhang, S.-Y.; He, G.; Nack, W.A.; Chen, G. Palladium-catalyzed picolinamide-directed halogenation of ortho C-H bonds of benzylamine substrates. Tetrahedron 2014, 70, 4197–4203. [Google Scholar] [CrossRef]
- Ma, X.-T.; Tian, S.-K. Palladium-Catalyzed Regioselective Halogenation of Aromatic Azo Compounds. Adv. Synth. Catal. 2013, 355, 337–340. [Google Scholar] [CrossRef]
- Sun, X.; Shan, G.; Sun, Y.; Rao, Y. Regio- and Chemoselective C-H Chlorination/Bromination of Electron-Deficient Arenes by Weak Coordination and Study of Relative Directing-Group Abilities. Angew. Chem. Int. Ed. 2013, 52, 4440–4444. [Google Scholar] [CrossRef] [PubMed]
- Organ, M.G.; Ghasemi, H. Metal-Catalyzed Coupling Reactions on an Olefin Template: The Total Synthesis of (13E,15E,18Z,20Z)-1-Hydroxypentacosa-13,15,18,20-tetraen-11-yn-4-one 1-Acetate. J. Org. Chem. 2004, 69, 695–700. [Google Scholar] [CrossRef] [PubMed]
- de Jong, G.T.; Kovács, A.; Bickelhaupt, F.M. Oxidative Addition of Hydrogen Halides and Dihalogens to Pd. Trends in Reactivity and Relativistic Effects. J. Phys. Chem. A 2006, 110, 7943–7951. [Google Scholar] [CrossRef] [PubMed]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451. [Google Scholar] [CrossRef]
- Albert, J.; Cadena, J.M.; González, A.; Granell, J.; Solans, X.; Font-Bardia, M. Deamination of the Amino Acid Fragment in Imine Metallacycles: Unexpected Synthesis of an NH-Aldimine Organometallic Compound. Chem. Eur. J. 2006, 12, 887–894. [Google Scholar] [CrossRef]
- Albert, J.; Cadena, J.M.; González, A.; Granell, J.; Solans, X.; Font-Bardia, M. The first NH aldimine organometallic compound. Isolation and crystal structure. Chem. Commun. 2003, 528–529. [Google Scholar] [CrossRef]
- García-Raso, A.; Deyá, P.M.; Saá, J.M. Oxidation of α-Amino Acids and α-Hydroxy Acids by Fremy’s Salt. A Model for Oxidases? J. Org. Chem. 1986, 51, 4285–4287. [Google Scholar] [CrossRef]
- Weast, R.C. CRC Handbook of Chemistry and Physics, 66th ed.; CRC Press, Inc.: Boca Raton, FL, USA, 1986. [Google Scholar]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Supplement. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J. Chem. Soc. Dalton Trans. 1989, S1–S83. [Google Scholar] [CrossRef]
- Wong, W.-K.; Zhang, L.-L.; Chen, Y.; Wong, W.-Y.; Wong, W.-T.; Xue, F.; Mak, T.C.W. Reactivity of chiral diiminodiphosphine ligands towards PdCl2(PhCN)2: Synthesis and crystal structures of two unexpected dinuclear palladium (II) complexes. J. Chem. Soc. Dalton Trans. 2000, 1397–1398. [Google Scholar] [CrossRef]
- Besenyei, G.; Párkányi, L.; Szalontai, G.; Holly, S.; Pápai, I.; Keresztury, G.; Nagy, A. N-benzoylimido complexes of palladium. Synthesis, structural characterisation and structure–reactivity relationship. Dalton Trans. 2004, 13, 2041–2050. [Google Scholar] [CrossRef]
- Ruiz, J.; Rodríguez, V.; Cutillas, N.; Hoffmann, A.; Chamayou, A.-C.; Kazmierczak, K.; Janiak, C. Structure–solid-state CPMAS 13C NMR correlation in palladacycle solvates (pseudo-polymorphs) with a transformation from Z′ = 1 to Z′ = 2. CrystEngComm 2008, 10, 1928–1938. [Google Scholar] [CrossRef]
- Fomina, I.G.; Sidorov, A.A.; Aleksandrov, G.G.; Nefedov, S.E.; Eremenko, I.L.; Moiseev, I.I. Deprotonated N-phenyl-o-phenylenediimine as a bridging ligand. J. Organomet. Chem. 2001, 636, 157–163. [Google Scholar] [CrossRef]
- Ruiz, J.; Rodríguez, V.; Cutillas, N.; Florenciano, F.; Pérez, J.; López, G. First complex containing a Pd2(μ2-N=CPh2)2 functional group. Inorg. Chem. Commun. 2001, 4, 23–25. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- Vicente, J.; Abad, J.A.; López-Sáez, M.-J.; Jones, P.G. Reactivity of ortho-Palladated Phenol Derivatives with Unsaturated Molecules: Insertion of Nitriles into a Late-Transition-Metal–Carbon Bond. Angew. Chem. Int. Ed. 2005, 44, 6001–6004. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, W.-H.; Liu, G.; Meng, F.-K.; Chen, J.-H.; Yang, Z.; Shi, Z.-J. Direct Imidation to Construct 1H-Benzo[d]imidazole through PdII-Catalyzed C-H Activation Promoted by Thiourea. Chem. Eur. J. 2009, 15, 7292–7296. [Google Scholar] [CrossRef]
- Masui, H. Metalloaromaticity. Coord. Chem. Rev. 2001, 219–221, 957–992. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. Metalloaromaticity. Wiley Interdiscip Rev. Comput. Mol. Sci. 2012, 3, 105–122. [Google Scholar] [CrossRef]
- SAINT Software Reference Manuals, Version V8.40B in APEX4; Bruker Analytical Xray Systems, Inc.: Madison, WI, USA, 2016.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-86, Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. 1990, 46, 467–473. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS 97 and SHELXL 97, Program for Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97, A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laga, E.; Nieto, S.; Cativiela, C.; Urriolabeitia, E.P. o-Halogenation and -Alkoxylation of Phenylglycine Derivatives by Pd-Mediated C-H Functionalization: Scope and Limitations. Molecules 2025, 30, 236. https://doi.org/10.3390/molecules30020236
Laga E, Nieto S, Cativiela C, Urriolabeitia EP. o-Halogenation and -Alkoxylation of Phenylglycine Derivatives by Pd-Mediated C-H Functionalization: Scope and Limitations. Molecules. 2025; 30(2):236. https://doi.org/10.3390/molecules30020236
Chicago/Turabian StyleLaga, Eduardo, Sonia Nieto, Carlos Cativiela, and Esteban P. Urriolabeitia. 2025. "o-Halogenation and -Alkoxylation of Phenylglycine Derivatives by Pd-Mediated C-H Functionalization: Scope and Limitations" Molecules 30, no. 2: 236. https://doi.org/10.3390/molecules30020236
APA StyleLaga, E., Nieto, S., Cativiela, C., & Urriolabeitia, E. P. (2025). o-Halogenation and -Alkoxylation of Phenylglycine Derivatives by Pd-Mediated C-H Functionalization: Scope and Limitations. Molecules, 30(2), 236. https://doi.org/10.3390/molecules30020236