
Small-Molecule Tyrosinase Inhibitors for Treatment of Hyperpigmentation



Abstract
:
1. Introduction
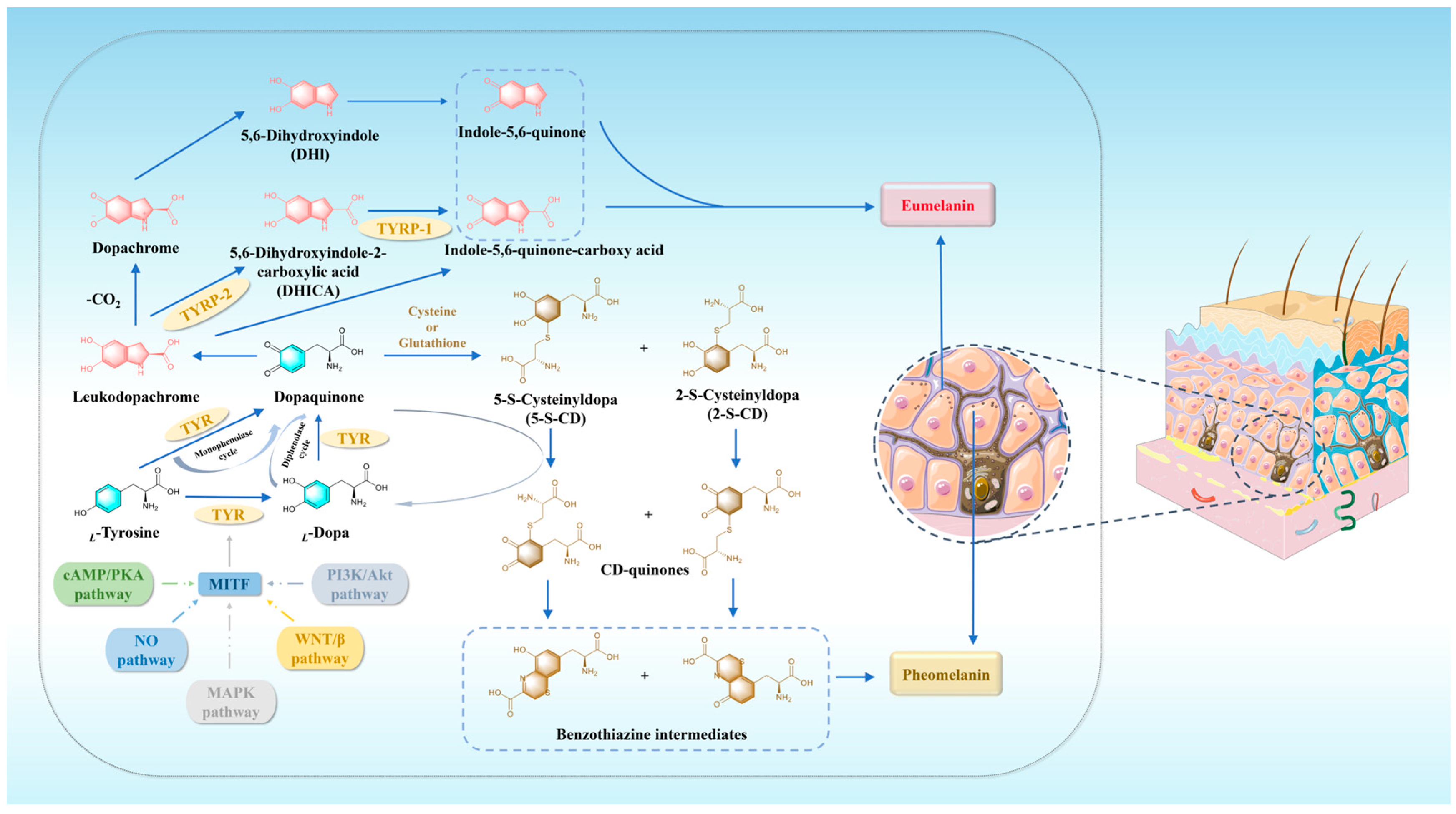
2. Melanin: Related Pathways and Biosynthesis Process
3. TYR: Structural Features and Catalytic Mechanism
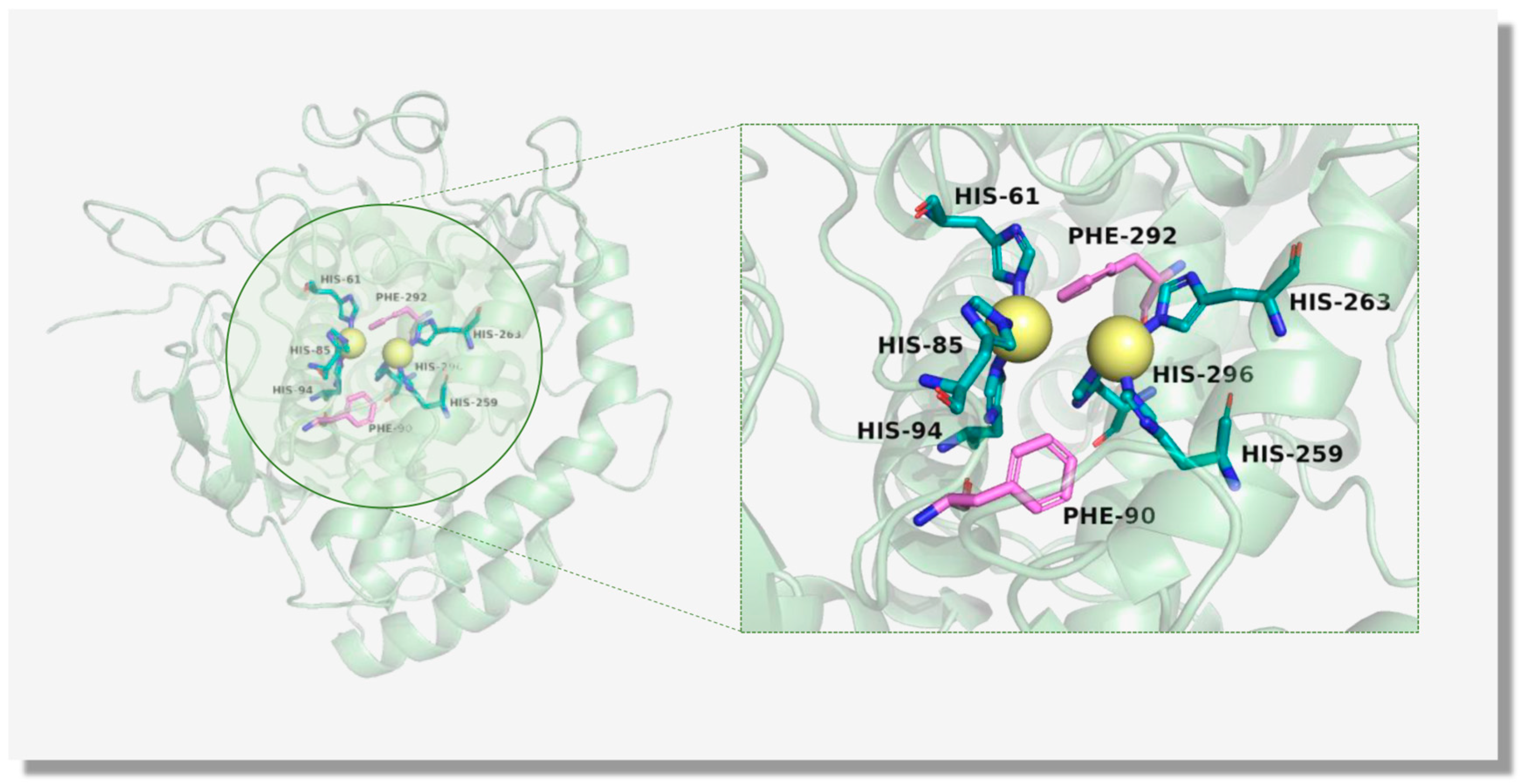
3.1. The Structure of TYR
3.2. The Catalytic Mechanism of TYR
4. Advanced Research Progress in TYR Inhibitors: Natural Products and Small-Molecule Compounds
4.1. TYR Inhibitors from Natural Products
4.2. Small-Molecule TYR Inhibitors in the Drug Discovery Phase
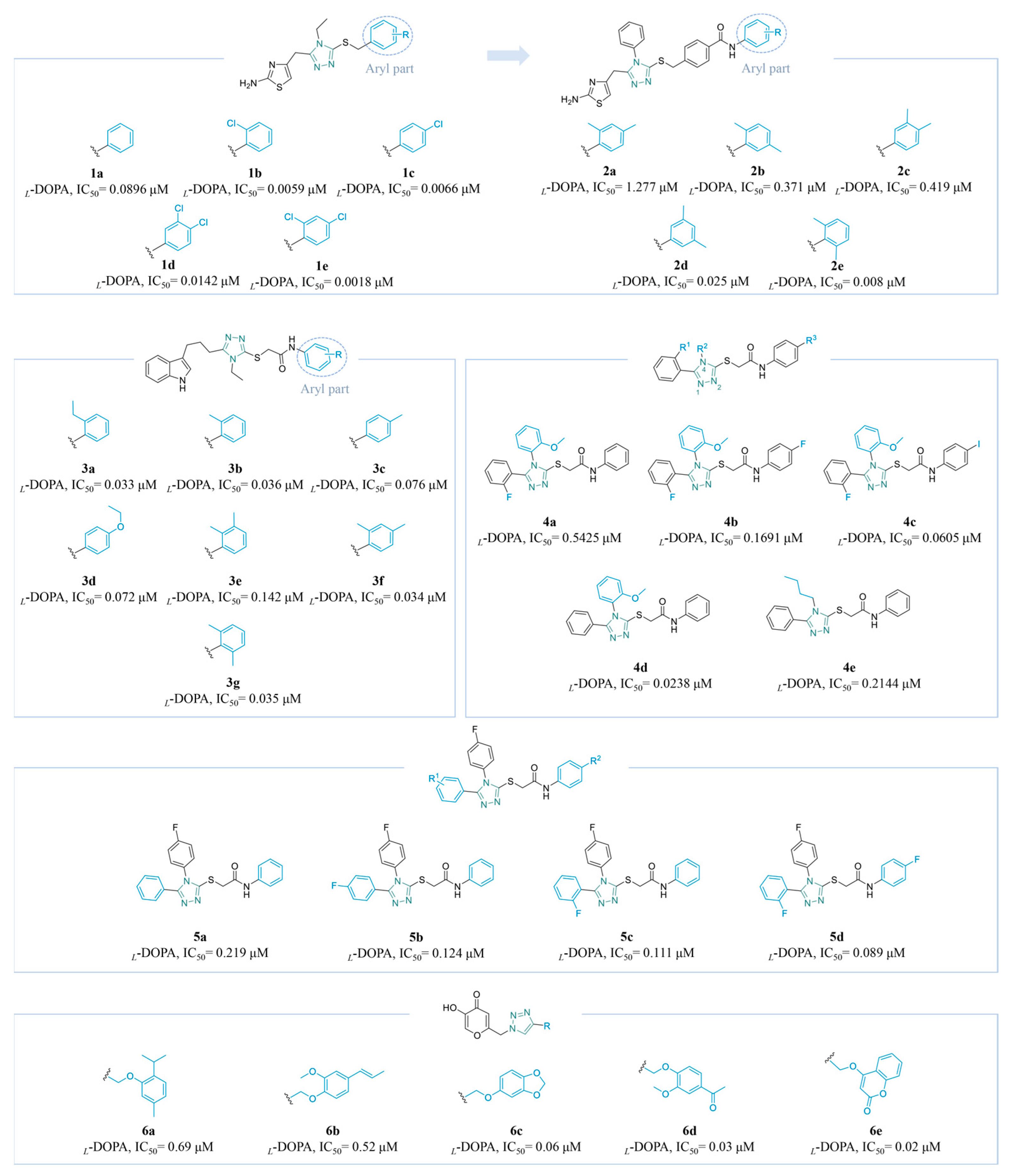
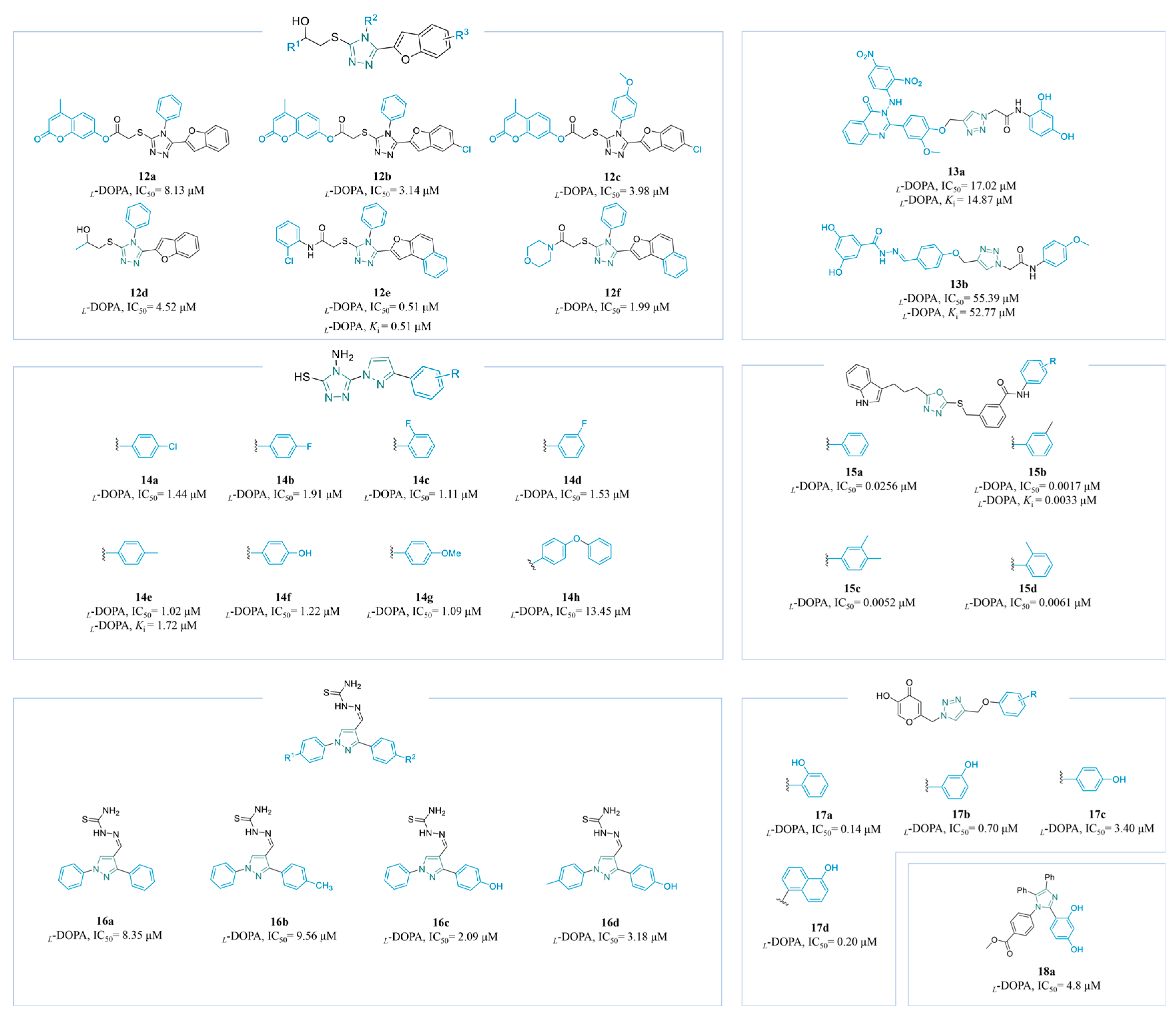
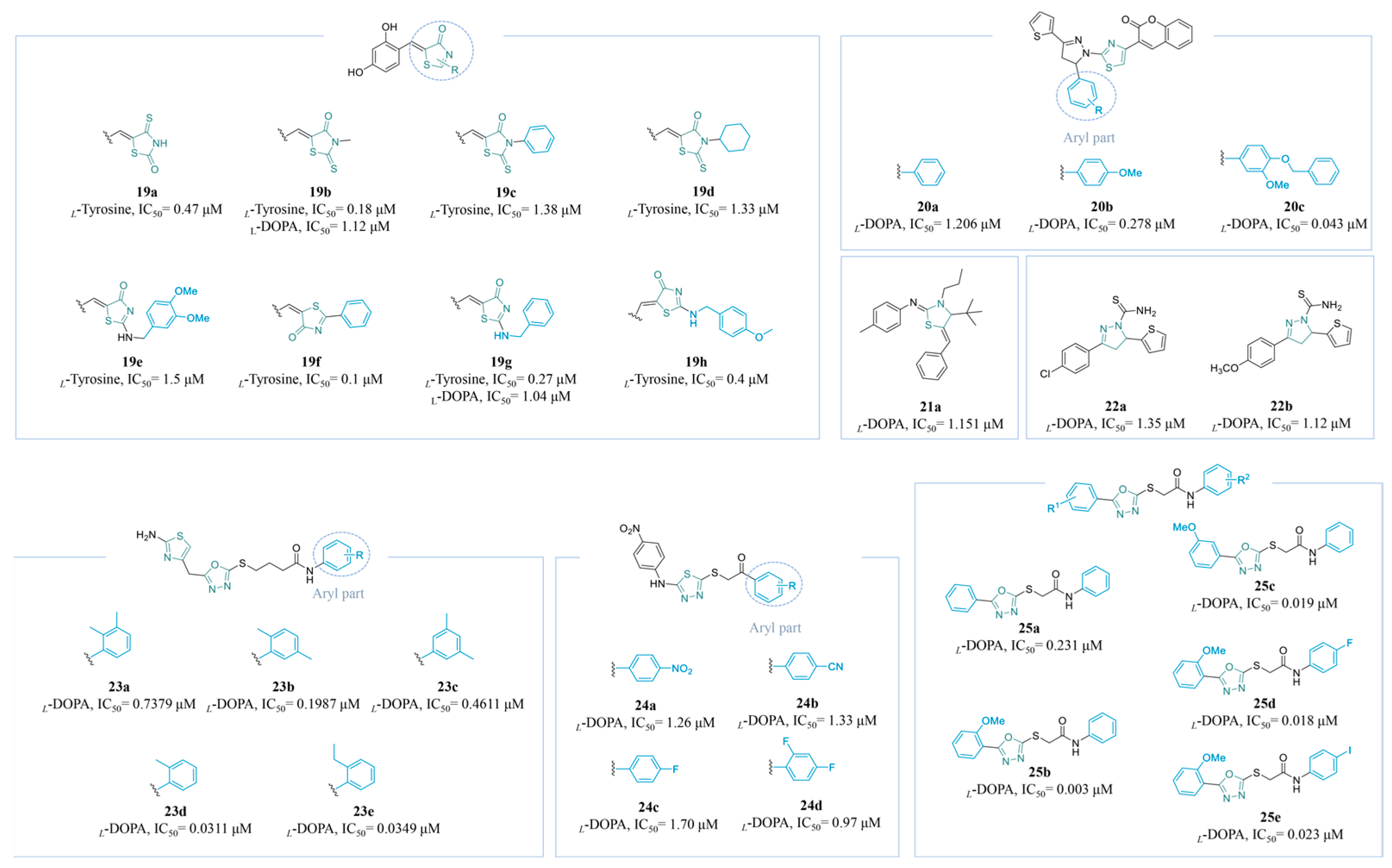
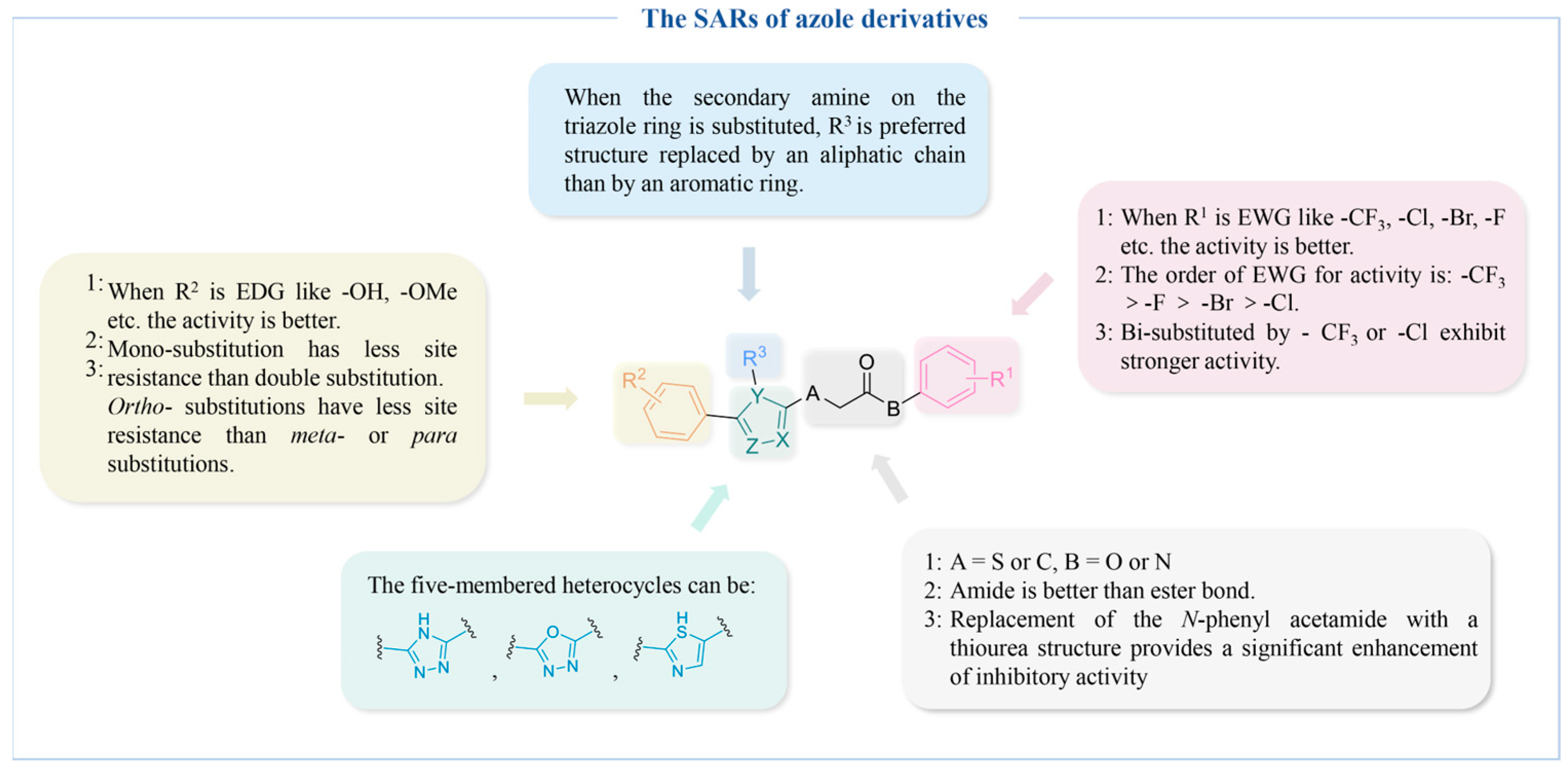
4.2.1. Azole Derivatives
Triazole Derivatives
Thiazole Derivatives
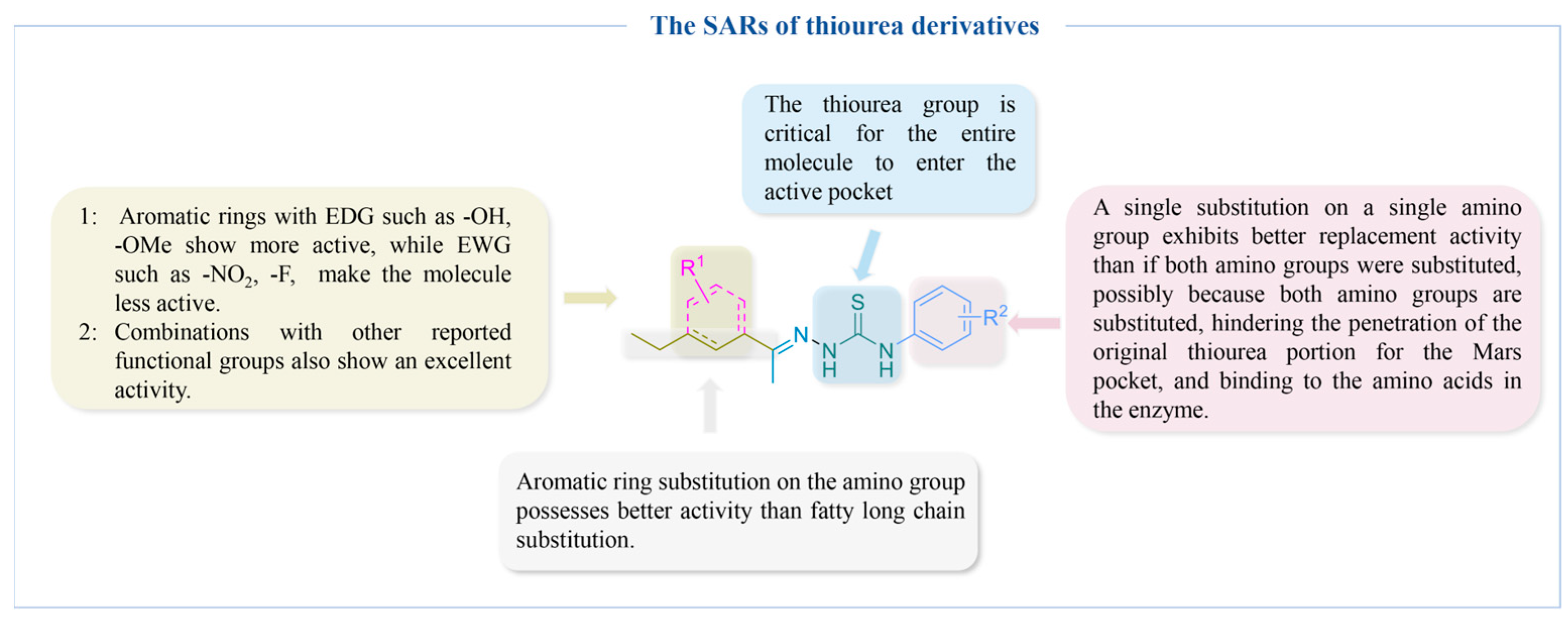
4.2.2. Thiourea Derivatives
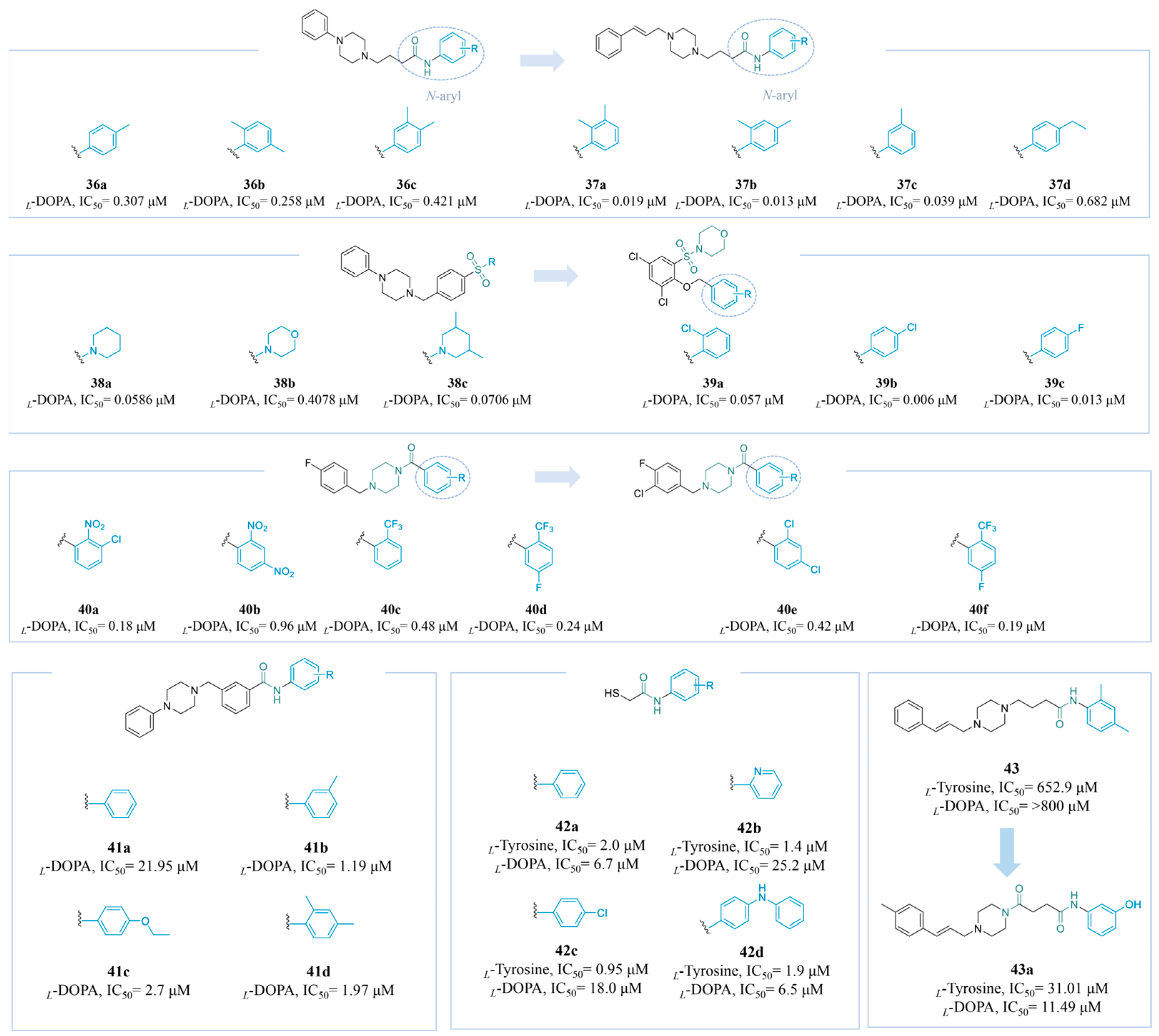
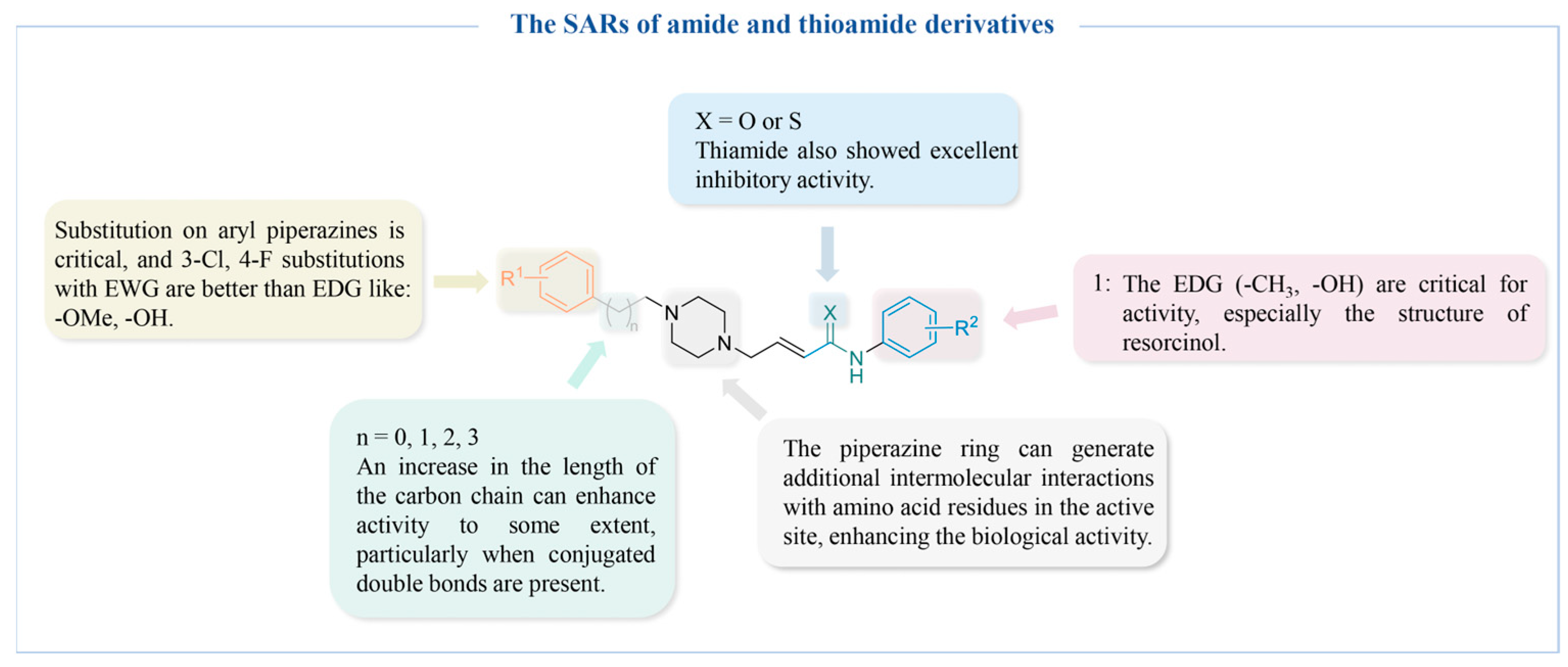
4.2.3. Amide and Thioamide Derivatives
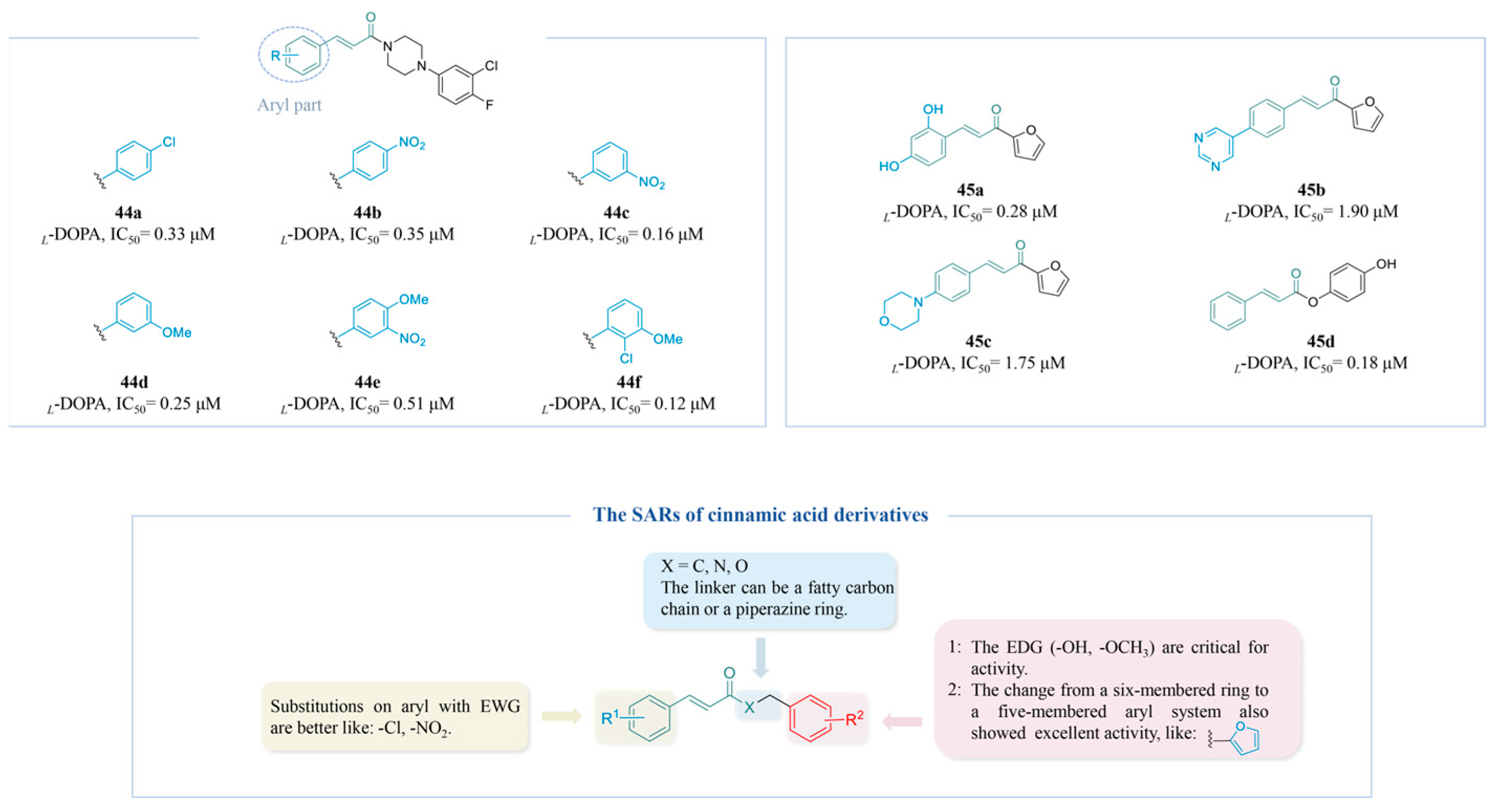
4.2.4. Cinnamic Acid Derivatives
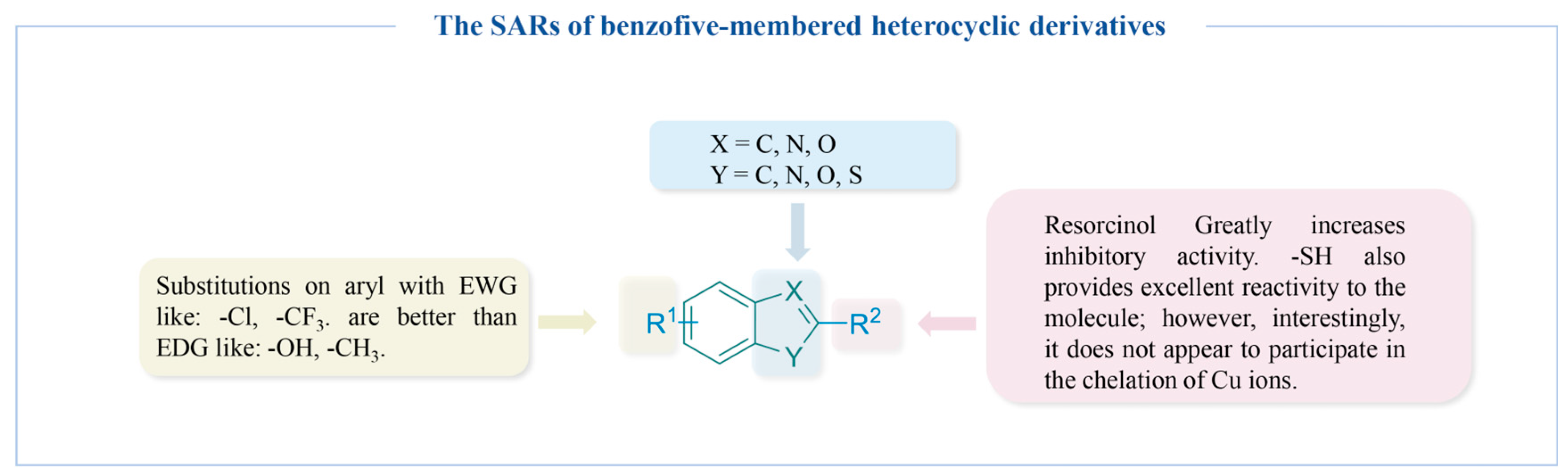
4.2.5. Benzo Five-Membered Heterocyclic Derivatives
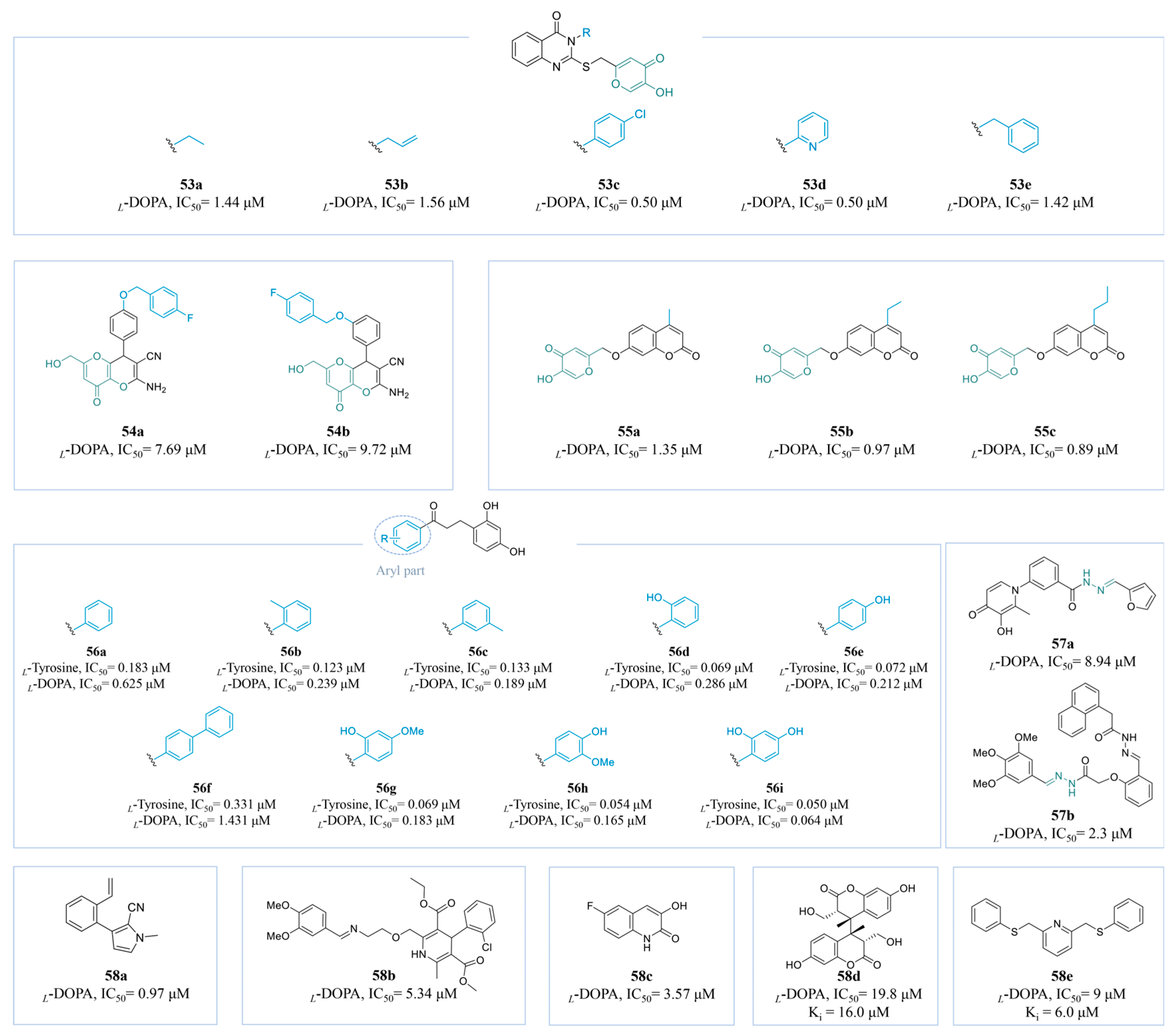
4.2.6. Other Derivatives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Xie, W.; Pakdel, E.; Liang, Y.; Kim, Y.J.; Liu, D.; Sun, L.; Wang, X. Natural Eumelanin and Its Derivatives as Multifunctional Materials for Bioinspired Applications: A Review. Biomacromolecules 2019, 20, 4312–4331. [Google Scholar] [CrossRef]
- Meredith, P.; Sarna, T. The physical and chemical properties of eumelanin. Pigment Cell Res. 2006, 19, 572–594. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Sarna, T.; Płonka, P.M.; Raman, C.; Brożyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496. [Google Scholar]
- Yoon, D.; Kang, M.K.; Jung, H.J.; Ullah, S.; Lee, J.; Jeong, Y.; Noh, S.G.; Kang, D.; Park, Y.; Chun, P.; et al. Design, Synthesis, In Vitro, and In Silico Insights of 5-(Substituted benzylidene)-2-phenylthiazol-4 (5H)-one Derivatives: A Novel Class of Anti-Melanogenic Compounds. Molecules 2023, 28, 3239. [Google Scholar] [CrossRef]
- Costin, G.E.; Hearing, V.J. Human skin pigmentation: Melanocytes modulate skin color in response to stress. Faseb J. 2007, 21, 976–994. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Jung, S.H. Recent development of signaling pathways inhibitors of melanogenesis. Cell Signal. 2017, 40, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Cestari, T.F.; Dantas, L.P.; Boza, J.C. Acquired hyperpigmentations. An. Bras. Dermatol. 2014, 89, 1–25. [Google Scholar] [CrossRef]
- Unver, N.; Freyschmidt-Paul, P.; Hörster, S.; Wenck, H.; Stäb, F.; Blatt, T.; Elsässer, H.P. Alterations in the epidermal-dermal melanin axis and factor XIIIa melanophages in senile lentigo and ageing skin. Br. J. Dermatol. 2006, 155, 119–128. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Qiao, G.; Zhang, Y. Treatment of Combined Freckles with Chloasma Using Q-Switched 1064 nm Laser. Int. J. Clin. Pract. 2023, 2023, 4081427. [Google Scholar] [CrossRef]
- Ali, Z.; Yousaf, N.; Larkin, J. Melanoma epidemiology, biology and prognosis. EJC. Suppl. 2013, 11, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Pang, Y.B.; Li, W.Q.; He, Q.Y.; Zhang, X.E.; Liu, E.; Guo, J. Global research trends on melasma: A bibliometric and visualized study from 2014 to 2023. Front. Pharmacol. 2024, 15, 1421499. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, E.J.; Ahn, Y.; Park, S.; Kim, S.H.; Oh, S.H. A chemical compound from fruit extract of Juglans mandshurica inhibits melanogenesis through p-ERK-associated MITF degradation. Phytomedicine 2019, 57, 57–64. [Google Scholar] [CrossRef]
- Ko, D.; Wang, R.F.; Ozog, D.; Lim, H.W.; Mohammad, T.F. Disorders of hyperpigmentation. Part II. Review of management and treatment options for hyperpigmentation. J. Am. Acad. Dermatol. 2023, 88, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.A.; Smit, S.E.; Barnes, C.C.; Pedley, J.; Parsons, P.G.; Sturm, R.A. Activation of the cAMP pathway by variant human MC1R alleles expressed in HEK and in melanoma cells. Peptides 2005, 26, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Hałdys, K.; Latajka, R. Thiosemicarbazones with tyrosinase inhibitory activity. Medchemcomm 2019, 10, 378–389. [Google Scholar] [CrossRef]
- Yu, F.; Pan, Z.; Qu, B.; Yu, X.; Xu, K.; Deng, Y.; Liang, F. Identification of a tyrosinase gene and its functional analysis in melanin synthesis of Pteria penguin. Gene 2018, 656, 1–8. [Google Scholar] [CrossRef]
- Baber, M.A.; Crist, C.M.; Devolve, N.L.; Patrone, J.D. Tyrosinase Inhibitors: A Perspective. Molecules 2023, 28, 5762. [Google Scholar] [CrossRef] [PubMed]
- Roulier, B.; Pérès, B.; Haudecoeur, R. Advances in the Design of Genuine Human Tyrosinase Inhibitors for Targeting Melanogenesis and Related Pigmentations. J. Med. Chem. 2020, 63, 13428–13443. [Google Scholar] [CrossRef]
- Jeon, H.J.; Kim, K.; Kim, C.; Lee, S.E. Antimelanogenic Effects of Curcumin and Its Dimethoxy Derivatives: Mechanistic Investigation Using B16F10 Melanoma Cells and Zebrafish (Danio rerio) Embryos. Foods 2023, 12, 926. [Google Scholar] [CrossRef] [PubMed]
- Niki, Y.; Adachi, N.; Fukata, M.; Fukata, Y.; Oku, S.; Makino-Okamura, C.; Takeuchi, S.; Wakamatsu, K.; Ito, S.; Declercq, L.; et al. S-Palmitoylation of Tyrosinase at Cysteine(500) Regulates Melanogenesis. J. Investig. Dermatol. 2023, 143, 317–327.e6. [Google Scholar] [CrossRef]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Kim, T.K.; Janjetovic, Z.; Brożyna, A.A.; Podgorska, E.; Dixon, K.M.; Mason, R.S.; Tuckey, R.C.; Sharma, R.; Crossman, D.K.; et al. Malignant Melanoma: An Overview, New Perspectives, and Vitamin D Signaling. Cancers 2024, 16, 2262. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Jóźwicki, W.; Carlson, J.A.; Slominski, A.T. Melanogenesis affects overall and disease-free survival in patients with stage III and IV melanoma. Hum. Pathol. 2013, 44, 2071–2074. [Google Scholar] [CrossRef]
- Buitrago, E.; Hardré, R.; Haudecoeur, R.; Jamet, H.; Belle, C.; Boumendjel, A.; Bubacco, L.; Réglier, M. Are Human Tyrosinase and Related Proteins Suitable Targets for Melanoma Therapy? Curr. Top. Med. Chem. 2016, 16, 3033–3047. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Zmijewski, M.A.; Slominski, A.T. The role of melanin pigment in melanoma. Exp. Dermatol. 2015, 24, 258–259. [Google Scholar] [CrossRef]
- Brozyna, A.A.; VanMiddlesworth, L.; Slominski, A.T. Inhibition of melanogenesis as a radiation sensitizer for melanoma therapy. Int. J. Cancer. 2008, 123, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Roulier, B.; Rush, I.; Lazinski, L.M.; Pérès, B.; Olleik, H.; Royal, G.; Fishman, A.; Maresca, M.; Haudecoeur, R. Resorcinol-based hemiindigoid derivatives as human tyrosinase inhibitors and melanogenesis suppressors in human melanoma cells. Eur. J. Med. Chem. 2023, 246, 114972. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Jóźwicki, W.; Roszkowski, K.; Filipiak, J.; Slominski, A.T. Melanin content in melanoma metastases affects the outcome of radiotherapy. Oncotarget 2016, 7, 17844–17853. [Google Scholar] [CrossRef]
- Slominski, A.; Zbytek, B.; Slominski, R. Inhibitors of melanogenesis increase toxicity of cyclophosphamide and lymphocytes against melanoma cells. Int. J. Cancer. 2009, 124, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Logesh, R.; Prasad, S.R.; Chipurupalli, S.; Robinson, N.; Mohankumar, S.K. Natural tyrosinase enzyme inhibitors: A path from melanin to melanoma and its reported pharmacological activities. Biochim. Biophys. Acta. Rev. Cancer 2023, 1878, 188968. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzyme Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef]
- Zolghadri, S.; Beygi, M.; Mohammad, T.F.; Alijanianzadeh, M.; Pillaiyar, T.; Garcia-Molina, P.; Garcia-Canovas, F.; Munoz-Munoz, J.; Saboury, A.A. Targeting tyrosinase in hyperpigmentation: Current status, limitations and future promises. Biochem. Pharmacol. 2023, 212, 115574. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A comprehensive review on tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Hassan, M.; Shahzadi, S.; Kloczkowski, A. Tyrosinase Inhibitors Naturally Present in Plants and Synthetic Modifications of These Natural Products as Anti-Melanogenic Agents: A Review. Molecules 2023, 28, 378. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Choi, H.; Abekura, F.; Park, J.Y.; Yang, W.S.; Yang, S.H.; Kim, C.H. Naturally-Occurring Tyrosinase Inhibitors Classified by Enzyme Kinetics and Copper Chelation. Int. J. Mol. Sci. 2023, 24, 8226. [Google Scholar] [CrossRef]
- Rolff, M.; Schottenheim, J.; Decker, H.; Tuczek, F. Copper-O2 reactivity of tyrosinase models towards external monophenolic substrates: Molecular mechanism and comparison with the enzyme. Chem. Soc. Rev. 2011, 40, 4077–4098. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure and Function of Human Tyrosinase and Tyrosinase-Related Proteins. Chemistry. 2018, 24, 47–55. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angew. Chem. Int. Ed. Engl. 2017, 56, 9812–9815. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viennet, C.; Robin, S.; Berthon, J.Y.; He, L.; Humbert, P. Precise role of dermal fibroblasts on melanocyte pigmentation. J. Dermatol. Sci. 2017, 88, 159–166. [Google Scholar] [CrossRef]
- Newton, R.A.; Roberts, D.W.; Leonard, J.H.; Sturm, R.A. Human melanocytes expressing MC1R variant alleles show impaired activation of multiple signaling pathways. Peptides 2007, 28, 2387–2396. [Google Scholar] [CrossRef]
- Kovacs, D.; Migliano, E.; Muscardin, L.; Silipo, V.; Catricalà, C.; Picardo, M.; Bellei, B. The role of Wnt/β-catenin signaling pathway in melanoma epithelial-to-mesenchymal-like switching: Evidences from patients-derived cell lines. Oncotarget 2016, 7, 43295–43314. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.F.; Ye, Y.; Tai, W.C.; Chou, G.X.; Leung, A.K.; Yu, Z.L.; Hsiao, W.L. Inhibition of the p38 and PKA signaling pathways is associated with the anti-melanogenic activity of Qian-wang-hong-bai-san, a Chinese herbal formula, in B16 cells. J. Ethnopharmacol 2012, 141, 622–628. [Google Scholar] [CrossRef]
- Park, H.Y.; Kosmadaki, M.; Yaar, M.; Gilchrest, B.A. Cellular mechanisms regulating human melanogenesis. Cell. Mol. Life. Sci. 2009, 66, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Chu, J.H.; Wang, H.; Xu, H.; Chou, G.X.; Leung, A.K.; Fong, W.F.; Yu, Z.L. Involvement of p38 MAPK signaling pathway in the anti-melanogenic effect of San-bai-tang, a Chinese herbal formula, in B16 cells. J. Ethnopharmacol 2010, 132, 533–535. [Google Scholar] [CrossRef]
- Tachibana, M. Cochlear melanocytes and MITF signaling. J. Investig. Dermatol. Symp. Proc. 2001, 6, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Borovanský, J. “Transcription physiology” of pigment formation in melanocytes: Central role of MITF. Exp. Dermatol. 2010, 19, 617–627. [Google Scholar] [CrossRef]
- Qu, Y.; Zhan, Q.; Du, S.; Ding, Y.; Fang, B.; Du, W.; Wu, Q.; Yu, H.; Li, L.; Huang, W. Catalysis-based specific detection and inhibition of tyrosinase and their application. J. Pharm. Anal. 2020, 10, 414–425. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Namasivayam, V.; Manickam, M.; Jung, S.H. Inhibitors of Melanogenesis: An Updated Review. J. Med. Chem. 2018, 61, 7395–7418. [Google Scholar] [CrossRef]
- Ortiz-Ruiz, C.V.; Maria-Solano, M.A.; Garcia-Molina Mdel, M.; Varon, R.; Tudela, J.; Tomas, V.; Garcia-Canovas, F. Kinetic characterization of substrate-analogous inhibitors of tyrosinase. IUBMB Life 2015, 67, 757–767. [Google Scholar] [CrossRef]
- Mendes, E.; Perry Mde, J.; Francisco, A.P. Design and discovery of mushroom tyrosinase inhibitors and their therapeutic applications. Expert Opin. Drug Discov. 2014, 9, 533–554. [Google Scholar] [CrossRef] [PubMed]
- Olivares, C.; García-Borrón, J.C.; Solano, F. Identification of active site residues involved in metal cofactor binding and stereospecific substrate recognition in Mammalian tyrosinase. Implications to the catalytic cycle. Biochemistry 2002, 41, 679–686. [Google Scholar] [CrossRef]
- Mayer, A.M. Polyphenol oxidases in plants and fungi: Going places? A review. Phytochemistry 2006, 67, 2318–2331. [Google Scholar] [CrossRef]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Mermer, A.; Demirci, S. Recent advances in triazoles as tyrosinase inhibitors. Eur. J. Med. Chem. 2023, 259, 115655. [Google Scholar] [CrossRef]
- Sanjust, E.; Cecchini, G.; Sollai, F.; Curreli, N.; Rescigno, A. 3-hydroxykynurenine as a substrate/activator for mushroom tyrosinase. Arch. Biochem. Biophys. 2003, 412, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Ahn, S.; Chang, H.; Cho, N.S.; Joo, K.; Lee, B.G.; Chang, I.; Hwang, J.S. Influence of N-glycan processing disruption on tyrosinase and melanin synthesis in HM3KO melanoma cells. Exp. Dermatol. 2007, 16, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, L.; Liu, L.; Wang, F.; Ouyang, L.; Zhang, L.; Hu, X.; Wang, G. Recent advances in the design and discovery of synthetic tyrosinase inhibitors. Eur. J. Med. Chem. 2021, 224, 113744. [Google Scholar] [CrossRef] [PubMed]
- McLarin, M.A.; Leung, I.K.H. Substrate specificity of polyphenol oxidase. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 274–308. [Google Scholar] [CrossRef]
- Olivares, C.; Solano, F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigment Cell Melanoma Res. 2009, 22, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wendt, M.; Klebe, G.; Röhm, K.H. On the interpretation of tyrosinase inhibition kinetics. J. Enzym. Inhib. Med. Chem. 2014, 29, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 22388–22395. [Google Scholar] [CrossRef]
- Min, K.; Park, G.W.; Yoo, Y.J.; Lee, J.S. A perspective on the biotechnological applications of the versatile tyrosinase. Bioresour. Technol. 2019, 289, 121730. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Fang, Z.; Zhang, P. The melanin inhibitory effect of plants and phytochemicals: A systematic review. Phytomedicine 2022, 107, 154449. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, D.H.; Lee, K.W.; Kim, K.D.; Shah, A.B.; Zhumanova, K.; Park, K.H. Tyrosinase Inhibition and Kinetic Details of Puerol A Having But-2-Enolide Structure from Amorpha fruticosa. Molecules 2020, 25, 2344. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.X.; Lin, M.; Lu, S.S.; Qi, X.Y.; Zhang, R.X.; Zhang, Y.Y. Curcumin inhibits melanogenesis in human melanocytes. Phytother. Res. 2012, 26, 174–179. [Google Scholar] [CrossRef]
- Zeng, H.J.; Li, Q.Y.; Ma, J.; Yang, R.; Qu, L.B. A comparative study on the effects of resveratrol and oxyresveratrol against tyrosinase activity and their inhibitory mechanism. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2021, 251, 119405. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Jeong, H.O.; Lee, E.K.; Kim, S.J.; Chun, P.; Chung, H.Y.; Moon, H.R. Evaluation of Antimelanogenic Activity and Mechanism of Galangin in Silico and in Vivo. Biol. Pharm. Bull. 2018, 41, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Lee, S.H. Sesamol decreases melanin biosynthesis in melanocyte cells and zebrafish: Possible involvement of MITF via the intracellular cAMP and p38/JNK signalling pathways. Exp. Dermatol. 2015, 24, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hwang, I.H.; Kim, J.H.; Kim, M.A.; Hwang, J.S.; Kim, Y.H.; Na, M. Quinoxaline-, dopamine-, and amino acid-derived metabolites from the edible insect Protaetia brevitarsis seulensis. Arch. Pharm. Res. 2017, 40, 1064–1070. [Google Scholar] [CrossRef]
- Ren, H.; Xu, Q.L.; Zhang, M.; Dong, L.M.; Zhang, Q.; Luo, B.; Luo, Q.W.; Tan, J.W. Bioactive caffeic acid derivatives from Wedelia trilobat. Phytochemistry Lett. 2017, 19, 18–22. [Google Scholar] [CrossRef]
- Xie, D.; Fu, W.; Yuan, T.; Han, K.; Lv, Y.; Wang, Q.; Jiang, Q.; Zhang, Y.; Zhu, G.; Xu, J.; et al. 6′-O-Caffeoylarbutin from Quezui Tea: A Highly Effective and Safe Tyrosinase Inhibitor. Int. J. Mol. Sci. 2024, 25, 972. [Google Scholar] [CrossRef]
- He, X.R.; Xu, L.Y.; Jin, C.; Yue, P.F.; Zhou, Z.W.; Liang, X.L. Tamariscinols U-W, new dihydrobenzofuran-type norneolignans with tyrosinase inhibitory activity from Selaginella tamariscina. Phytochem. Lett. 2019, 34, 79–83. [Google Scholar] [CrossRef]
- Lien, G.T.K.; Van, D.T.T.; Cuong, D.H.; Yen, P.H.; Tai, B.H.; Kiem, P.V. A New Phenolic Constituent from Carica papaya Flowers and Its Tyrosinase Inhibitory Activity. Nat. Prod. Commun. 2019, 14, 1934578X19850987. [Google Scholar]
- Paudel, P.; Wagle, A.; Seong, S.H.; Park, H.J.; Jung, H.A.; Choi, J.S. A New Tyrosinase Inhibitor from the Red Alga Symphyocladia latiuscula (Harvey) Yamada (Rhodomelaceae). Mar. Drugs 2019, 17, 295. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.I.; Chabán, M.F.; Lanza, P.A.; Joray, M.B.; Palacios, S.M.; Vera, D.M.A.M.; Carpinella, C. Inhibitory effects of compounds isolated from Lepechinia meyenii on tyrosinase. Food. Chem. Toxicol. 2019, 125, 383–391. [Google Scholar] [CrossRef]
- Kim, D.H.; Lee, J.H. Comparative evaluation of phenolic phytochemicals from perilla seeds of diverse species and screening for their tyrosinase inhibitory and antioxidant properties. S. Afr. J. Bot. 2019, 123, 341–350. [Google Scholar] [CrossRef]
- Honisch, C.; Osto, A.; Dupas de Matos, A.; Vincenzi, S.; Ruzza, P. Isolation of a tyrosinase inhibitor from unripe grapes juice: A spectrophotometric study. Food Chem. 2020, 305, 125506. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.T.; Liang, Y.Q.; Chai, W.M.; Wei, Q.M.; Yu, Z.Y.; Wang, L.J. Effect of ascorbic acid on tyrosinase and its anti-browning activity in fresh-cut Fuji apple. J. Food Bio. 2021, 45, e13995. [Google Scholar] [CrossRef]
- Yalo, M.; Makhaba, M.; Hussein, A.A.; Sharma, R.; Koki, M.; Nako, N.; Mabusela, W.T. Characterization of Four New Compounds from Protea cynaroide Leaves and Their Tyrosinase Inhibitory Potential. Plants 2022, 11, 1751. [Google Scholar] [CrossRef]
- Shang, C.; Zhang, Y.; You, X.; Guo, N.; Wang, Y.; Fan, Y.; Liu, W. The effect of 7,8,4’-trihydroxyflavone on tyrosinase activity and conformation: Spectroscopy and docking studies. Luminescence 2018, 33, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Song, X.; Ouyang, M.; El-kott, A.F.; Bani-Fwaz, M.Z.; Yu, Z. Anti-HMG-CoA Reductase, Anti-diabetic, Anti-urease, Anti-tyrosinase and Anti-leukemia Cancer Potentials of Panicolin as a Natural Compound: In vitro and in silico Study. J. Oleo Sci. 2022, 71, 1469–1480. [Google Scholar] [CrossRef]
- Tian, J.L.; Liu, T.L.; Xue, J.J.; Hong, W.; Zhang, Y.; Zhang, D.X.; Cui, C.C.; Liu, M.C.; Niu, S.L. Flavanoids derivatives from the root bark of Broussonetia papyrifera as a tyrosinase inhibitor. Ind. Crops. Prod. 2019, 138, 111445. [Google Scholar] [CrossRef]
- Nguyen, M.T.T.; Le, T.H.; Nguyen, H.X.; Dang, P.H.; Do, T.N.V.; Abe, M.; Takagi, R.; Nguyen, N.T. Artocarmins G-M, Prenylated 4-Chromenones from the Stems of Artocarpus rigid and Their Tyrosinase Inhibitory Activities. J. Nat. Prod. 2017, 80, 3173–3179. [Google Scholar] [CrossRef]
- Nguyen, H.X.; Nguyen, N.T.; Nguyen, M.H.K.; Le, T.H.; Do, T.N.V.; Hung, T.M.; Nguyen, M.T.T. Tyrosinase inhibitory activity of flavonoids from Artocarpus heterophyllous. Chem. Cent. J. 2016, 10, 2. [Google Scholar] [CrossRef]
- Kim, J.H.; Cho, I.S.; So, Y.K.; Kim, H.H.; Kim, Y.H. Kushenol A and 8-prenylkaempferol, tyrosinase inhibitors, derived from Sophora flavescens. J. Enzym. Inhib. Med. Chem. 2018, 33, 1048–1054. [Google Scholar] [CrossRef]
- Fan, F.; Chen, L.; Chen, C.; Ang, S.; Gutkowski, J.; Seeram, N.P.; Ma, H.; Li, D. Prenylated flavonoids from Sophora flavescens inhibit mushroom tyrosinase activity and modulate melanogenesis in murine melanoma cells and zebrafish. Front. Pharmacol. 2024, 15, 1422310. [Google Scholar] [CrossRef]
- Kim, D.W.; Woo, H.S.; Kim, J.Y.; Ryuk, J.A.; Park, K.H.; Ko, B.S. Phenols displaying tyrosinase inhibition from Humulus lupulus. J. Enzyme Inhib. Med. Chem. 2016, 31, 742–747. [Google Scholar] [CrossRef]
- Solimine, J.; Garo, E.; Wedler, J.; Rusanov, K.; Fertig, O.; Hamburger, M.; Atanassov, I.; Butterweck, V. Tyrosinase inhibitory constituents from a polyphenol enriched fraction of rose oil distillation wastewater. Fitoterapia 2016, 108, 13–19. [Google Scholar] [CrossRef]
- Omar, S.H.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. Biophenols: Enzymes (β-secretase, Cholinesterases, histone deacetylase and tyrosinase) inhibitors from olive (Olea europaea L.). Fitoterapia 2018, 128, 118–129. [Google Scholar] [CrossRef]
- Chaita, E.; Lambrinidis, G.; Cheimonidi, C.; Agalou, A.; Beis, D.; Trougakos, I.; Mikros, E.; Skaltsounis, A.L.; Aligiannis, N. Anti-Melanogenic Properties of Greek Plants. A Novel Depigmenting Agent from Morus alba Wood. Molecules 2017, 22, 514. [Google Scholar] [CrossRef]
- Ding, H.Y.; Chiang, C.M.; Tzeng, W.M.; Chang, T.S. Identification of 3’-hydroxygenistein as a potent melanogenesis inhibitor from biotransformation of genistein by recombinant Pichia pastoris. Process Biochem. 2015, 50, 1614–1617. [Google Scholar] [CrossRef]
- Morgan, A.M.A.; Jeon, M.N.; Jeong, M.H.; Yang, S.Y.; Kim, Y.H. Chemical Components from the Stems of Pueraria lobata and Their Tyrosinase Inhibitory Activity. Nat. Prod. Sci. 2016, 22, 111–116. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.Y.; Kang, S.Y.; Kim, J.B.; Kim, Y.H.; Jin, C.H. Chemical Constituents from Apios american and Their Inhibitory Activity on Tyrosinase. Molecules 2018, 23, 232. [Google Scholar] [CrossRef]
- Wagle, A.; Seong, S.H.; Jung, H.A.; Choi, J.S. Identifying an isoflavone from the root of Pueraria lobat as a potent tyrosinase inhibitor. Food Chem. 2019, 276, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Kuang, Y.; Li, K.; Wang, S.; Song, W.; Qiao, X.; Sabir, G.; Ye, M. Screening for bioactive natural products from a 67-compound library of Glycyrrhiza inflat. Bioor. Med. Chem. 2017, 25, 3706–3713. [Google Scholar] [CrossRef]
- Zhu, J.J.; Yan, G.R.; Xu, Z.J.; Hu, X.; Wang, G.H.; Wang, T.; Zhu, W.L.; Hou, A.J.; Wang, H.Y. Inhibitory Effects of (2’R)-2’, 3’-dihydro-2’-(1-hydroxy-1-methylethyl)-2,6’-bibenzofuran-6,4’-diol on Mushroom Tyrosinase and Melanogenesis in B16-F10 Melanoma Cells. Phytother Res. 2015, 29, 1040–1045. [Google Scholar] [CrossRef]
- Zhang, L.; Tao, G.J.; Chen, J.; Zheng, Z.P. Characterization of a New Flavone and Tyrosinase Inhibition Constituents from the Twigs of Morus alb L. Molecules 2016, 21, 1130. [Google Scholar] [CrossRef]
- Le, T.H.; Nguyen, H.X.; Do, T.V.N.; Dang, P.H.; Nguyen, N.T.; Nguyen, M.T.T.; Moracin, V.N. A New Tyrosinase and Xanthine Oxidase Inhibitor from the Woods of Artocarpus heterophyllu. Nat. Prod. Commun. 2017, 12, 925–927. [Google Scholar]
- Xue, Y.L.; Miyakawa, T.; Hayashi, Y.; Okamoto, K.; Hu, F.Y.; Mitani, N.; Furihata, K.; Sawano, Y.; Tanokura, M. Isolation and Tyrosinase Inhibitory Effects of Polyphenols from the Leaves of Persimmon. Diospyros kaki. J. Agri. Food Chem. 2011, 59, 6011–6017. [Google Scholar] [CrossRef]
- Yang, S.Y.; Kim, J.H.; Su, X.D.; Kim, J.A. The Luteolinidin and Petunidin 3-O-Glucoside: A Competitive Inhibitor of Tyrosinase. Molecules 2022, 27, 5703. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.N.; Cazier, E.A.; Gormally, E.; Lawrence, P. Valorization of biomass polyphenols as potential tyrosinase inhibitors. Drug Discov. Today 2024, 29, 103843. [Google Scholar] [CrossRef]
- Tan, X.; Song, Y.H.; Park, C.; Lee, K.W.; Kim, J.Y.; Kim, D.W.; Kim, K.D.; Lee, K.; Curtis-Long, W.M.J.; Park, K.H. Highly potent tyrosinase inhibitor, neorauflavane from Campylotropis hirtella and inhibitory mechanism with molecular docking. Bioorg. Med. Chem. 2016, 24, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.H.; Oh, K.E.; Jo, Y.H.; Ahn, J.H.; Liu, Q.; Turk, A.; Jang, J.Y.; Hwang, B.Y.; Lee, K.Y.; Lee, M.K. Characterization of tyrosinase inhibitory constituents from the aerial parts of Humulus japonicus using LC-MS/MS coupled online assay. Bioorg. Med. Chem. 2018, 26, 509–515. [Google Scholar] [CrossRef]
- Yang, Y.F.; Sun, X.; Ni, H.; Du, X.P.; Chen, F.; Jiang, Z.D.; Li, Q.B. Identification and Characterization of the Tyrosinase Inhibitory Activity of Caffeine from Camellia Pollen. J. Agri. Food Chem. 2019, 67, 12741–12751. [Google Scholar] [CrossRef]
- Georgousaki, K.; Tsafantakis, N.; Gumeni, S.; Gonzalez, I.; Mackenzie, T.A.; Reyes, F.; Lambert, C.; Trougakos, I.P.; Genilloud, O.; Fokialakis, N. Screening for tyrosinase inhibitors from actinomycetes; identification of trichostatin derivatives from Streptomyces sp. CA-129531 and scale up production in bioreactor. Bioorg. Med. Chem. Lett. 2020, 30, 126952. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Leem, H.H.; Lee, G.Y. The Guanidine Pseudoalkaloids 10-Methoxy-Leonurine and Leonurine Act as Competitive Inhibitors of Tyrosinase. Biomolecules 2020, 10, 174. [Google Scholar] [CrossRef]
- Zeng, H.J.; Sun, D.Q.; Chu, S.H.; Zhang, J.J.; Hu, G.Z.; Yang, R. Inhibitory effects of four anthraquinones on tyrosinase activity: Insight from spectroscopic analysis and molecular docking. Int. J. Biol. Macromol. 2020, 160, 153–163. [Google Scholar] [CrossRef]
- Butt, A.R.S.; Abbasi, M.A.; Aziz Ur, R.; Siddiqui, S.Z.; Raza, H.; Hassan, M.; Shah, S.A.A.; Seo, S.Y. Synthesis, Kinetics, Binding Conformations and Structure-activity Relationship of Potent Tyrosinase Inhibitors: Aralkylated 2-aminothiazole-ethyltriazole Hybrids. Iran. J. Pharm. Res. 2021, 20, 206–228. [Google Scholar]
- Khan, F.M.; Abbasi, M.A.; Rehman, A.U.; Siddiqui, S.Z.; Butt, A.R.S.; Raza, H.; Hassan, M.; Shah, S.A.A.; Shahid, M.; Kim, S.J. Design of potent tyrosinase inhibiting N-arylated-4-yl-benzamides bearing 2-aminothiazole-triazole bi-heterocycles: Mechanistic insight through enzyme inhibition, kinetics and computational studies. Rsc Adv. 2024, 14, 16546–16559. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.A.; Siddiqui, S.Z.; Nazir, M.; Raza, H.; Zafar, A.; Shah, S.A.A.; Shahid, M.S.; Seo, Y.; Shakila, M.; Aziz-ur-Rehman, S.Y. Multi-step synthesis of indole-N-ethyltriazole hybrids amalgamated with N-arylated ethanamides: Structure-activity relationship and mechanistic explorations through tyrosinase inhibition, kinetics and computational ascriptions. J. Mol. Struct. 2022, 1261, 132953. [Google Scholar]
- Vanjare, B.D.; Mahajan, P.G.; Dige, N.C.; Raza, H.; Hassan, M.; Han, Y.; Kim, S.J.; Seo, S.Y.; Lee, K.H. Novel 1,2,4-triazole analogues as mushroom tyrosinase inhibitors: Synthesis, kinetic mechanism, cytotoxicity and computational studies. Mol. Divers 2021, 25, 2089–2106. [Google Scholar] [CrossRef]
- Hassan, M.; Vanjare, B.D.; Sim, K.Y.; Raza, H.; Lee, K.H.; Shahzadi, S.; Kloczkowski, A. Biological and Cheminformatics Studies of Newly Designed Triazole Based Derivatives as Potent Inhibitors against Mushroom Tyrosinase. Molecules 2022, 27, 1731. [Google Scholar] [CrossRef]
- Ashooriha, M.; Khoshneviszadeh, M.; Khoshneviszadeh, M.; Rafiei, A.; Kardan, M.; Yazdian-Robati, R.; Emami, S. Kojic acid-natural product conjugates as mushroom tyrosinase inhibitors. Eur. J. Med. Chem. 2020, 201, 112480. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, G.; Zeng, Q.H.; Li, Y.; Wu, Y.; Liu, H.; Wang, J.J.; Zhao, Y. Synthesis, antioxidant and anti-tyrosinase activity of 1,2,4-triazole hydrazones as antibrowning agents. Food Chem. 2021, 341, 128265. [Google Scholar] [CrossRef]
- Hosseinpoor, H.; Farid, S.M.; Iraji, A.; Askari, S.; Edraki, N.; Hosseini, S.; Jamshidzadeh, A.; Larijani, B.; Attarroshan, M.; Pirhadi, S.; et al. Anti-melanogenesis and anti-tyrosinase properties of aryl-substituted acetamides of phenoxy methyl triazole conjugated with thiosemicarbazide: Design, synthesis and biological evaluations. Bioorg. Chem. 2021, 114, 104979. [Google Scholar] [CrossRef]
- Divar, M.; Tadayyon, S.; Khoshneviszadeh, M.; Pirhadi, S.; Attarroshan, M.; Mobaraki, K.; Damghani, T.; Mirfazli, S.; Edraki, N. Benzyl-Triazole Derivatives of Hydrazinecarbothiamide Derivatives as Potent Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Structure-Activity Relationship and Docking Study. Chemistryselect 2023, 8, e202203382. [Google Scholar] [CrossRef]
- Kausar, R.; Zahoor, A.F.; Tabassum, H.; Kamal, S.; Ahmad Bhat, M. Synergistic Biomedical Potential and Molecular Docking Analyses of Coumarin-Triazole Hybrids as Tyrosinase Inhibitors: Design, Synthesis, In Vitro Profiling, and In Silico Studies. Pharmaceuticals 2024, 17, 532. [Google Scholar] [CrossRef] [PubMed]
- Gultekin, E.; Bekircan, O.; Kolcuoglu, Y.; Akdemir, A. Synthesis of new 1,2,4-triazole-(thio)semicarbazide hybrid molecules: Their tyrosinase inhibitor activities and molecular docking analysis. Arch. Pharm. 2021, 354, e2100058. [Google Scholar] [CrossRef]
- Saeed, S.; Saif, M.J.; Zahoor, A.F.; Tabassum, H.; Kamal, S.; Faisal, S.; Ashraf, R.; Khan, S.G.; Nazeer, U.; Irfan, A.; et al. Discovery of novel 1,2,4-triazole tethered β-hydroxy sulfides as bacterial tyrosinase inhibitors: Synthesis and biophysical evaluation through in vitro and in silico approaches. RSC. Advances. 2024, 14, 15419–15430. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, A.; Ahmad, M.N.; Zahoor, A.F.; Kamal, S.; Ali, K.G.; Javid, J.; Parveen, B.; Nazeer, U.; Bhat, M.A. Design, CTAB-catalyzed ultrasound-assisted synthesis and tyrosinase inhibition potential of naphthofuran-triazole conjugates. RSC. Advances. 2024, 14, 37521–37538. [Google Scholar] [CrossRef]
- Bagheri, A.; Moradi, S.; Iraji, A.; Mahdavi, M. Structure-based development of 3,5-dihydroxybenzoyl-hydrazineylidene as tyrosinase inhibitor; in vitro and in silico study. Sci. Rep. 2024, 14, 1540. [Google Scholar] [CrossRef] [PubMed]
- Moghadam Farid, S.; Moradi Dehaghi, S.; Iraji, A.; Mahdavi, M.; Saeedi, M. Synthesis, biological evaluations, and in silico assessments of phenylamino quinazolinones as tyrosinase inhibitors. Sci. Rep. 2025, 15, 846. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, W.; He, M.; Peng, Z.; Wang, G. Development of novel pyrazole-1,2,4-triazole derivatives as tyrosinase inhibitors: Design, preparation, mechanism of action and anti-browning application. Food Chem. 2024, 460, 140722. [Google Scholar] [CrossRef] [PubMed]
- Nazir, M.; Khan, U.; Jahangir, M.; Hayat, K.; Bokhari, S.A.R.; Shakoor, A.; Ahmad, E.; Haider, H.M.F. Exploring the Mechanisms of Indole-Oxadiazole Benzamide Hybrids as Tyrosinase Inhibitors: Insights from Lineweaver-Burk Plot Analysis and Computational Studies. Russ. J. Bioorg. Chem. 2024, 50, 2325–2343. [Google Scholar] [CrossRef]
- Azimi, F.; Mahdavi, M.; Khoshneviszadeh, M.; Shafiee, F.; Azimi, M.; Hassanzadeh, F.; Haji Ashrafee, F. Kinetic studies, molecular docking, and antioxidant activity of novel 1,3-diphenyl pyrazole-thiosemicarbazone with anti-tyrosinase and anti-melanogenesis properties. Bioorg. Chem. 2024, 152, 107722. [Google Scholar] [CrossRef]
- Ashooriha, M.; Khoshneviszadeh, M.; Kabiri, M.; Dehshahri, A.; Hassani, B.; Ansari, M.; Emami, S. Multi-functional tyrosinase inhibitors derived from kojic acid and hydroquinone-like diphenols for treatment of hyperpigmentation: Synthesis and in vitro biological evaluation. Arch. Pharm. 2024, 357, e2400380. [Google Scholar] [CrossRef]
- Naseem, M.; Rafique, H.; Roshan, S.; Ashraf, Z.; Perveen, F.; Tayyab, M. One-pot, Four-component Synthesis, Molecular Docking and Pharmacokinetic Studies of Tetra-substituted Imidazole Derivatives as Potential Mushroom Tyrosinase Inhibitors. Curr. Pharm. Des. 2025. Online ahead of print. [Google Scholar] [CrossRef]
- Choi, H.; Ryu, I.Y.; Choi, I.; Ullah, S.; Jung, H.J.; Park, Y.; Jeong, Y.; Hwang, Y.; Hong, S.; Yoon, I.S.; et al. Novel Anti-Melanogenic Compounds, (Z)-5-(Substituted Benzylidene)-4-thioxothiazolidin-2-one Derivatives: In Vitro and In Silico Insights. Molecules 2021, 26, 4963. [Google Scholar] [CrossRef]
- Kim, H.J.; Jung, H.J.; Kim, Y.E.; Jeong, D.; Park, H.S.; Park, H.S.; Kang, D.; Park, Y.; Chun, P.; Chung, H.Y.; et al. Investigation of the Efficacy of Benzylidene-3-methyl-2-thioxothiazolidin-4-one Analogs with Antioxidant Activities on the Inhibition of Mushroom and Mammal Tyrosinases. Molecules 2024, 2, 2887. [Google Scholar] [CrossRef]
- Jeong, Y.; Hong, S.; Jung, H.J.; Ullah, S.; Hwang, Y.; Choi, H.; Ko, J.; Lee, J.; Chun, P.; Chung, H.Y.; et al. Identification of a Novel Class of Anti-Melanogenic Compounds, (Z)-5-(Substituted benzylidene)-3-phenyl-2-thioxothiazolidin-4-one Derivatives, and Their Reactive Oxygen Species Scavenging Activities. Antioxidants 2022, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Lee, J.; Jung, H.J.; Ullah, S.; Jeong, Y.; Hong, S.; Kang, M.K.; Park, Y.J.; Hwang, Y.; Kang, D.; et al. Design and Synthesis of (Z)-5-(Substituted benzylidene)-3-cyclohexyl-2-thioxothiazolidin-4-one Analogues as Anti-Tyrosinase and Antioxidant Compounds: In Vitro and In Silico Insights. Antioxidants 2022, 11, 1918. [Google Scholar] [CrossRef]
- Kang, M.K.; Yoon, D.; Jung, H.J.; Ullah, S.; Lee, J.; Park, H.S.; Kim, H.J.; Kang, D.; Park, Y.; Chun, P.; et al. Identification and molecular mechanism of novel 5-alkenyl-2-benzylaminothiazol-4(5H)-one analogs as anti-melanogenic and antioxidant agents. Bioorg. Chem. 2023, 140, 106763. [Google Scholar] [CrossRef]
- Lee, J.; Park, Y.J.; Jung, H.J.; Ullah, S.; Yoon, D.; Jeong, Y.; Kim, G.Y.; Kang, M.K.; Kang, D.; Park, Y.; et al. Design and Synthesis of (Z)-2-(Benzylamino)-5-benzylidenethiazol-4(5H)-one Derivatives as Tyrosinase Inhibitors and Their Anti-Melanogenic and Antioxidant Effects. Molecules 2023, 28, 848. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Jung, H.J.; Kim, H.J.; Park, H.S.; Lee, J.; Yoon, D.; Kang, M.K.; Kim, G.Y.; Ullah, S.; Kang, D.; et al. Thiazol-4(5H)-one analogs as potent tyrosinase inhibitors: Synthesis, tyrosinase inhibition, antimelanogenic effect, antioxidant activity, and in silico docking simulation. Bioorg. Med. Chem. 2024, 98, 117578. [Google Scholar]
- Hosseini Nasab, N.; Raza, H.; Eom, Y.S.; Hassan, M.; Kloczkowski, A.; Kim, S.J. Synthesis and discovery of potential tyrosinase inhibitor of new coumarin-based thiophenyl-pyrazolylthiazole nuclei: In vitro evaluation, cytotoxicity, kinetic, and computational studies. Chem. Biol. Drug. Des. 2023, 101, 1262–1272. [Google Scholar]
- Shehzadi, S.A.; Saeed, A.; Perveen, F.; Channar, P.A.; Arshad, I.; Abbas, Q.; Kalsoom, S.; Yousaf, S.; Simpson, J. Identification of two novel thiazolidin-2-imines as tyrosinase inhibitors: Synthesis, crystal structure, molecular docking and DFT studies. Heliyon 2022, 8, e10098. [Google Scholar]
- Kisa, D.; Koc, E.; Bas Topcu, K.S.; Imamoglu, R. Heterocyclic compounds with different moieties: Synthesis and evaluation of biological activities assisted with the computational study. J. Biomol. Struct. Dyn. 2023, 42, 12144–12153. [Google Scholar] [CrossRef]
- Raza, H.; Rehman Sadiq Butt, A.; Athar Abbasi, M.; Aziz-ur-Rehman, S.; Zahra Siddiqui, S.; Hassan, M.; Adnan Ali Shah, S.; Ja Kim, S. 2-Aminothiazole-Oxadiazole Bearing N-Arylated Butanamides: Convergent Synthesis, Tyrosinase Inhibition, Kinetics, Structure-Activity Relationship, and Binding Conformations. Chem. Biodivers. 2023, 20, e202201019. [Google Scholar] [CrossRef]
- Ghani, U.; Cevik, U.A.; Rudrapal, M.; Rakshit, G.; Kaplancikli, Z.A. Synthesis of thiadiazole derivatives as competitive inhibitors of α-glucosidase and tyrosinase. J. Mol. Struct. 2024, 1307, 138028. [Google Scholar] [CrossRef]
- Vanjare, B.D.; Choi, N.G.; Mahajan, P.G.; Raza, H.; Hassan, M.; Han, Y.; Yu, S.M.; Kim, S.J.; Seo, S.Y.; Lee, K.H. Novel 1,3,4-oxadiazole compounds inhibit the tyrosinase and melanin level: Synthesis, in-vitro, and in-silico studies. Bioorg. Med. Chem. 2021, 41, 116222. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpoor, H.; Iraji, A.; Edraki, N.; Pirhadi, S.; Attarroshan, M.; Khoshneviszadeh, M.; Khoshneviszadeh, M. A Series of Benzylidenes Linked to Hydrazine-1-carbothioamide as Tyrosinase Inhibitors: Synthesis, Biological Evaluation and Structure-Activity Relationship. Chem. Biodivers. 2020, 17, e2000285. [Google Scholar] [CrossRef] [PubMed]
- Haldys, K.; Goldeman, W.; Anger-Gora, N.; Rossowska, J.; Latajka, R. Monosubstituted Acetophenone Thiosemicarbazones as Potent Inhibitors of Tyrosinase: Synthesis, Inhibitory Studies, and Molecular Docking. Pharmaceuticals 2021, 14, 74. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, G.; Wang, J.J.; Zhao, Y. Anti-browning and antibacterial dual functions of novel hydroxypyranone-thiosemicarbazone derivatives as shrimp preservative agents: Synthesis, bio-evaluation, mechanism, and application. Food Chem. 2023, 419, 136106. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Ejaz, S.A.; Khalid, A.; Channar, P.A.; Aziz, M.; Abbas, Q.; Wani, T.A.; Alsaif, N.A.; Alanazi, M.M.; Al-Hossaini, A.M.; et al. Acetophenone-Based 3,4-Dihydropyrimidine-2(1H)-Thione as Potential Inhibitor of Tyrosinase and Ribonucleotide Reductase: Facile Synthesis, Crystal Structure, In-Vitro and In-Silico Investigations. Int. J. Mol. Sci. 2022, 23, 13164. [Google Scholar] [CrossRef]
- Yari Boroujeni, S.; Haghighijoo, Z.; Mohammadi-Khanaposhtani, M.; Mosadeghkhah, A.; Moazzam, A.; Yavari, A.; Hajimahmoodi, M.; Sabourian, R.; Hosseini, S.; Larijani, B.; et al. Design, Synthesis, in Vitro, and in Silico Evaluation of N-Phenylacetamide-Oxindole-Thiosemicarbazide Hybrids as New Potential Tyrosinase Inhibitors. Chem. Biodivers. 2022, 19, e202100666. [Google Scholar] [CrossRef]
- Batool, Z.; Ullah, S.; Khan, A.; Siddique, F.; Nadeem, S.; Alshammari, A.; Albekairi, N.A.; Talib, R.; Al-Harrasi, A.; Shafiq, Z. Design, synthesis, and in vitro and in silico study of 1-benzyl-indole hybrid thiosemicarbazones as competitive tyrosinase inhibitors. RSC. Adv. 2024, 14, 28524–28542. [Google Scholar] [CrossRef] [PubMed]
- Batool, Z.; Ullah, S.; Khan, A.; Mali, S.N.; Gurav, S.S.; Jawarkar, R.D.; Alshammari, A.; Albekairi, N.A.; Al-Harrasi, A.; Shafiq, Z. Design, synthesis, QSAR modelling and molecular dynamic simulations of N-tosyl-indole hybrid thiosemicarbazones as competitive tyrosinase inhibitors. Sci. Rep. 2024, 14, 25754. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, J.; He, M.; Wang, G. Inhibitory activity and mechanism of thiosemicarbazide derivatives on tyrosinase and their anti-browning activity in fresh apple juice. Int. J. Biol. Macromo. 2024, 280, 135631. [Google Scholar] [CrossRef]
- Masuri, S.; Era, B.; Pintus, F.; Floris, S.; Meloni, F.; Pettinau, F.; Podda, E.; Cabiddu, M.G.; Fais, A.; Pivetta, T. Design, Synthesis, Structural Insights, Tyrosinase Inhibition, and Sun Protection Factor of New Thiosemicarbazone Derivatives. Molecules 2024, 29, 5629. [Google Scholar] [CrossRef]
- Xu, Y.; Liang, X.; Hyun, C.G. Discovery of Indole-Thiourea Derivatives as Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Kinetic Studies, and In Silico Analysis. Int. J. Mol. Sci. 2024, 25, 9636. [Google Scholar] [CrossRef]
- Raza, H.; Abbasi, M.A.; Aziz ur, R.; Siddiqui, S.Z.; Hassan, M.; Shah, S.A.A.; Shahid, M.; Hong, H.; Seo, S.Y. Design, synthesis and computational studies of N-(substituted-phenyl)-4-(4-phenyl-1-piperazinyl)butanamides as potent anti-melanogenic and tyrosinase inhibitors. J. Mol. Struct. 2020, 1210, 127969. [Google Scholar] [CrossRef]
- Raza, H.; Abbasi, M.A.; Aziz ur, R.; Siddiqui, S.Z.; Hassan, M.; Abbas, Q.; Hong, H.; Shah, S.A.A.; Shahid, M.; Seo, S.Y. Synthesis, molecular docking, dynamic simulations, kinetic mechanism, cytotoxicity evaluation of N-(substituted-phenyl)-4-{(4-(E)-3-phenyl-2-propenyl -1-piperazinyl} butanamides as tyrosinase and melanin inhibitors: In vitro, in vivo and in silico approaches. Bioorg. Chem. 2020, 94, 103445. [Google Scholar]
- Abbasi, M.A.; Zia ur, R.; Aziz ur, R.; Siddiqui, S.Z.; Nazir, M.; Hassan, M.; Raza, H.; Shah, S.A.A.; Seo, S.Y. Synthesis of Bi-Heterocyclic Sulfonamides as Tyrosinase Inhibitors: Lineweaver-Burk Plot Evaluation and Computational Ascriptions. Acta. Chim. Slov. 2020, 67, 403–414. [Google Scholar] [CrossRef]
- Abbasi, M.A.; Raza, H.; Aziz-ur-Rehman, S.Z.; Siddiqui, S.Z.; Muhammad, S.; Khan, F.M.; Shah, S.A.A.; Al-Sehemi, A.G.; Kim, S.J. Synthesis and Computational Exploration of Morpholine Bearing Halogenated Sulfonamides as Potential Tyrosinase Inhibitors. Chem. Biodivers. 2023, 20, e202300257. [Google Scholar] [CrossRef]
- Mirabile, S.; Vittorio, S.; Paola Germano, M.; Adornato, I.; Ielo, L.; Rapisarda, A.; Gitto, R.; Pintus, F.; Fais, A.; De Luca, L. Evaluation of 4-(4-Fluorobenzyl)piperazin-1-yl -Based Compounds as Competitive Tyrosinase Inhibitors Endowed with Antimelanogenic Effects. Chemmedchem 2021, 16, 3083–3093. [Google Scholar] [CrossRef]
- Mirabile, S.; Ielo, L.; Lombardo, L.; Ricci, F.; Gitto, R.; Germano, M.P.; Pace, V.; De Luca, L. Leveraging the 3-Chloro-4-fluorophenyl Motif to Identify Inhibitors of Tyrosinase from Agaricus bisporus. Int. J. Mol. Sci. 2023, 24, 7944. [Google Scholar] [CrossRef]
- Zeb, A.; Abbasi, M.A.; Aziz-Ur-Rehman; Siddiqui, S.Z.; Hassan, M.; Javed, Q.; Rafiq, M.; Ali, A.; Shah, S.A.A.; Kloczkowski, A. Synthesis, Kinetics and Computational Explorations of 4-Phenylpiperazine Bearing N-(Aryl)-3-substituted-benzamides as Auspicious Tyrosinase Inhibitors. Chem. Biodivers 2024, 21, e202400133. [Google Scholar] [CrossRef]
- Jung, H.J.; Park, H.S.; Kim, H.J.; Park, H.S.; Kim, Y.E.; Jeong, D.E.; Noh, S.G.; Park, Y.; Chun, P.; Chung, H.Y.; et al. Exploring 2-mercapto-N-arylacetamide analogs as promising anti-melanogenic agents: In vitro and in vivo evaluation. Org. Biomo. Chem. 2024, 22, 7671–7689. [Google Scholar] [CrossRef]
- Cai, H.; Chen, W.; Jiang, J.; Wen, H.; Luo, X.; Li, J.; Lu, L.; Zhao, R.; Ni, X.; Sun, Y.; et al. Artificial Intelligence-Assisted Optimization of Antipigmentation Tyrosinase Inhibitors: De Novo Molecular Generation Based on a Low Activity Lead Compound. J. Med. Chem. 2024, 67, 7260–7275. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Oliva, P.; Prencipe, F.; Manfredini, S.; Ricci, F.; Corallo, D.; Aveic, S.; Mariotto, E.; Viola, G.; Bortolozzi, R.; et al. De Cinnamic acid derivatives linked to arylpiperazines as novel potent inhibitors of tyrosinase activity and melanin synthesis. Eur. J. Med. Chem. 2022, 231, 114147. [Google Scholar] [CrossRef]
- Jung, H.J.; Noh, S.G.; Ryu, I.Y.; Park, C.; Lee, J.Y.; Chun, P.; Moon, H.R.; Chung, H.Y. (E)-1-(Furan-2-yl)-(substituted phenyl)prop-2-en-1-one Derivatives as Tyrosinase Inhibitors and Melanogenesis Inhibition: An In Vitro and In Silico Study. Molecules 2020, 25, 5460. [Google Scholar] [CrossRef]
- Sicak, Y.; Kekecmuhammed, H.; Karakucuk-Iyidogan, A.; Taskin-Tok, T.; Oruc-Emre, E.E.; Ozturk, M. Chalcones bearing nitrogen-containing heterocyclics as multi-targeted inhibitors: Design, synthesis, biological evaluation and molecular docking studies. J. Mol. Recognit. 2023, 36, e3020. [Google Scholar] [CrossRef]
- Xie, D.; Han, K.; Jiang, Q.; Xie, S.; Zhou, J.; Zhang, Y.; Xu, J.; He, Y.; Zhao, P.; Yang, X. Design, synthesis, and inhibitory activity of hydroquinone ester derivatives against mushroom tyrosinase. Rsc. Adv. 2024, 14, 6085–6095. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, H.S.; Jung, H.J.; Park, Y.J.; Kang, M.K.; Kim, H.J.; Yoon, D.; Ullah, S.; Kang, D.; Park, Y.; et al. Anti-Browning Effect of 2-Mercaptobenzo [d] imidazole Analogs with Antioxidant Activity on Freshly-Cut Apple Slices and Their Highly Potent Tyrosinase Inhibitory Activity. Antioxidants 2023, 12, 1814. [Google Scholar] [CrossRef]
- Yoon, D.; Jung, H.J.; Lee, J.; Kim, H.J.; Park, H.S.; Park, Y.J.; Kang, M.K.; Kim, G.Y.; Kang, D.; Park, Y.; et al. In vitro and in vivo anti-pigmentation effects of 2-mercaptobenzimidazoles as nanomolar tyrosinase inhibitors on mammalian cells and zebrafish embryos: Preparation of pigment-free zebrafish embryos. Eur. J. Med. Chem. 2024, 266, 116136. [Google Scholar] [CrossRef]
- Jin Jung, H.; Jin Kim, H.; Soo Park, H.; Young Kim, G.; Jung Park, Y.; Lee, J.; Kyung Kang, M.; Yoon, D.; Kang, D.; Park, Y.; et al. Highly potent anti-melanogenic effect of 2-thiobenzothiazole derivatives through nanomolar tyrosinase activity inhibition. Bioorg. Chem. 2024, 150, 107586. [Google Scholar] [CrossRef]
- Jung, H.J.; Park, H.S.; Park, H.S.; Kim, H.J.; Yoon, D.; Park, Y.; Chun, P.; Chung, H.Y.; Moon, H.R. Exploration of Compounds with 2-Phenylbenzo[d]oxazole Scaffold as Potential Skin-Lightening Agents through Inhibition of Melanin Biosynthesis and Tyrosinase Activity. Molecules 2024, 29, 4162. [Google Scholar] [CrossRef]
- Park, Y.J.; Jung, H.J.; Kang, M.K.; Lee, J.; Yoon, D.; Park, H.S.; Jin Kim, H.; Kim, G.Y.; Kang, D.; Park, Y.; et al. Design, synthesis, and anti-melanogenic efficacy of 2-mercaptobenzoxazoles with nanomolar tyrosinase activity inhibition. Bioorg. Med. Chem. 2024, 110, 117832. [Google Scholar] [CrossRef]
- Lazinski, L.M.; Beaumet, M.; Roulier, B.; Gay, R.; Royal, G.; Maresca, M.; Haudecoeur, R. Design and synthesis of 4-amino-2’,4’-dihydroxyindanone derivatives as potent inhibitors of tyrosinase and melanin biosynthesis in human melanoma cells. Eur. J. Med. Chem. 2024, 266, 116165. [Google Scholar] [CrossRef]
- Hwang, Y.; Lee, J.; Jung, H.J.; Ullah, S.; Ko, J.; Jeong, Y.; Park, Y.J.; Kang, M.K.; Yun, H.; Kim, M.S.; et al. A Novel Class of Potent Anti-Tyrosinase Compounds with Antioxidant Activity, 2-(Substituted phenyl)-5-(trifluoromethyl)benzo [d] thiazoles: In Vitro and In Silico Insights. Antioxidants 2022, 11, 1375. [Google Scholar] [CrossRef]
- Iraji, A.; Sheikhi, N.; Attarroshan, M.; Ardani, G.R.S.; Kabiri, M.; Bafghi, A.N.; Kobarfard, F.; Rezaei, Z.; Khoshneviszadeh, M.; Foroumadi, A.; et al. Design, synthesis, spectroscopic characterization, in vitro tyrosinase inhibition, antioxidant evaluation, in silico and kinetic studies of substituted indole-carbohydrazides. Bioorg. Chem. 2022, 129, 106140. [Google Scholar] [CrossRef]
- Sepehri, N.; Iraji, A.; Yavari, A.; Asgari, M.S.; Zamani, S.; Hosseini, S.; Bahadorikhalili, S.; Pirhadi, S.; Larijani, B.; Khoshneviszadeh, M.; et al. The natural-based optimization of kojic acid conjugated to different thio-quinazolinones as potential anti-melanogenesis agents with tyrosinase inhibitory activity. Bioorg. Med. Chem. 2021, 36, 116044. [Google Scholar] [CrossRef]
- Najafi, Z.; Zandi Haramabadi, M.; Chehardoli, G.; Ebadi, A.; Iraji, A. Design, synthesis, and molecular dynamics simulation studies of some novel kojic acid fused 2-amino-3-cyano-4H-pyran derivatives as tyrosinase inhibitors. BMC Chem. 2024, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, J.; Li, N.; Chen, L.; He, Y.; Peng, Z.; Wang, G. Synthesis, anti-browning effect and mechanism research of kojic acid-coumarin derivatives as anti-tyrosinase inhibitors. Food Chem. X. 2024, 21, 101128. [Google Scholar] [CrossRef]
- Xue, S.; Li, Z.; Ze, X.; Wu, X.; He, C.; Shuai, W.; Marlow, M.; Chen, J.; Scurr, D.; Zhu, Z.; et al. Design, Synthesis, and Biological Evaluation of Novel Hybrids Containing Dihydrochalcone as Tyrosinase Inhibitors to Treat Skin Hyperpigmentation. J. Med. Chem. 2023, 66, 5099–5117. [Google Scholar] [CrossRef]
- Shah, T.A.; Alam, A.; Zainab; Khan, M.; Elhenawy, A.A.; Ayaz, M.; Ali, M.; Latif, A.; Shah, S.A.A.; Ahmad, M. Experimental and computational profiling of novel bis-Schiff base derivatives bearing α-naphthalene moiety as potential tyrosinase inhibitors. J. Mol. Struct. 2025, 1321, 139919. [Google Scholar] [CrossRef]
- Fazel, R.; Hassani, B.; Zare, F.; Jokar Darzi, H.; Khoshneviszadeh, M.; Poustforoosh, A.; Behrouz, M.; Sabet, R.; Sadeghpour, H. Design, synthesis, in silico ADME, DFT, molecular dynamics simulation, anti-tyrosinase, and antioxidant activity of some of the 3-hydroxypyridin-4-one hybrids in combination with acylhydrazone derivatives. J.Biomol.Struct.Dyn. 2024, 4, 9518–9528. [Google Scholar] [CrossRef]
- Hu, Y.G.; Gao, Z.P.; Zheng, Y.Y.; Hu, C.M.; Lin, J.; Wu, X.Z.; Zhang, X.; Zhou, Y.S.; Xiong, Z.; Zhu, D.Y. Synthesis and Biological Activity Evaluation of 2-Cyanopyrrole Derivatives as Potential Tyrosinase Inhibitors. Front. Chem. 2022, 10, 914944. [Google Scholar] [CrossRef]
- Ahmad, I.; Parveen, W.; Noor, S.; Udin, Z.; Ali, A.; Ali, I.; Ullah, R.; Ali, H. Design and synthesis of novel dihydropyridine- and benzylideneimine-based tyrosinase inhibitors. Front. Pharmacol. 2024, 15, 1332184. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.E.; Petzer, J.P.; Pretorius, J.; Cloete, S.J.; Crous, C.; Petzer, A. Synthesis and evaluation of 3-hydroxyquinolin-2(1H)-one derivatives as inhibitors of tyrosinase. Bioorg. Med. Chem. Lett. 2024, 109, 129823. [Google Scholar] [CrossRef]
- Jeong, G.H.; Yadav, M.; Lee, S.S.; Chung, B.Y.; Cho, J.H.; Lee, I.C.; Bai, H.W.; Kim, T.H. Novel Dihydrocoumarins Induced by Radiolysis as Potent Tyrosinase Inhibitors. Molecules 2024, 29, 341. [Google Scholar] [CrossRef]
- Lotfi Shahpar, E.; Mahdavi, A.; Mohamadnia, Z. Inhibitory Effects, Fluorescence Studies, and Molecular Docking Analysis of Some Novel Pyridine-Based Compounds on Mushroom Tyrosinase. Biochemistry 2024, 63, 2063–2074. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Jung, S.H. Downregulation of melanogenesis: Drug discovery and therapeutic options. Drug Discov. Today 2017, 22, 282–298. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Source | Chemical Structure | Inhibition Mechanism | TYR Inhibition (IC50) a | Ref. | |
|---|---|---|---|---|---|---|
| L-Tyrosine | L-DOPA | |||||
| Polyphenols | ||||||
| Sesamol (1) | Sesamum indicum |  | - | - | 0.6 μM | [72] |
| N-Acetyldopamine (2) | Protaetia brevitarsis seulensis | 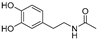 | - | - | 44.8 μM | [73] |
| (3) | Wedelia trilobata | 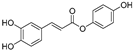 | - | - | 2.0 μM | [74] |
| 6′-O-Caffeoylarbutin (4) | Quezui Tea | 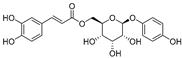 | Competitive | 1.1 μM | >50 μM | [75] |
| Tamariscinol U (5) | Selaginella tamariscina |  | - | - | 5.8 μM | [76] |
| (6) | Carica papaya | 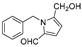 | - | 25.5 μM | - | [77] |
| Lariciresinol (7) | Carica papaya |  | - | 19.8 μM | - | [77] |
| (8) | Symphyocladia latiuscula |  | Competitive | 10.8 μM | >50 μM | [78] |
| (9) | Symphyocladia latiuscula | 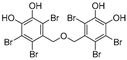 | Competitive | 2.9 μM | >50 μM | [78] |
| Puerol A (10) | Amorpha fruticosa | 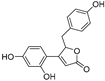 | Competitive | 2.2 μM | 3.9 μM | [68] |
| Phenolic acids | ||||||
| p-Coumaric acid (11) | Lepechinia meyenii | 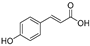 | Non-competitive | 0.3 μM | 0.6 μM | [79] |
| Caffeic acid (12) | Lepechinia meyenii | 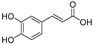 | Non-competitive | 1.5 μM | 2.3 μM | [79] |
| Rosmarinic acid (13) | Lepechinia meyenii | 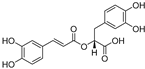 | Non-competitive | 4.1 μM | 8.6 μM | [79,80] |
| Caftaric acid (14) | Lepechinia meyenii |  | Competitive | - | 30.0 μM | [81] |
| Ascorbic acid (15) | Fuji apple | 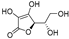 | - | - | 13.4 μM | [82] |
| (16) | Protea cynaroides |  | Competitive | 0.88 μg/mL | - | [83] |
| (17) | Protea cynaroides | 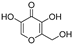 | Competitive | 0.72 μg/mL | - | [83] |
| Flavonoids—flavones | ||||||
| Luteolin (18) | Perilla seeds | 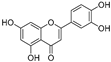 | - | - | 24.6 μM | [80] |
| Apigenin (19) | Perilla seeds | 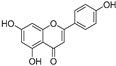 | - | - | 49.3 μM | [80] |
| Chrysoeriol (20) | Perilla seeds | 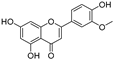 | - | - | 35.8 μM | [80] |
| (21) | Petals and foliage | 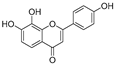 | Non-competitive | - | 10.3 μM | [84] |
| Panicolin (22) | Petals and foliage |  | Competitive | - | 2.75 μg/mL | [85] |
| Broussoflavonol H (23) | Broussonetia papyrifera | 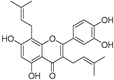 | - | 13.7 μM | - | [86] |
| Norartocarpetin (24) | Artocarpus rigida | 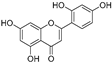 | - | 0.02 μM | - | [87] |
| Flavonoids—flavanones | ||||||
| Artocarpanone (25) | Artocarpus heterophyllous |  | - | -- | 2.0 μM | [88] |
| Liquiritigenin (26) | Artocarpus heterophyllous | 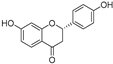 | - | - | 22.0 μM | [88] |
| Steppogenin (27) | Artocarpus heterophyllous | 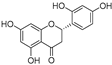 | - | - | 7.5 μM | [88] |
| Kushenol A (28) | Sophora flavescens | 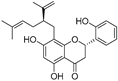 | Non-competitive | 1.1 μM | - | [89] |
| Kurarinone (29) | Sophora flavescens | 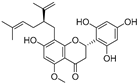 | Mixed | 7.1 μM | - | [90] |
| Sophoraflavanone G (30) | Sophora flavescens | 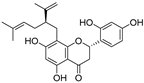 | Mixed | 66.7 μM | - | [90] |
| 6-Prenylanringenin (31) | Humulus lupulus | 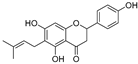 | Mixed | 38.1 μM | >50 μM | [91] |
| Flavonoids—flavonols | ||||||
| 8-Prenylkaempferol (32) | Sophora flavescens |  | Competitive | 2.4 μM | - | [89] |
| Kushenol (33) | Sophora flavescens | 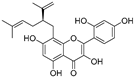 | Non-competitive | 24.1 μM | - | [89] |
| Lsoanhydroicaritin (34) | Sophora flavescens | 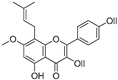 | Mixed | 0.7 μM | - | [90] |
| Quercetin (35) | Rose flowers | 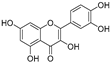 | Competitive | 4.2 μM | 10.7 μM | [92,93] |
| Kaempferol (36) | Rose flowers | 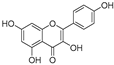 | Competitive | 5.5 μM | - | [92] |
| Galangin (37) | Alpinia officinarum | 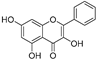 | Competitive | - | 3.6 μM | [71] |
| Broussoflavonol I (38) | Broussonetia papyrifera | 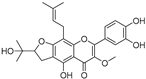 | - | 29.6 μM | - | [86] |
| Broussoflavonol K (39) | Broussonetia papyrifera | 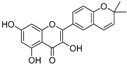 | - | 17.6 μM | - | [86] |
| Glycyrrhiza flavonol A (40) | Broussonetia papyrifera | 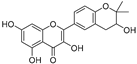 | - | 20.7 μM | - | [86] |
| Papyriflavonol A (41) | Broussonetia papyrifera | 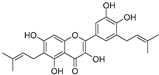 | - | 29.6 μM | - | [86] |
| Broussoflavonol F (42) | Broussonetia papyrifera |  | - | 29.7 μM | - | [86] |
| Broussoflavonol B (43) | Broussonetia papyrifera | 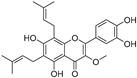 | - | 31.7 μM | - | [86] |
| Isolicofavonol (44) | Broussonetia papyrifera | 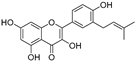 | - | 24.7 μM | - | [86] |
| Flavonoids—flavanonols | ||||||
| Trans-dihydromorin (45) | Morus alba | 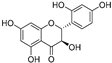 | - | - | 9.4 μM | [94] |
| Broussoflavonol J (46) | