
Computation of the pKa Values of Gallic Acid and Its Anionic Forms in Aqueous Solution: A Self-Similar Transformation Approach for Accurate Proton Hydration Free Energy Estimation


Abstract
:
1. Introduction
- (i)
- The accuracy of pKa reproduction is contingent upon the computational method employed to determine G(RH) and G(R−); it varies within an MAE (mean absolute error) range of 0.51–2.86 pKa for monoprotic acids, depending on the method used in the calculations [5].
- (ii)
- (iii)
- (i)
- To employ a modified version of the method proposed by Dutra et al. [6] to determine the pKa1,2,3,4 parameters for the four-stage deprotonation of GA, within the bounds of experimental errors of 0.01–0.40 pKa units.
- (ii)
- (iii)
- To select the experimental data that most accurately represent the pKa values for GA within the framework of the proposed approach.
- (iv)
- To demonstrate that the accurate reproduction of the pKa of GA, within the range of experimental errors, can be achieved using the DFT method and LSDA/QZVP level of theory combined with the SMD solvation model, without the explicit inclusion of a water molecule.
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gregory, M.T.; Gao, Y.; Cui, Q.; Yang, W. Multiple deprotonation paths of the nucleophile 3′-OH in the DNA synthesis reaction. Proc. Natl. Acad. Sci. USA 2021, 118, e2103990118. [Google Scholar] [CrossRef] [PubMed]
- Alongi, K.S.; Shields, G.S. Theoretical Calculations of Acid Dissociation Constants: A Review Article. Annu. Rep. Comput. Chem. 2010, 6, 113–138. [Google Scholar] [CrossRef]
- Marino, T.; Galano, A.; Russo, N. Radical scavenging ability of gallic acid toward OH and OOH radicals. Reaction mechanism and rate constants from the density functional theory. J. Phys. Chem. B 2014, 118, 10380–10389. [Google Scholar] [CrossRef] [PubMed]
- Molski, M. Theoretical study on the radical scavenging activity of gallic acid. Heliyon 2023, 9, e12806. [Google Scholar] [CrossRef]
- De Souza Silva, C.d.S.; Custodio, R. Assessment of pKa determination for monocarboxylic acids with an accurate theoretical composite method: G4CEP. J. Phys. Chem. A 2019, 123, 8314–8320. [Google Scholar] [CrossRef]
- Dutra, F.R.; Silva, C.d.S.; Custodio, R. On the accuracy of the direct method to calculate pKa from electronic structure calculations. J. Phys. Chem. A 2021, 125, 65–73. [Google Scholar] [CrossRef]
- Walton-Raaby, M.; Floen, T.; García-Díez, G.; Mora-Diez, N. Calculating the aqueous pKa of phenols: Predictions for antioxidants and cannabinoids. Antioxidants 2023, 12, 1420. [Google Scholar] [CrossRef]
- Morency, M.; Néron, S.; Iftimie, R.; Wuest, J.D. Predicting pKa values of quinols and related aromatic compounds with multiple OH groups. J. Org. Chem. 2021, 86, 14444–14460. [Google Scholar] [CrossRef]
- Badhani, B.; Kakkar, R. Influence of intrinsic and extrinsic factors on the antiradical activity of gallic acid: A theoretical study. Struct. Chem. 2018, 29, 359–373. [Google Scholar] [CrossRef]
- Masoud, M.S.; Ali, A.E.; Haggag, S.S.; Nasr, N.M. Spectroscopic studies on gallic acid and its azo derivatives and their iron(III) complexes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 120, 505–511. [Google Scholar] [CrossRef]
- Agrawal, M.D.; Bhandari, C.S.; Dixit, M.K.; Sogani, N.C. 3,4,5-Trihydroxybenzoesäure als Chelatbildner, 1. Mitt.: Praseodym. Monatsh. Chem. 1976, 713, 75–82. [Google Scholar] [CrossRef]
- Huguenin, J.; Hamady, S.O.S.; Bourson, P. Monitoring deprotonation of gallic acid by Raman spectroscopy. J. Raman Spectrosc. 2015, 46, 1062–1066. [Google Scholar] [CrossRef]
- Kipton, H.; Powell, J.; Taylor, M.C. Interactions of iron(II) and iron(III) with gallic acid and its homologues: A potentiometric and spectrophotometric study. Aust. J. Chem. 1982, 35, 739–756. [Google Scholar] [CrossRef]
- Loginova, L.F.; Medyntsev, V.V.; Khomutov, B.I. Acidic properties of gallic acid and stability constants of iron complexes. Zh. Obshch. Him. 1972, 42, 739–742. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Xu, L.; Coote, M.L. Methods To Improve the Calculations of Solvation Model Density Solvation Free Energies and Associated Aqueous pKa Values: Comparison between Choosing an Optimal Theoretical Level, Solute Cavity Scaling, and Using Explicit Solvent Molecules. J. Phys. Chem. A 2019, 123, 7430–7438. [Google Scholar] [CrossRef]
