
Mechanistic Pathways in Cyanide-Mediated Benzoin Condensation: A Comprehensive Electron Localisation Function (ELF) and Catastrophe Theory Analysis of the Umpolung Reaction


Abstract
1. Introduction
2. Results and Discussion
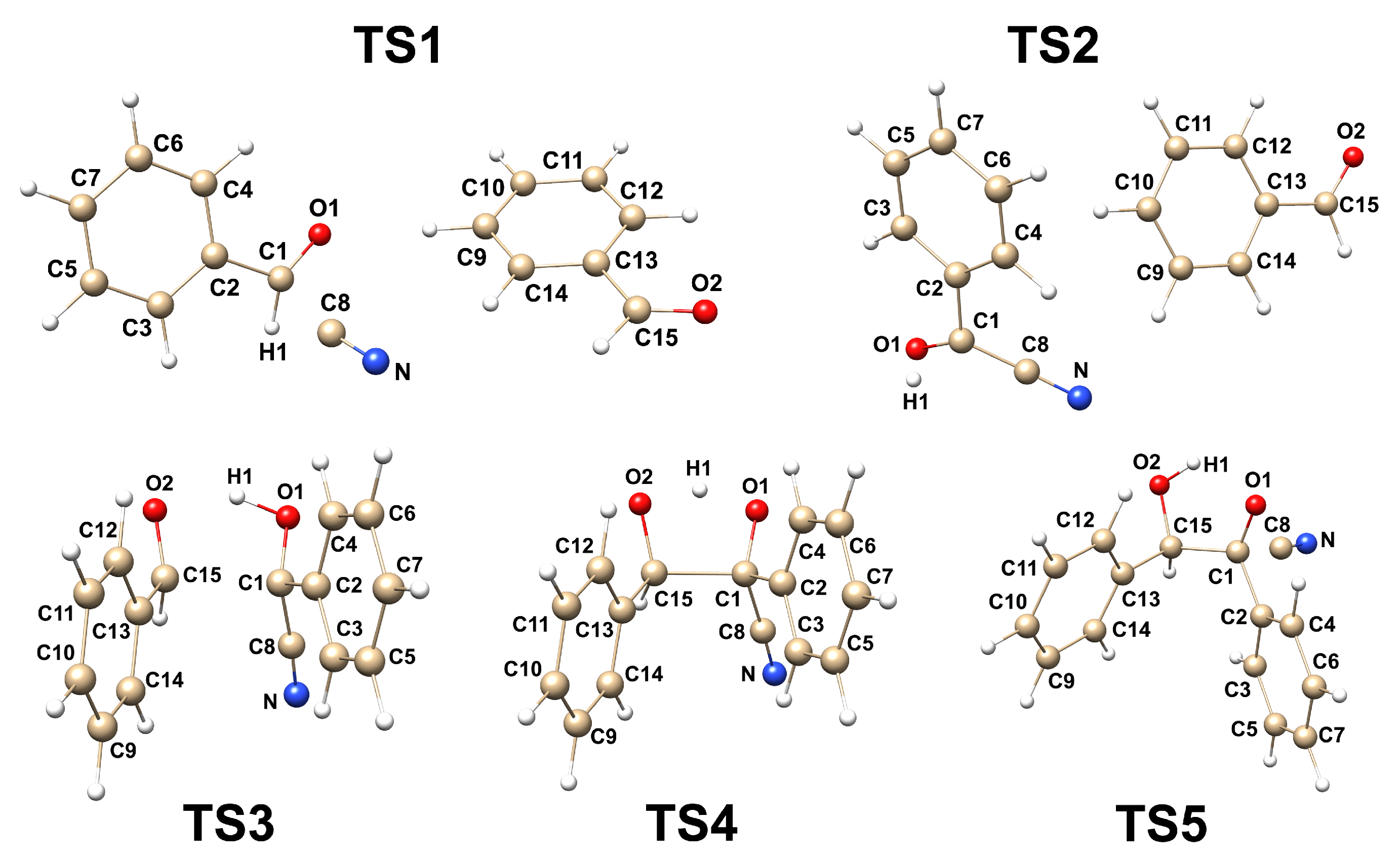
2.1. The Energetic of the Reaction and Geometrical Analysis
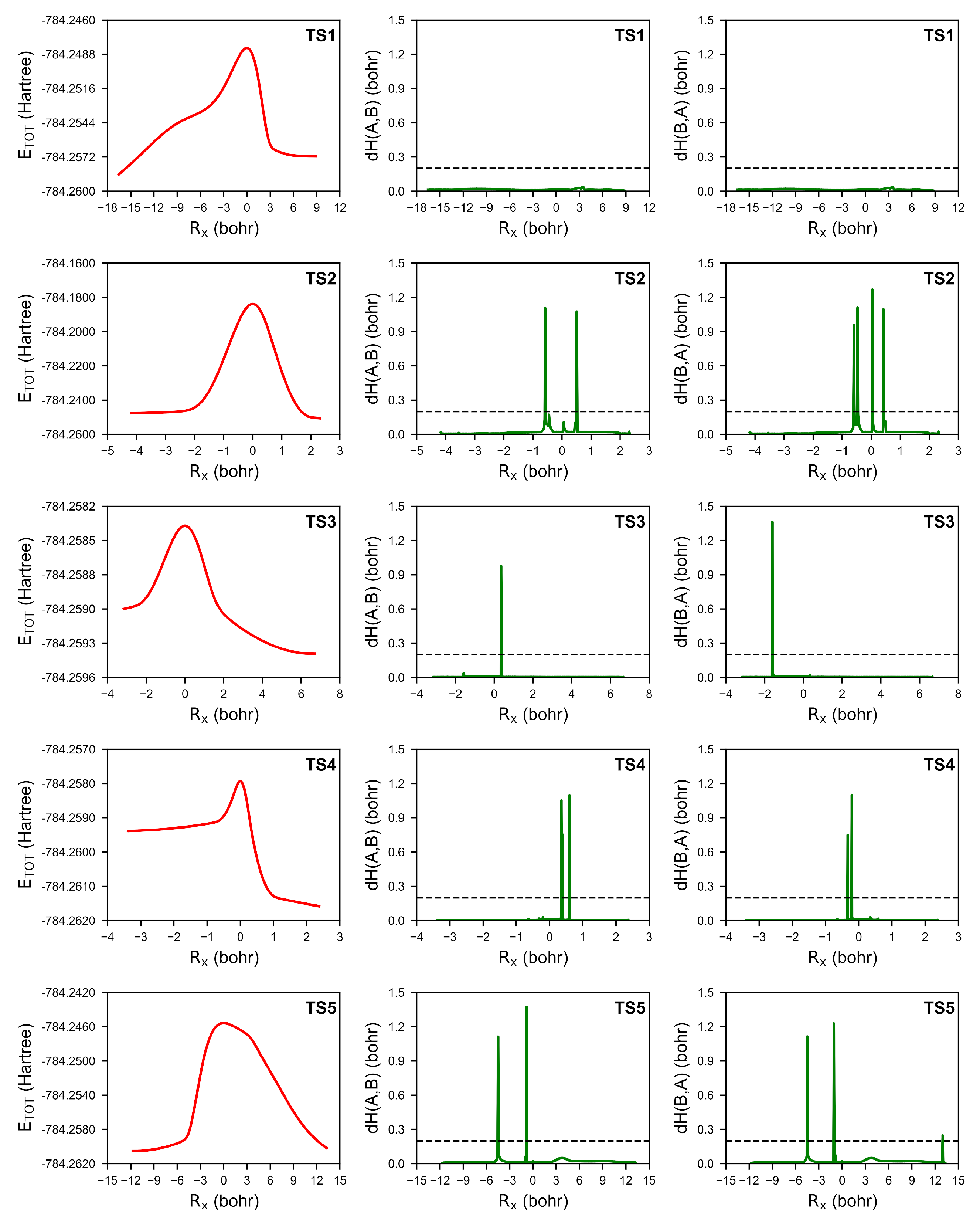
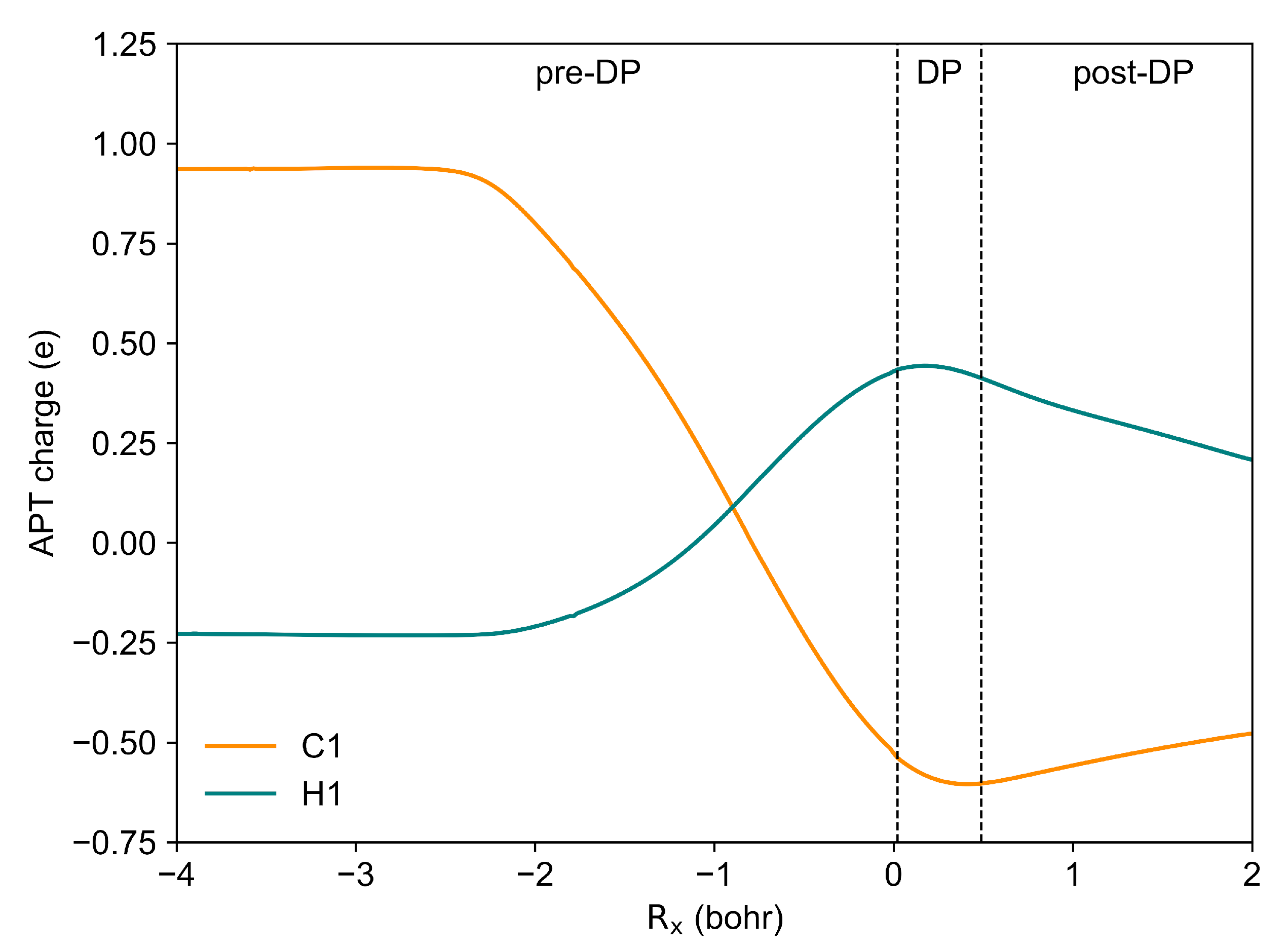
2.2. The Mechanism of the Reaction—BET Analysis
3. Materials and Methods
4. Conclusions
- The umpolung mechanism in the gas phase and without solvent assistance proceeds through five transition state structures. The formation of Lapworth’s cyanohydrin is not spontaneous, as evidenced by the positive value. Among these steps, the formation of the Lapworth’s cyanohydrin has the highest value, making it the rate-determining step. Additionally, it was observed that the proton transfer in TS4 does not occur simultaneously with C-C bond formation and has a barrier-less character, as indicated by the negative value.
- The evolution of the electron localisation function (ELF) field in the studied umpolung mechanism is characterised by 17 distinct catastrophes. While some changes within the ELF field lack straightforward chemical interpretation, the application of topological analysis alongside catastrophe theory has enabled a detailed mapping of electron density evolution. In particular, a comprehensive analysis of ELF catastrophes in the cyanide-type umpolung reaction reveals that the formation of the C1-C8 bond occurs through a shift in the synapticity of the V(C8) basin to a V(C1,C8) basin at a distance of 2.029 Å. This was followed by proton transfer, resulting in the formation of Lapworth’s cyanohydrin and the appearance of additional electron density near the C1 atom, as indicated by the V(C1) basin. The formation of the C1-C15 bond in TS3 is characterised by two distinct ELF catastrophes: one describing the creation of non-bonded electron density near the C15 atom and the other describing the formation of the C1-C15 bond through the annihilation of the V(C1) and V(C15) basins. The proton transfer between the O1 and O2 atoms is found to involve the creation of a dressed-atom-type V(H1) basin. Finally, the dissociation of the C1-C8 bond and the elimination of the cyanide group occur through the annihilation of the disynaptic V(C1,C8) basin and the creation of monosynaptic V(C1) and V(C8) basins.
