
Chiroptical Spectroscopy, Theoretical Calculations, and Symmetry of a Chiral Transition Metal Complex with Low-Lying Electronic States


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
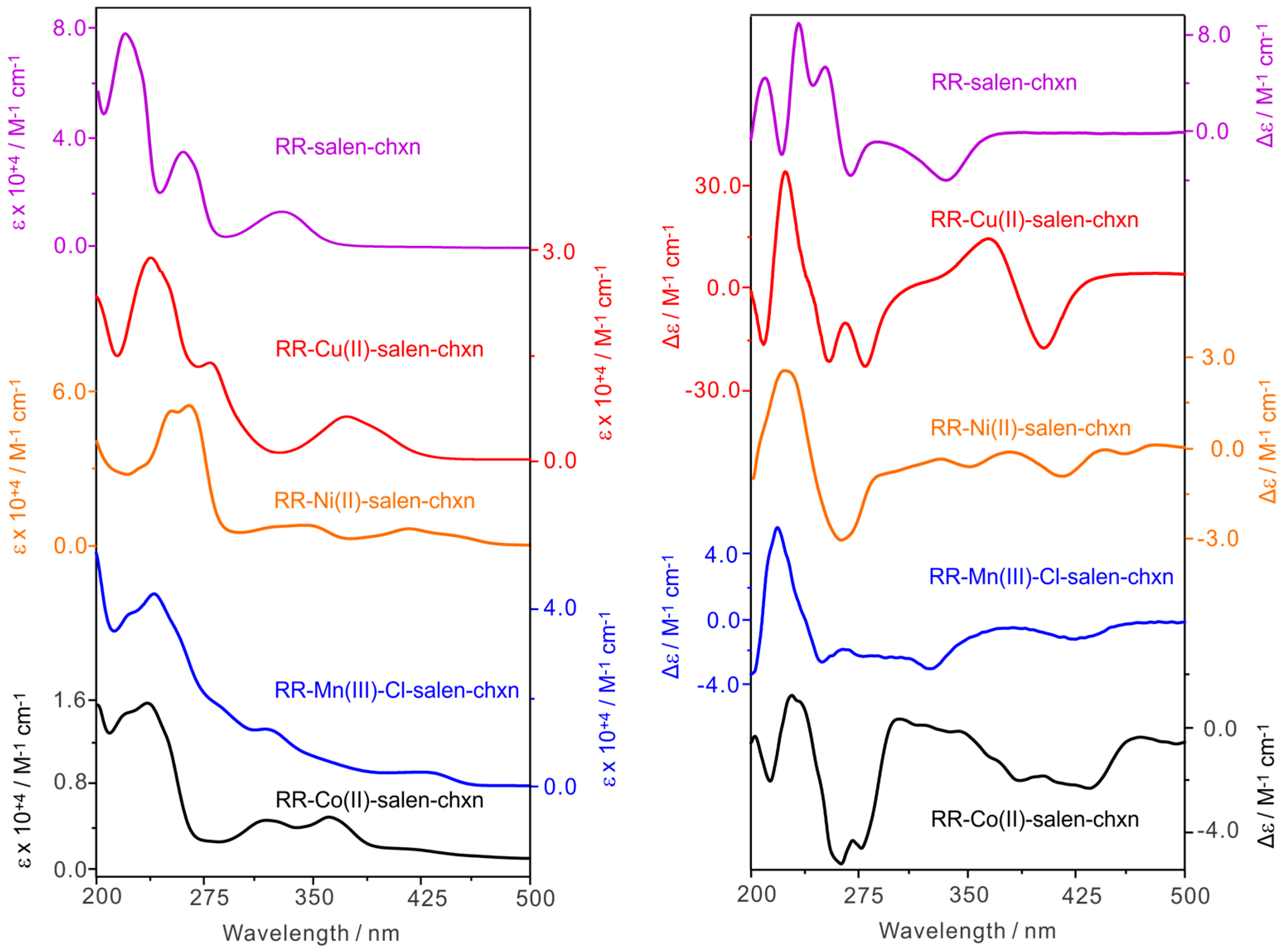
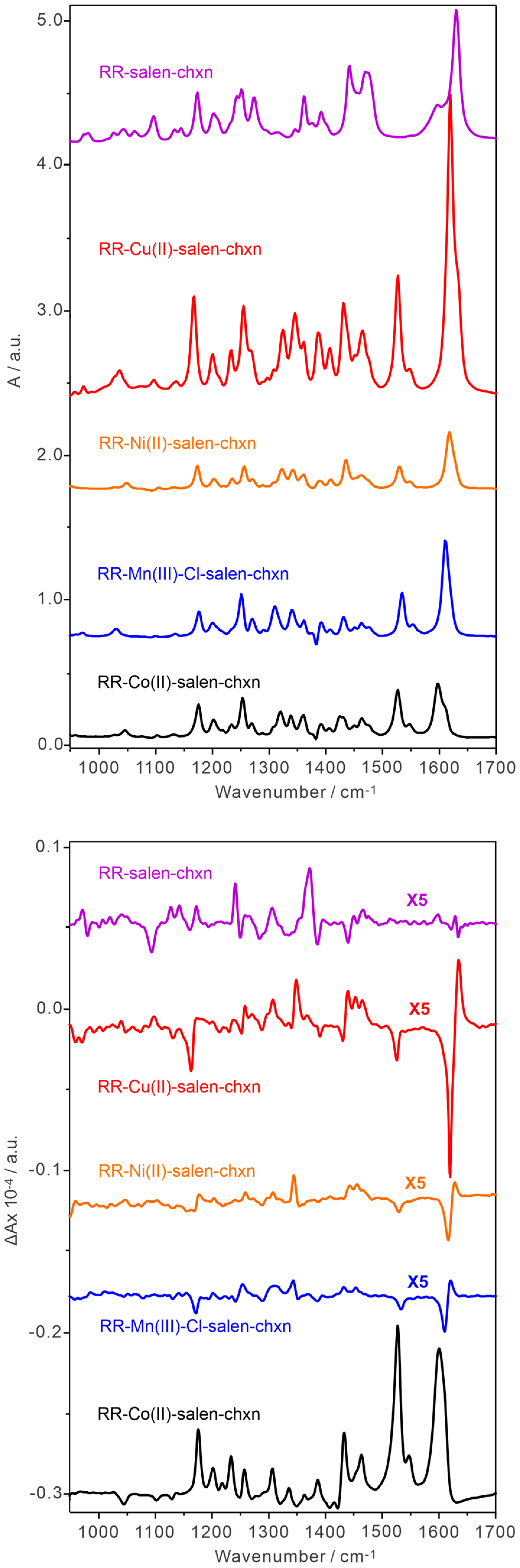
2.1. Comparison of the Experimental UV-Vis, ECD, IR, and VCD Spectra of the Co(II)-Salen-Chxn Complex with Those of Its Ligand and Other Related Complexes
2.2. The Low-Energy Conformers of the Co(II)-Salen-Chxn Complex
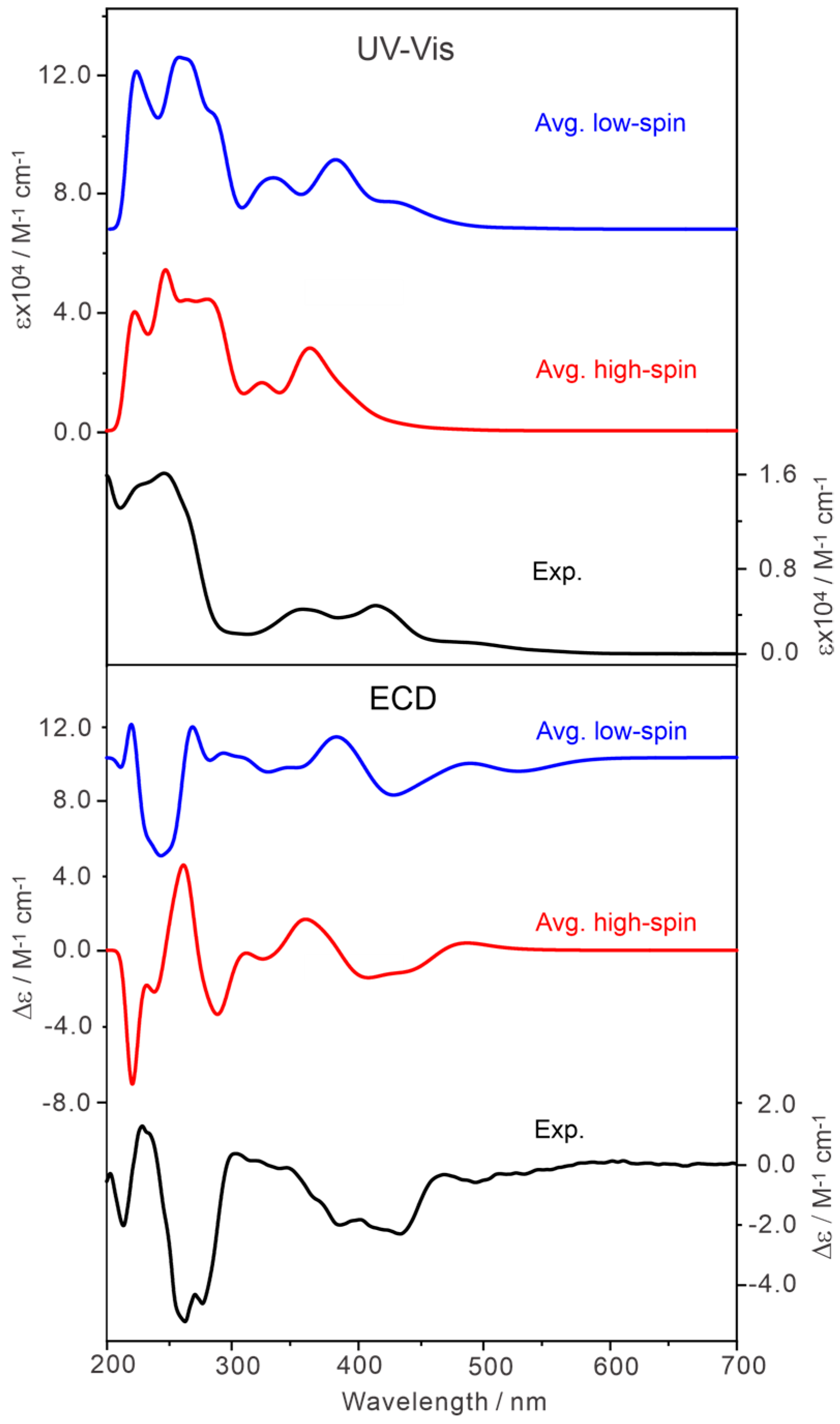
2.3. Simulated UV-Vis and ECD Spectra and the Experimental Results
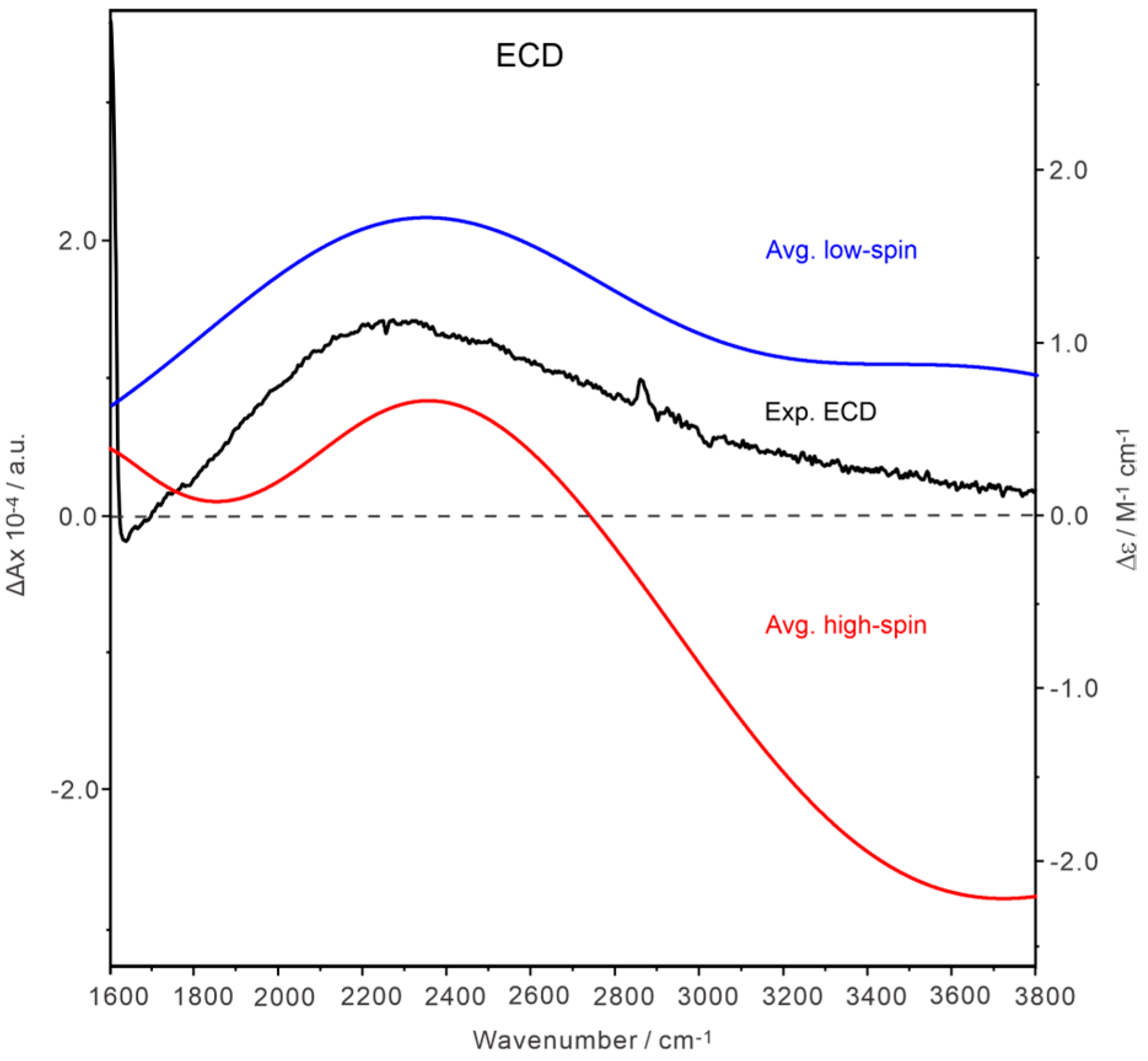
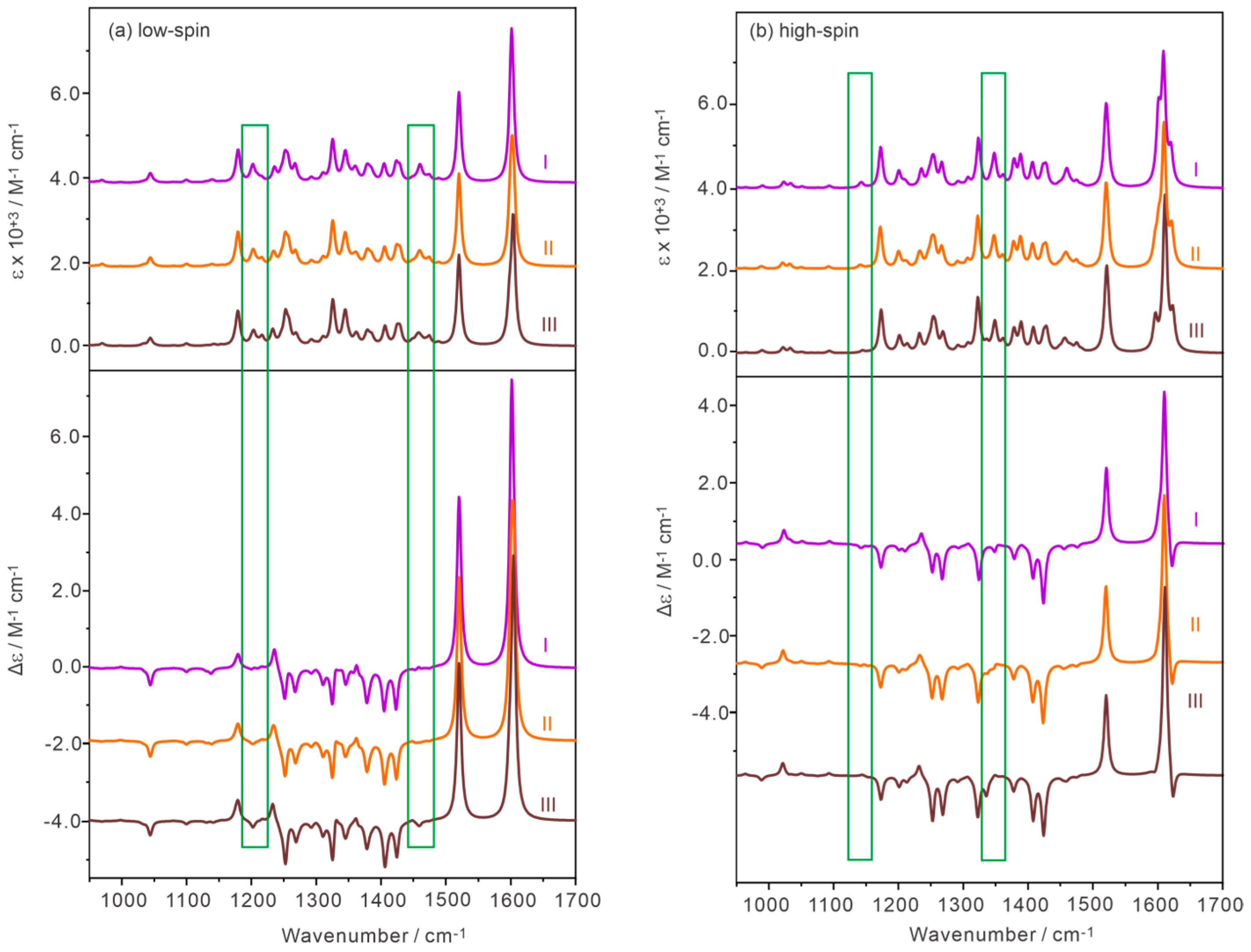
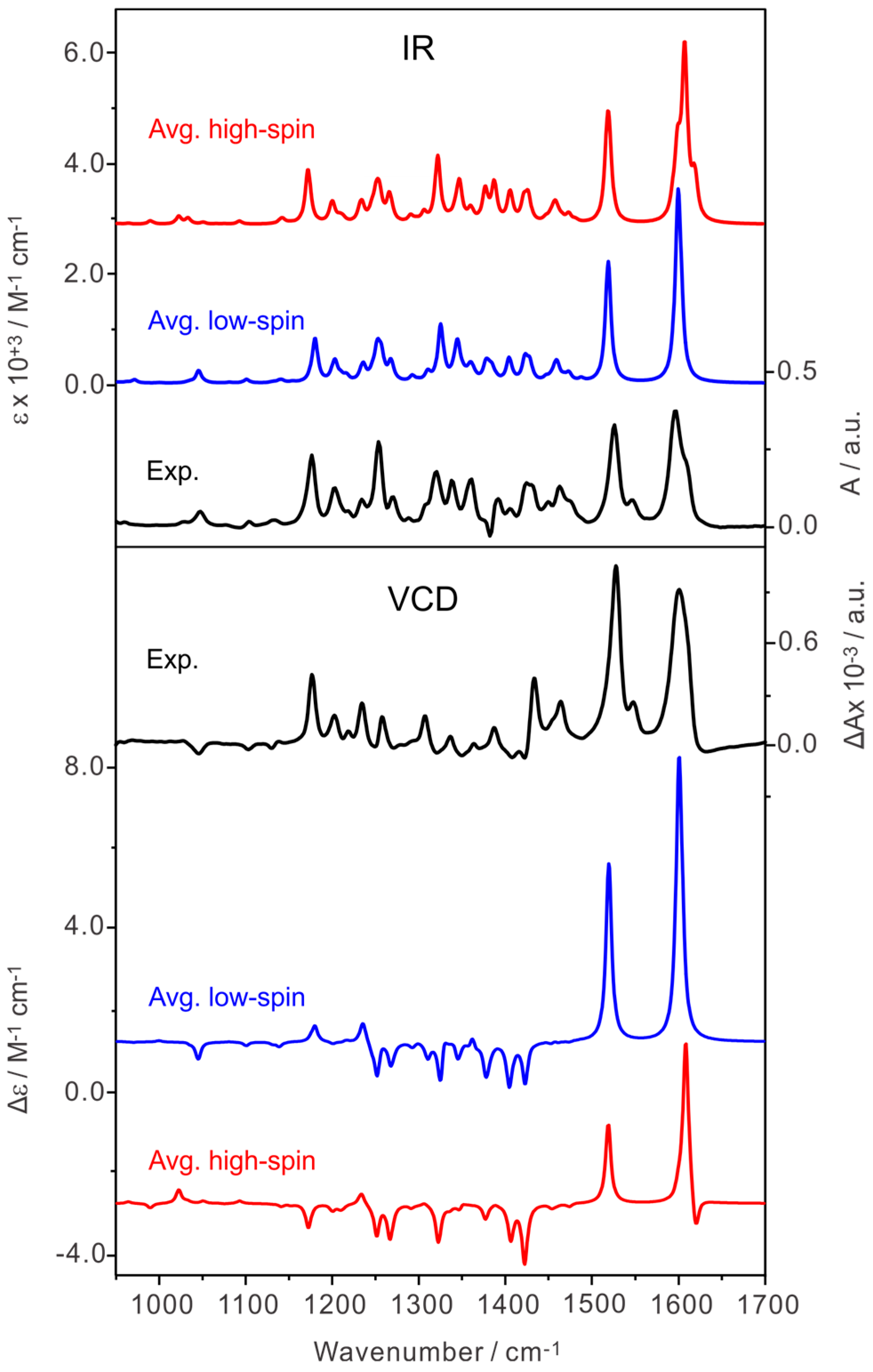
2.4. Comparison of the Experimental and Simulated IR and VCD Spectra
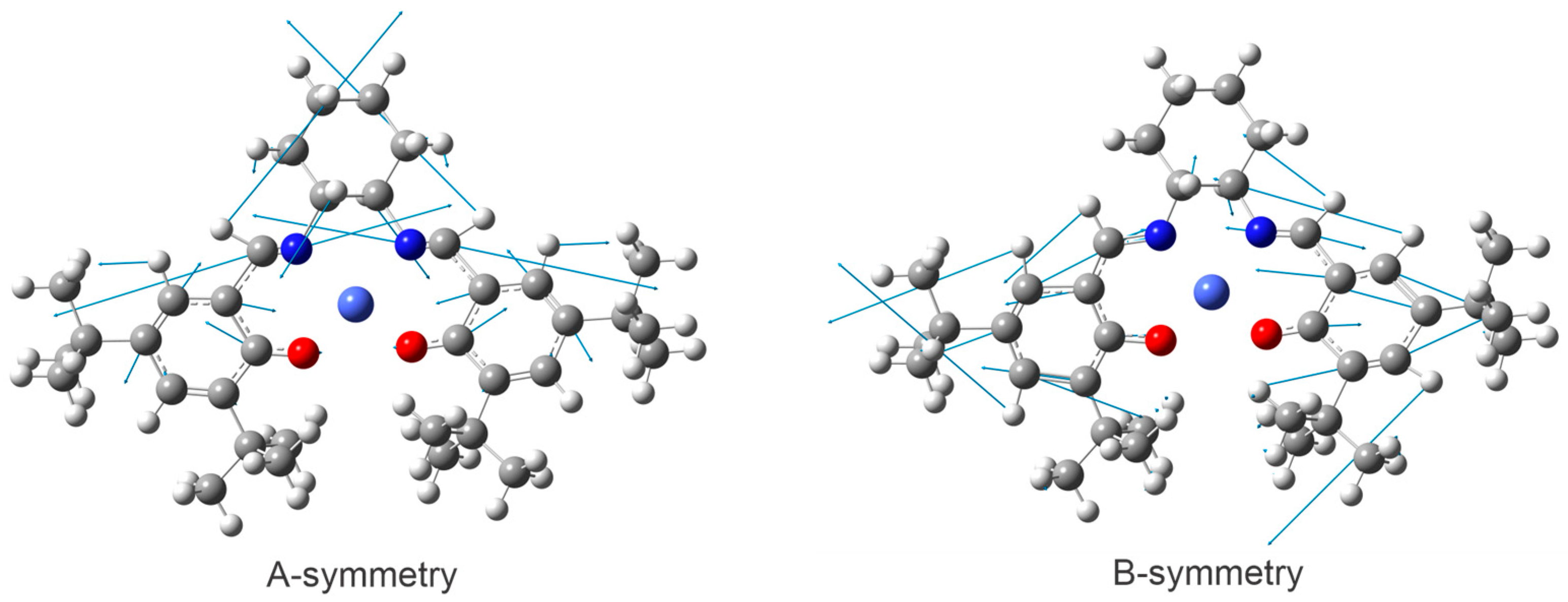
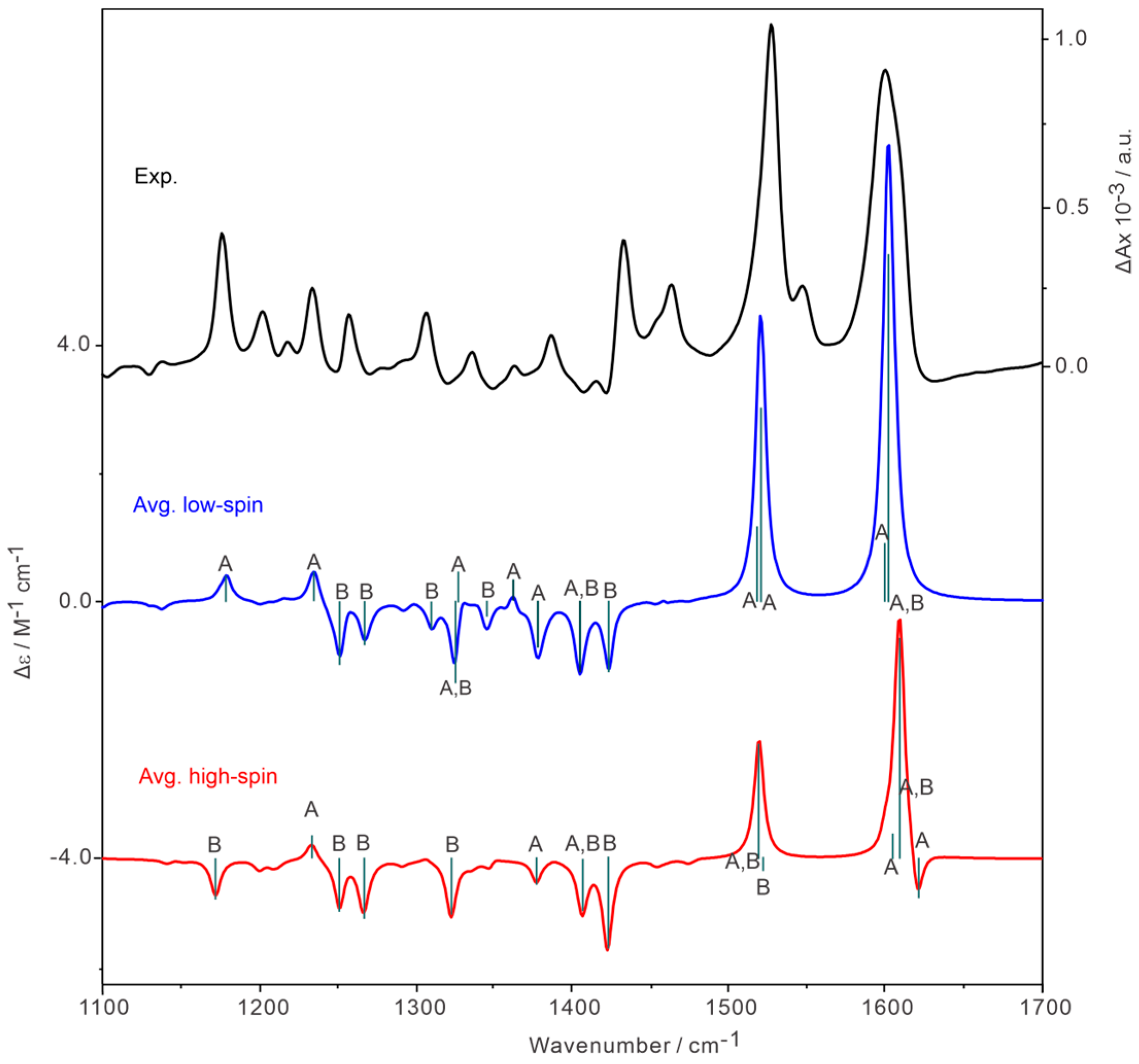
2.5. Discussion on Symmetry-Dependent VCD Enhancement
3. Materials and Methods
3.1. Experimental
3.2. Theoretical
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soroceanu, A.; Bargan, A. Advanced and biomedical applications of Schiff-base ligands and their metal complexes: A review. Crystals 2022, 12, 1436. [Google Scholar] [CrossRef]
- More, M.S.; Joshi, P.G.; Mishra, Y.K.; Khanna, P.K. Metal complexes driven from Schiff bases and semicarbazones for biomedical and allied applications: A review. Mater. Today Chem. 2019, 14, 100195. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Manzur, C.; Novoa, N.; Celedón, S.; Carrillo, D.; Hamon, J.R. Multidentate unsymmetrically-substituted Schiff bases and their metal complexes: Synthesis, functional materials properties, and applications to catalysis. Coord. Chem. Rev. 2018, 357, 144–172. [Google Scholar] [CrossRef]
- Goshisht, M.K.; Patra, G.K.; Tripathi, N. Fluorescent Schiff base sensors as a versatile tool for metal ion detection: Strategies, mechanistic insights, and applications. Mater. Adv. 2022, 3, 2612–2669. [Google Scholar] [CrossRef]