- Pezzola, S.; Venanzi, M.; Galloni, P.; Conte, V.; Sabuzi, F. Towards the “Eldorado” of pKa Determination: A Reliable and Rapid DFT Model. Molecules 2024, 29, 1255. [Google Scholar] [CrossRef]
- Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A. Calculation of solvation free energies of charged solutes using mixed cluster/continuum models. J. Phys. Chem. B 2008, 112, 9709–9719. [Google Scholar] [CrossRef]
- Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Adding explicit solvent molecules to continuum solvent calculations for the calculation of aqueous acid dissociation constants. J. Phys. Chem. A 2006, 110, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Fifen, J.J.; Dhaouadi, Z.; Nsangou, M. Revision of the thermodynamics of the proton in gas phase. J. Phys. Chem. A 2014, 118, 11090–11097. [Google Scholar] [CrossRef]
- Lim, C.; Bashford, D.; Karplus, M. Absolute pKa calculations with continuum dielectric methods. J. Phys. Chem. 1991, 95, 5610–5620. [Google Scholar] [CrossRef]
- Tissandier, M.D.; Cowen, K.A.; Feng, W.Y.; Gundlach, E.; Cohen, M.H.; Earhart, A.D.; Coe, J.V.; Tuttle, T.R. The Proton’s Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data. J. Phys. Chem. A 1998, 102, 7787–7794. [Google Scholar] [CrossRef]
- Klots, C.E. Solubility of protons in water. J. Phys. Chem. 1981, 85, 3585–3588. [Google Scholar] [CrossRef]
- Tawa, G.J.; Topol, I.A.; Burt, S.K.; Caldwell, R.A.; Rashin, A.A. Calculation of the aqueous solvation free energy of the proton. J. Chem. Phys. 1998, 109, 4852–4863. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Dixon, D.A. Absolute Hydration Free Energy of the Proton from First-Principles Electronic Structure Calculations. J. Phys. Chem. A 2001, 105, 11534–11540. [Google Scholar] [CrossRef]
- Marković, Z.; Tošović, J.; Milenkovic, D.; Markovic, S. Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]