- The applications of topological analysis of ELF and catastrophe theory confirm the well-established Lapworth mechanism for this reaction.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lapworth, A. XCVI.—Reactions involving the addition of hydrogen cyanide to carbon compounds. J. Chem. Soc. Trans. 1903, 83, 995–1005. [Google Scholar] [CrossRef]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- Juhl, M.; Lee, J.W. Umpolung Reactivity of Aldehydes toward Carbon Dioxide. Angew. Chem. Int. Ed. 2018, 57, 12318–12322. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, B.Y.; Sršen, M.; König, B. Umpolung Difunctionalization of Carbonyls via Visible-Light Photoredox Catalytic Radical-Carbanion Relay. J. Am. Chem. Soc. 2020, 142, 7524–7531. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Duan, M.; Lin, S.L.; Liu, Y.W.; Cheng, J.K.; Xiang, S.H.; Yu, P.; Houk, K.N.; Tan, B. Organocatalytic aromatization-promoted umpolung reaction of imines. Nat. Chem. 2024, 16, 408–416. [Google Scholar] [CrossRef]
- Bai, D.; Guo, X.; Wang, X.; Xu, W.; Cheng, R.; Wei, D.; Lan, Y.; Chang, J. Umpolung reactivity of strained C–C σ-bonds without transition-metal catalysis. Nat. Commun. 2024, 15, 2833. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, W.H. Reactivity Umpolung of Amides in Organic Synthesis. Asian J. Org. Chem. 2023, 12, e202300473. [Google Scholar] [CrossRef]
- Wiberg, K.B. The Deuterium Isotope Effect of Some Ionic Reactions of Benzaldehyde. J. Am. Chem. Soc. 1954, 76, 5371–5375. [Google Scholar] [CrossRef]
- Wiberg, K.B. The Deuterium Isotope Effect of the Methanolysis of Some Organometallic Compounds. J. Am. Chem. Soc. 1955, 77, 5987–5990. [Google Scholar] [CrossRef]
- Kool, E.T.; Breslow, R. Dichotomous salt effects in the hydrophobic acceleration of the benzoin condensation. J. Am. Chem. Soc. 1988, 110, 1596–1597. [Google Scholar] [CrossRef]
- Breslow, R. Hydrophobic effects on simple organic reactions in water. Accounts Chem. Res. 1991, 24, 159–164. [Google Scholar] [CrossRef]
- He, Y.; Xue, Y. Mechanism Insight into the Cyanide-Catalyzed Benzoin Condensation: A Density Functional Theory Study. J. Phys. Chem. A 2010, 114, 9222–9230. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Savin, A.; Becke, A.D.; Flad, J.; Nesper, R.; Preuss, H.; von Schnering, H.G. A New Look at Electron Localization. Angew. Chem. Int. Ed. Engl. 1991, 30, 409–412. [Google Scholar] [CrossRef]
- Thom, R. Structural Stability and Morphogenesis; Taylor & Francis Group LLC: London, UK, 1972. [Google Scholar]
- Polo, V.; Andres, J.; Berski, S.; Domingo, L.R.; Silvi, B. Understanding Reaction Mechanisms in Organic Chemistry from Catastrophe Theory Applied to the Electron Localization Function Topology. J. Phys. Chem. A 2008, 112, 7128–7136. [Google Scholar] [CrossRef]
- Berski, S.; Andrés, J.; Silvi, B.; Domingo, L.R. The Joint Use of Catastrophe Theory and Electron Localization Function to Characterize Molecular Mechanisms. A Density Functional Study of the Diels-Alder Reaction between Ethylene and 1,3-Butadiene. J. Phys. Chem. A 2003, 107, 6014–6024. [Google Scholar] [CrossRef]
- Berski, S.; Andrés, J.; Silvi, B.; Domingo, L.R. New Findings on the Diels-Alder Reactions. An Analysis Based on the Bonding Evolution Theory. J. Phys. Chem. A 2006, 110, 13939–13947. [Google Scholar] [CrossRef]
- Polo, V.; Andres, J.; Castillo, R.; Berski, S.; Silvi, B. Understanding the Molecular Mechanism of the 1,3-Dipolar Cycloaddition between Fulminic Acid and Acetylene in Terms of the Electron Localization Function and Catastrophe Theory. Chem.— Eur. J. 2004, 10, 5165–5172. [Google Scholar] [CrossRef]
- Andrés, J.; Berski, S.; Silvi, B. Curly arrows meet electron density transfers in chemical reaction mechanisms: From electron localization function (ELF) analysis to valence-shell electron-pair repulsion (VSEPR) inspired interpretation. Chem. Commun. 2016, 52, 8183–8195. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Silvi, B. The synaptic order: A key concept to understand multicenter bonding. J. Mol. Struct. 2002, 614, 3–10. [Google Scholar] [CrossRef]
- Hausdorff, F. Grundzüge der Mengenlehre; Göschens Lehrbücherei/Gruppe I: Reine und Angewandte Mathematik Series; Von Veit: Leipzig, Germany, 1914. [Google Scholar]
- Michalski, M.; Berski, S. Understanding the molecular mechanism of the chlorine atom transfer between ammonia and hypochlorous acid with electron localisation function (ELF). Mol. Phys. 2021, 119, e1961035. [Google Scholar] [CrossRef]
- Han, G.; Liu, X.; Cao, Z.; Sun, Y. Photocatalytic Pinacol C–C Coupling and Jet Fuel Precursor Production on ZnIn2S4 Nanosheets. ACS Catal. 2020, 10, 9346–9355. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B 2002, 58, 389–397. [Google Scholar] [CrossRef]
- Revell, L.E.; Williamson, B.E. Why Are Some Reactions Slower at Higher Temperatures? J. Chem. Educ. 2013, 90, 1024–1027. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Shukla, P.B.; Mishra, P.; Baruah, T.; Zope, R.R.; Jackson, K.A.; Johnson, J.K. How Do Self-Interaction Errors Associated with Stretched Bonds Affect Barrier Height Predictions? J. Phys. Chem. A 2023, 127, 1750–1759. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Accounts Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Michalski, M. AyudaTop. 2024. Available online: https://github.com/Parecido/AyudaTop (accessed on 6 July 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy | TS1 | TS2 | TS3 | TS4 | TS5 |
|---|---|---|---|---|---|
| 10.88 | 43.03 | 1.48 | −1.04 | 7.60 | |
| 4.17 | 40.53 | 0.26 | 0.05 | 17.77 | |
| 6.71 | 2.50 | 1.22 | −1.09 | −10.17 |
| Energy | B3LYP | M06-2X | BHHLYP | wB97XD | PBE0 |
|---|---|---|---|---|---|
| 6-31+G(d,p) | |||||
| −1.16 | −1.15 | −1.02 | −1.36 | −1.08 | |
| −0.31 | 0.15 | 1.32 | 0.55 | −0.40 | |
| −0.85 | −1.30 | −2.34 | −1.91 | −0.68 | |
| 6-311++G(d,p) | |||||
| −1.04 | −1.10 | −0.71 | −1.01 | −1.20 | |
| 0.05 | 0.68 | 1.72 | 0.77 | −0.27 | |
| −1.09 | −1.78 | −2.43 | −1.78 | −0.93 | |
| 6-311++G(2df,p) | |||||
| −0.93 | −1.14 | −0.86 | −1.16 | −0.86 | |
| −0.30 | 0.39 | 1.38 | 0.60 | 1.38 | |
| −0.63 | 0.75 | −2.24 | −1.76 | −2.24 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalski, M.; Berski, S. Mechanistic Pathways in Cyanide-Mediated Benzoin Condensation: A Comprehensive Electron Localisation Function (ELF) and Catastrophe Theory Analysis of the Umpolung Reaction. Molecules 2025, 30, 378. https://doi.org/10.3390/molecules30020378
Michalski M, Berski S. Mechanistic Pathways in Cyanide-Mediated Benzoin Condensation: A Comprehensive Electron Localisation Function (ELF) and Catastrophe Theory Analysis of the Umpolung Reaction. Molecules. 2025; 30(2):378. https://doi.org/10.3390/molecules30020378
Chicago/Turabian StyleMichalski, Michal, and Slawomir Berski. 2025. "Mechanistic Pathways in Cyanide-Mediated Benzoin Condensation: A Comprehensive Electron Localisation Function (ELF) and Catastrophe Theory Analysis of the Umpolung Reaction" Molecules 30, no. 2: 378. https://doi.org/10.3390/molecules30020378
APA StyleMichalski, M., & Berski, S. (2025). Mechanistic Pathways in Cyanide-Mediated Benzoin Condensation: A Comprehensive Electron Localisation Function (ELF) and Catastrophe Theory Analysis of the Umpolung Reaction. Molecules, 30(2), 378. https://doi.org/10.3390/molecules30020378