- Górecki, M.; Enamullah, M.; Islam, M.A.; Islam, M.K.; Höfert, S.-P.; Woschko, D.; Janiak, C.; Pescitelli, G. Synthesis and characterization of Bis[(R or S)-N-1-(X-C6H4)ethyl-2-oxo-1-naphthaldiminato-κ2N,O]-Λ/Δ-cobalt(II) (X = H, p-CH3O, p-Br) with symmetry- and distance-dependent vibrational circular dichroism enhancement and sign inversion. Inorg. Chem. 2021, 60, 14116–14131. [Google Scholar] [CrossRef]
- Pescitelli, G.; Lüdeke, S.; Gorecki, M.; Di Bari, L. Symmetry-dependent vibrational circular dichroism enhancement in Co (II) salicylaldiminato complexes. J. Phys. Chem. Lett. 2019, 10, 650–654. [Google Scholar] [CrossRef]
- Dezhahang, Z.; Poopari, M.R.; Xu, Y. Vibrational circular dichroism spectroscopy of three multidentate nitrogen donor ligands: Conformational flexibility and solvent effects. Chem. Asian J. 2013, 8, 1205–1212. [Google Scholar] [CrossRef]
- Fusè, M.; Mazzeo, G.; Abbate, S.; Ruzziconi, R.; Bloino, J.; Longhi, G. Mid-IR and CH stretching vibrational circular dichroism spectroscopy to distinguish various sources of chirality: The case of quinophaneoxazoline based ruthenium(II) complexes. Chirality 2024, 36, e23649. [Google Scholar] [CrossRef]
- Sato, H.; Yamagishi, A. VCD studies on chiral characters of metal complex oligomers. Int. J. Mol. Sci. 2013, 14, 964–978. [Google Scholar] [CrossRef]
- Berardozzi, R.; Badetti, E.; dos Santos, N.A.C.; Wurst, K.; Licini, G.; Pescitelli, G.; Zonta, C.; Di Bari, L. Co(ii)-induced giant vibrational CD provides a new design of methods for rapid and sensitive chirality recognition. Chem. Commun. 2016, 52, 8428–8431. [Google Scholar] [CrossRef]
- Zhu, P.; Yang, G.; Poopari, M.R.; Bie, Z.; Xu, Y. Conformations of serine in aqueous solutions as revealed by vibrational circular dichroism. ChemPhysChem 2012, 13, 1272–1281. [Google Scholar] [CrossRef]
- Poopari, M.R.; Zhu, P.; Dezhahang, Z.; Xu, Y. Vibrational absorption and vibrational circular dichroism spectra of leucine in water under different pH conditions: Hydrogen-bonding interactions with water. J. Chem. Phys. 2012, 137, 194308. [Google Scholar] [CrossRef]
- Chamayou, A.C.; Makhloufi, G.; Nafie, L.A.; Janiak, C.; Lüdeke, S. Solvation-induced helicity inversion of pseudotetrahedral chiral copper(II) complexes. Inorg. Chem. 2015, 54, 2193–2203. [Google Scholar]
- Le Barbu-Debus, K.; Bowles, J.; Jähnigen, S.; Clavaguéra, C.; Calvo, F.; Vuilleumier, R.; Zehnacker, A. Assessing cluster models of solvation for the description of vibrational circular dichroism spectra: Synergy between static and dynamic approaches. Phys. Chem. Chem. Phys. 2020, 22, 26047–26068. [Google Scholar] [CrossRef]
- Puente, A.R.; Polavarapu, P.L. Influence of microsolvation on vibrational circular dichroism spectra in dimethyl sulfoxide solvent: A bottom-up approach using quantum cluster growth. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 303, 123231. [Google Scholar] [CrossRef]
- Merten, C.; Berger, C.J.; McDonald, R.; Xu, Y. Evidence of dihydrogen bonding of a chiral amine–borane complex in solution by VCD spectroscopy. Angew. Chem. Int. Ed. 2014, 53, 9940–9943. [Google Scholar] [CrossRef]
- Ghidinelli, S.; Abbate, S.; Koshoubu, J.; Araki, Y.; Wada, T.; Longhi, G. Solvent effects and aggregation phenomena studied by vibrational optical activity and molecular dynamics: The case of pantolactone. J. Phys. Chem. B 2020, 124, 4512–4526. [Google Scholar] [CrossRef]
- Nafie, L.A. Vibrational optical activity: From discovery and development to future challenges. Chirality 2020, 32, 667–692. [Google Scholar] [CrossRef]
- Bloino, J.; Jähnigen, S.; Merten, C. After 50 years of vibrational circular dichroism spectroscopy: Challenges and opportunities of increasingly accurate and complex experiments and computations. J. Phys. Chem. Lett. 2024, 15, 8813–8828. [Google Scholar] [CrossRef]
- Krupová, M.; Kessler, J.; Bouř, P. Recent trends in chiroptical spectroscopy: Theory and applications of vibrational circular dichroism and Raman optical activity. ChemPlusChem 2020, 85, 561–575. [Google Scholar] [CrossRef]
- Pescitelli, G.; Di Bari, L. The phenomenon of vibrational circular dichroism enhancement: A systematic survey of literature data. J. Phys. Chem. B 2024, 128, 9043–9060. [Google Scholar] [CrossRef]
- Sato, H. A new horizon for vibrational circular dichroism spectroscopy: A challenge for supramolecular chirality. Phys. Chem. Chem. Phys. 2020, 22, 7671–7679. [Google Scholar] [CrossRef]
- Krupová, M.; Andrushchenko, V. Vibrational circular dichroism beyond solutions, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2025; in press. [Google Scholar]
- Wu, T.; Pelc, R.; Bouř, P. Molecular properties of 3d and 4f coordination compounds deciphered by Raman optical activity spectroscopy. ChemPlusChem 2023, 88, e202300385. [Google Scholar] [CrossRef]
- Merten, C.; Xu, Y. Chirality transfer in the methyl lactate-ammonia complex observed by matrix-isolation vibrational circular dichroism spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 2073–2076. [Google Scholar] [CrossRef]
- Yang, Q.; Bloino, J.; Šestáková, H.; Šebestík, J.; Kessler, J.; Hudecová, J.; Kapitán, J.; Bouř, P. Combination of resonance and non-resonance chiral Raman scattering in a cobalt(III) complex. Angew. Chem. Int. Ed. 2023, 62, e202312521. [Google Scholar] [CrossRef]
- Nafie, L.A. Theory of vibrational circular dichroism and infrared absorption: Extension to molecules with low-lying excited electronic states. J. Phys. Chem. A 2004, 108, 7222–7231. [Google Scholar] [CrossRef]
- Li, G.; Alshalalfeh, M.; Yang, Y.; Cheeseman, J.R.; Bouř, P.; Xu, Y. Can one measure resonance Raman optical activity? Angew. Chem. Int. Ed. 2021, 60, 22004–22009. [Google Scholar] [CrossRef]
- Machalska, E.; Zajac, G.; Wierzba, A.J.; Kapitán, J.; Andruniów, T.; Spiegel, M.; Gryko, D.; Bouř, P.; Baranska, M. Recognition of the true and false resonance Raman optical activity. Angew. Chem. Int. Ed. 2021, 60, 21205–21210. [Google Scholar] [CrossRef]
- Wu, T.; Li, G.; Kapitán, J.; Kessler, J.; Xu, Y.; Bouř, P. Two spectroscopies in one: Interference of circular dichroism and Raman optical activity. Angew. Chem. Int. Ed. 2020, 59, 21895–21898. [Google Scholar] [CrossRef]
- Domingos, S.R.; Hartl, F.; Buma, W.J.; Woutersen, S. Elucidating the structure of chiral molecules by using amplified vibrational circular dichroism: From theory to experimental realization. ChemPhysChem 2015, 16, 3363–3373. [Google Scholar] [CrossRef]
- He, Y.; Cao, X.; Nafie, L.A.; Freedman, T.B. Ab initio VCD calculation of a transition-metal containing molecule and a new intensity enhancement mechanism for VCD. J. Am. Chem. Soc. 2001, 123, 11320–11321. [Google Scholar] [CrossRef]
- Merten, C.; Hiller, K.; Xu, Y. Effects of electron configuration and coordination number on the vibrational circular dichroism spectra of metal complexes of trans-1,2-diaminocyclohexane. Phys. Chem. Chem. Phys. 2012, 14, 12884–12891. [Google Scholar] [CrossRef]
- Sato, H.; Takimoto, K.; Yoshida, J.; Yamagishi, A. Vibrational circular dichroism towards asymmetric catalysis: Chiral induction in substrates coordinated with copper(II) ions. Phys. Chem. Chem. Phys. 2020, 22, 24393–24398. [Google Scholar] [CrossRef]
- Merten, C.; Xu, Y. A comparative vibrational CD study of homo- and heteroleptic complexes of the type [Cu(trans-1,2-diaminocyclohexane)2L](ClO4)2. Dalton Trans. 2013, 42, 10572–10578. [Google Scholar] [CrossRef]
- Bhattacharya, S.R.; Bürgi, T. Amplified vibrational circular dichroism as a manifestation of the interaction between a water soluble gold nanocluster and cobalt salt. Nanoscale 2019, 11, 23226–23233. [Google Scholar] [CrossRef]
- Nafie, L.A. Vibrational Optical Activity: Principles and Applications; John Wiley: New York, NY, USA, 2011. [Google Scholar]
- Tomeček, J.; Bouř, P. Density Functional Computations of Vibrational Circular Dichroism Spectra beyond the Born–Oppenheimer Approximation. J. Chem. Theory Comput. 2020, 16, 2627–2634. [Google Scholar] [CrossRef]
- Fusè, M.; Egidi, F.; Bloino, J. Vibrational circular dichroism under the quantum magnifying glass: From the electronic flow to the spectroscopic observable. Phys. Chem. Chem. Phys. 2019, 21, 4224–4239. [Google Scholar] [CrossRef]
- He, Y. Enhanced VCD in Transition Metal Complexes and Metalloproteins. Ph.D. Thesis, Syracuse University, Syracuse, NY, USA, 2005. [Google Scholar]
- Li, G.; Li, D.; Alshalalfeh, M.; Cheramy, J.; Zhang, H.; Xu, Y. Stereochemical properties of two Schiff-base transition metal complexes and their ligand by using multiple chiroptical spectroscopic tools and DFT calculations. Molecules 2023, 28, 2571. [Google Scholar] [CrossRef]
- Lassen, P.R. Structural Characterization of Chiral Molecules Using Vibrational Circular Dichroism Spectroscopy. Ph.D. Thesis, Tech University, Lyngby, Denmark, 2006. [Google Scholar]
- Hitchman, M.A. Electronic structure of low-spin cobalt(II) Schiff base complexes. Inorg. Chem. 1977, 16, 1985–1993. [Google Scholar] [CrossRef]
- Grimme, S. Exploration of chemical compound, conformer, and reaction space with meta-dynamics simulations based on tight-binding quantum chemical calculations. J. Chem. Theory Comput. 2019, 15, 2847–2862. [Google Scholar] [CrossRef]
- Pracht, P.; Grimme, S.; Bannwarth, C.; Bohle, F.; Ehlert, S.; Feldmann, G.; Gorges, J.; Müller, M.; Neudecker, T.; Plett, C.; et al. CREST—A program for the exploration of low-energy molecular chemical space. J. Chem. Phys. 2024, 160, 114110. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Lange, A.W.; Herbert, J.M. Improving generalized Born models by exploiting connections to polarizable continuum models. I. An improved effective Coulomb operator. J. Chem. Theory Comput. 2012, 8, 1999–2011. [Google Scholar] [CrossRef]
- Ehlert, S.; Stahn, M.; Spicher, S.; Grimme, S. Robust and efficient implicit solvation model for fast semiempirical methods. J. Chem. Theory Comput. 2021, 17, 4250–4261. [Google Scholar] [CrossRef]
- Xie, F.; Seifert, N.A.; Jäger, W.; Xu, Y. Conformational panorama and chirality controlled structure-energy relationship in a chiral carboxylic acid dimer. Angew. Chem. Int. Ed. 2020, 59, 15703–15710. [Google Scholar] [CrossRef]
- Hazrah, A.S.; Insausti, A.; Ma, J.; Al-Jabiri, M.H.; Jager, W.; Xu, Y. Wetting vs droplet aggregation: A broadband rotational spectroscopic study of 3-methylcatechol···water clusters. Angew. Chem. Int. Ed. 2023, 62, e202310610. [Google Scholar] [CrossRef]
- Seifert, N.A.; Thomas, J.; Jäger, W.; Xu, Y. Rotational spectra and theoretical study of tetramers and trimers of 2-fluoroethanol: Dramatic intermolecular compensation for intramolecular instability. Phys. Chem. Chem. Phys. 2018, 20, 27630–27637. [Google Scholar] [CrossRef]
- Yang, Y.; Cheramy, J.; Brehm, M.; Xu, Y. Raman optical activity of N-acetyl-L-cysteine in water and in methanol: The “clusters-in-a-liquid” model and ab initio molecular dynamics simulations. ChemPhysChem 2022, 23, e202200161. [Google Scholar] [CrossRef]
- Dezhahang, D.; Poopari, M.R.; Cheramy, J.; Xu, Y. Conservation of helicity in a chiral pyrrol-2-yl Schiff-base ligand and its transition metal complexes. Inorg. Chem. 2015, 54, 4539–4549. [Google Scholar] [CrossRef]
- Smura, C.F.; Parker, D.R.; Zbiri, M.; Johnson, M.R.; Gál, Z.A.; Clarke, S.J. High-spin cobalt (II) ions in square planar coordination: Structures and magnetism of the oxysulfides Sr2CoO2Cu2S2 and Ba2CoO2Cu2S2 and their solid solution. J. Am. Chem. Soc. 2011, 133, 2691–2705. [Google Scholar] [CrossRef]
- Kaya, B.; Akyüz, D.; Karakurt, T.; Şahin, O.; Koca, A.; Ülküseven, B. Cobalt(II)/(III) complexes bearing a tetradentate thiosemicarbazone: Synthesis, experimental and theoretical characterization, and electrochemical and antioxidant properties. Appl. Organomet. Chem. 2020, 34, e5930. [Google Scholar] [CrossRef]
- Izadnia, S.; LaForge, A.C.; Stienkemeier, F.; Cheeseman, J.R.; Bloino, J.; Cheramy, J.; Jäger, W.; Xu, Y. Unusual binary aggregates of perylene bisimide revealed by their electronic transitions in helium nanodroplets and DFT calculations. Phys. Chem. Chem. Phys. 2021, 23, 13862–13872. [Google Scholar] [CrossRef]
- Kaviani, S.; Izadyar, M.; Housaindokht, M.R. Solvent and spin state effects on molecular structure, IR spectra, binding energies and quantum chemical reactivity indices of deferiprone ferric complex: DFT study. Polyhedron 2016, 117, 623–627. [Google Scholar] [CrossRef]
- Ould-Hamouda, A.; Viquerat, B.; Degert, J.; Matar, S.F.; Létard, J.F.; Guillaume, F.; Freysz, E. Impact of spin state transition on vibrations of [Fe–(PM–BiA)2 (NCS)2] and [Fe–(PM–PEA)2 (NCS)2] spin crossover compounds: Experimental and theoretical far IR and Raman study. Eur. J. Inorg. Chem. 2018, 2018, 385–393. [Google Scholar] [CrossRef]
- Haikarainen, A.; Sipilä, J.; Pietikäinen, P.; Pajunen, A.; Mutikainen, I. Synthesis and characterization of bulky salen-type complexes of Co, Cu, Fe, Mn and Ni with amphiphilic solubility properties. J. Chem. Soc. Dalton Trans. 2001, 991–995. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Carlson, C.D.; D’Souza, M.; Ma, J.; Mort, A.N.; Insausti, A.; Xu, Y. Tunneling and conformational preferences in racemic- and meso-2,4-pentanediol: A rotational spectroscopic and theoretical investigation. J. Chem. Phys. 2024, 161, 244301. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.03; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Frisch, M.J.; Pople, J.A. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Pescitelli, G.; Lüdeke, S.; Chamayou, A.-C.; Marolt, M.; Justus, V.; Górecki, M.; Arrico, L.; Di Bari, L.; Islam, M.A.; Gruber, I.; et al. Broad-range spectral analysis for chiral metal coordination compounds: (chiro)optical superspectrum of cobalt(II) complexes. Inorg. Chem. 2018, 57, 13397–13408. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshalalfeh, M.; Xu, Y. Chiroptical Spectroscopy, Theoretical Calculations, and Symmetry of a Chiral Transition Metal Complex with Low-Lying Electronic States. Molecules 2025, 30, 804. https://doi.org/10.3390/molecules30040804
Alshalalfeh M, Xu Y. Chiroptical Spectroscopy, Theoretical Calculations, and Symmetry of a Chiral Transition Metal Complex with Low-Lying Electronic States. Molecules. 2025; 30(4):804. https://doi.org/10.3390/molecules30040804
Chicago/Turabian StyleAlshalalfeh, Mutasem, and Yunjie Xu. 2025. "Chiroptical Spectroscopy, Theoretical Calculations, and Symmetry of a Chiral Transition Metal Complex with Low-Lying Electronic States" Molecules 30, no. 4: 804. https://doi.org/10.3390/molecules30040804
APA StyleAlshalalfeh, M., & Xu, Y. (2025). Chiroptical Spectroscopy, Theoretical Calculations, and Symmetry of a Chiral Transition Metal Complex with Low-Lying Electronic States. Molecules, 30(4), 804. https://doi.org/10.3390/molecules30040804