- Rimarčík, J.; Lukeš, V.; Klein, E.; Ilčin, M. Study of the solvent effect on the enthalpies of homolytic and heterolytic N–H bond cleavage in p-phenylenediamine and tetracyano-p-phenylenediamine. J. Mol. Struct. THEOCHEM 2010, 952, 25–30. [Google Scholar] [CrossRef]
- Mejías, A.; Lago, S. Calculation of the absolute hydration enthalpy and free energy of H+ and OH−. J. Chem. Phys. 2000, 113, 7306–7316. [Google Scholar] [CrossRef]
- Galano, A.; Pérez-González, A.; Castañeda-Arriaga, R.; Muñoz-Rugeles, L.; Mendoza-Sarmiento, G.; Romero-Silva, A.; Ibarra-Escutia, A.; Rebollar-Zepeda, A.M.; León-Carmona, J.R.; Hernández-Olivares, M.A.; et al. Empirically Fitted Parameters for Calculating pKa Values with Small Deviations from Experiments Using a Simple Computational Strategy. J. Chem. Inf. Model. 2016, 56, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Pérez-González, A.; Castañeda-Arriaga, R.; Verastegui, B.; Carreón-González, M.; Alvarez-Idaboy, J.R.; Galano, A. Estimation of empirically fitted parameters for calculating pK a values of thiols in a fast and reliable way. Theor. Chem. Acc. 2018, 137, 5. [Google Scholar] [CrossRef]
- Miguel, E.L.M.; Silva, P.L.; Pliego, J.R. Theoretical Prediction of pKa in Methanol: Testing SM8 and SMD Models for Carboxylic Acids, Phenols, and Amines. J. Phys. Chem. B 2014, 118, 5730–5739. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Schröder, H.; Hühnert, J.; Schwabe, T. Evaluation of DFT-D3 dispersion corrections for various structural benchmark sets. J. Chem. Phys. 2017, 146, 044115. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA; Oxford, UK, 1989. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1. 2017. [Google Scholar]
- Braun, D.E.; Bhardwaj, R.M.; Florence, A.J.; Tocher, D.A.; Price, S.L. Complex Polymorphic System of Gallic Acid—Five Monohydrates, Three Anhydrates, and over 20 Solvates. Cryst. Growth Des. 2013, 13, 19–23. [Google Scholar] [CrossRef]
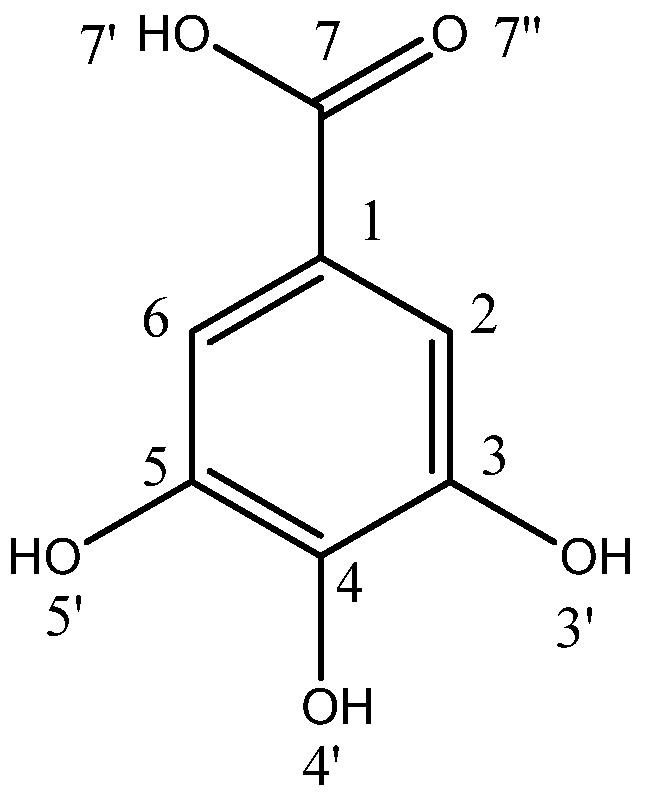
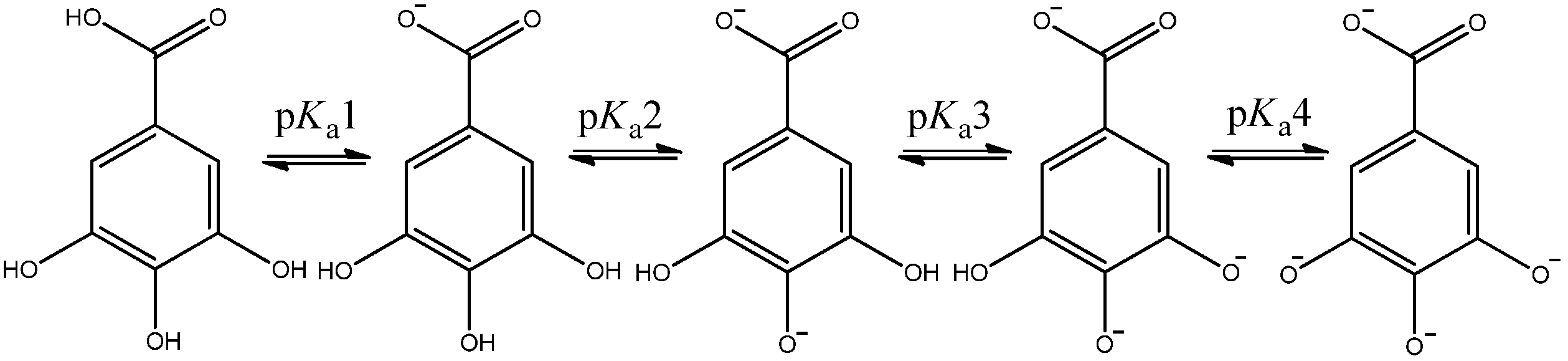


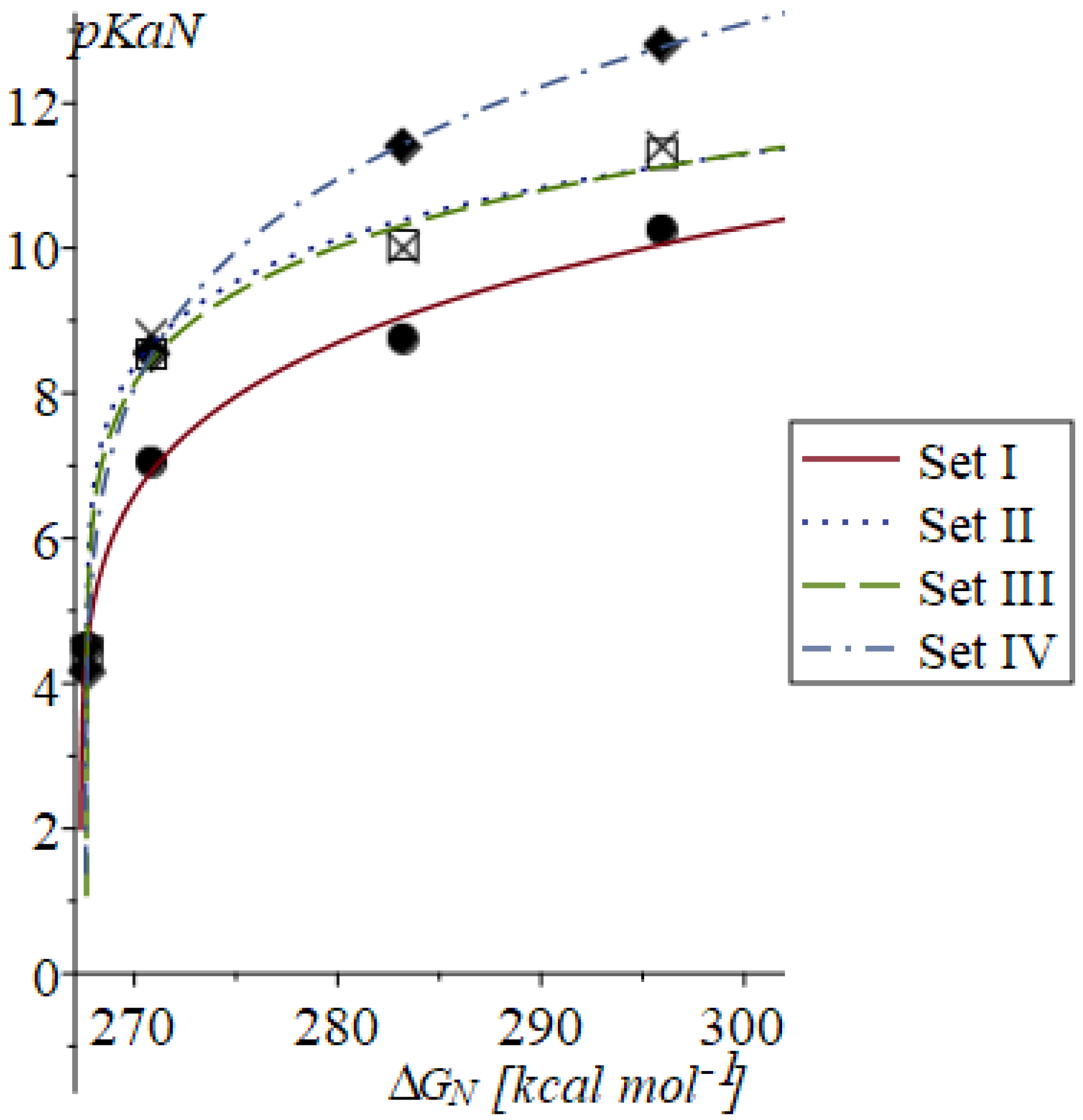

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∆GN [kcal mol−1] | LSDA | M062X | B3LYP | |
|---|---|---|---|---|
| ∆G1 | G(GA−1)aq–G(GA0)aq | 267.6773 | 270.6210 | 272.7539 |
| ∆G2 | G(GA−2)aq–G(GA−1)aq | 270.8450 | 279.5730 | 280.5099 |
| ∆G3 | G(GA−3)aq–G(GA−2)aq | 283.2258 | 289.4136 | 290.8582 |
| ∆G4 | G(GA−4)aq–G(GA−3)aq | 295.9617 | 297.0059 | 298.7058 |
| LSDA | M062X | B3LYP | LSDA | M062X | B3LYP | |
|---|---|---|---|---|---|---|
| Parameter | Set I | Set II | ||||
| G(H+)aq | −265.47(32) | −62.1(3.2) | −266.6(2.5) | −265.775(20) | −268.12(65) | −270.51(44) |
| a | 5.87(59) | 2.02(80) | 2.84(94) | 7.87(64) | 5.36(48) | 5.90(96) |
| c | 0.177(38) | 0.51(11) | 0.41(10) | 0.114(33) | 0.247(66) | 0.216(61) |
| SE | 0.3861 | 0.2031 | 0.2630 | 0.4959 | 0.4020 | 0.4383 |
| R2 | 0.9918 | 0.9977 | 0.9962 | 0.9910 | 0.9941 | 0.9930 |
| Parameter | Set III | Set IV | ||||
| G(H+)aq | −265.764(26) | −267.78(51) | −270.29(38) | −265.7155(75) | −266.81(45) | −269.63(15) |
| a | 7.59(83) | 4.91(57) | 5.48(60) | 7.271(57) | 3.60(34) | 4.33(16) |
| c | 0.126(23) | 0.275(41) | 0.240(40) | 0.1858(31) | 0.420(31) | 0.364(13) |
| SE | 0.3327 | 0.2261 | 0.2666 | 0.0454 | 0.1453 | 0.0766 |
| R2 | 0.9959 | 0.9981 | 0.9973 | 1.0000 | 0.9995 | 0.9994 |
| pKa (exp) | pKa | pKa (exp) | pKa | |||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Set I | LSDA | M062X | B3LYP | Set II | Set I | LSDA | M062X |
| pKa1 | 4.50 | 4.52 | 4.52 | 4.52 | 4.4(1) | 4.61 | 4.40 | 4.40 |
| pKa2 | 7.05 | 6.92 | 6.95 | 6.93 | 8.8(1) | 8.67 | 8.67 | 8.67 |
| pKa3 | 8.75 | 9.06 | 8.91 | 8.96 | 10.0(1) | 10.40 | 10.32 | 10.35 |
| pKa4 | 10.25 | 10.05 | 10.17 | 10.14 | 11.4(1) | 11.13 | 11.20 | 11.17 |
| MAE | 0.165 | 0.090 | 0.115 | 0.253 | 0.163 | 0.178 | ||
| Parameter | Set III | LSDA | M062X | B3LYP | Set IV | LSDA | M062X | B3LYP |
| pKa1 | 4.44 | 4.55 | 4.44 | 4.44 | 4.16(2) | 4.161 | 4.155 | 4.168 |
| pKa2 | 8.54 | 8.45 | 8.46 | 8.46 | 8.55(1) | 8.536 | 8.607 | 8.600 |
| pKa3 | 10.05 | 10.32 | 10.23 | 10.26 | 11.40(10) | 11.44 | 11.28 | 11.37 |
| pKa4 | 11.30 | 11.12 | 11.19 | 11.16 | 12.80(40) | 12.78 | 12.87 | 12.87 |
| MAE | 0.163 | 0.118 | 0.108 | 0.019 | 0.063 | 0.060 | ||
| Parameter | Set I | Set II | Set III | Set IV |
|---|---|---|---|---|
| ∆G(H+)aq | −259.19(32) | −259.486(20) | −259.476(26) | −259.4272(75) |
| a | 5.87(59) | 7.871(39) | 7.59(43) | 7.271(57) |
| c | 0.177(38) | 0.114(33) | 0.126(23) | 0.1858(31) |
| R2 | 0.9918 | 0.9910 | 0.9959 | 1.0000 |
| SE | 0.3861 | 0.4959 | 0.3327 | 0.0454 |
| pKa1 | 4.51 | 4.40 | 4.44 | 4.16 |
| pKa2 | 6.92 | 8.67 | 8.45 | 8.54 |
| pKa3 | 9.08 | 10.40 | 10.32 | 11.44 |
| pKa4 | 10.05 | 11.13 | 11.12 | 12.78 |
| MAE | 0.160 | 0.199 | 0.134 | 0.019 |
| NMAE | 1.992 | 0.463 |
| 6311++G(d,p) | aug-cc-pVQZ | QZVP | |
|---|---|---|---|
| ∆G(H+)aq | −257.1671(51) | −259.2358(72) | −259.4272(75) |
| a | 7.156(34) | 7.268(54) | 7.271(57) |
| c | 0.1934(19) | 0.1869(29) | 0.1858(31) |
| R2 | 1.0000 | 1.0000 | 1.0000 |
| SE | 0.0265 | 0.0430 | 0.0454 |
| pKa1 | 4.159 | 4.160 | 4.161 |
| pKa2 | 8.542 | 8.537 | 8.536 |
| pKa3 | 11.423 | 11.435 | 11.436 |
| pKa4 | 12.788 | 12.778 | 12.776 |
| MAE | 0.011 | 0.017 | 0.019 |
| NMAE | 0.268 | 0.427 | 0.463 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molski, M. Computation of the pKa Values of Gallic Acid and Its Anionic Forms in Aqueous Solution: A Self-Similar Transformation Approach for Accurate Proton Hydration Free Energy Estimation. Molecules 2025, 30, 742. https://doi.org/10.3390/molecules30030742
Molski M. Computation of the pKa Values of Gallic Acid and Its Anionic Forms in Aqueous Solution: A Self-Similar Transformation Approach for Accurate Proton Hydration Free Energy Estimation. Molecules. 2025; 30(3):742. https://doi.org/10.3390/molecules30030742
Chicago/Turabian StyleMolski, Marcin. 2025. "Computation of the pKa Values of Gallic Acid and Its Anionic Forms in Aqueous Solution: A Self-Similar Transformation Approach for Accurate Proton Hydration Free Energy Estimation" Molecules 30, no. 3: 742. https://doi.org/10.3390/molecules30030742
APA StyleMolski, M. (2025). Computation of the pKa Values of Gallic Acid and Its Anionic Forms in Aqueous Solution: A Self-Similar Transformation Approach for Accurate Proton Hydration Free Energy Estimation. Molecules, 30(3), 742. https://doi.org/10.3390/molecules30030742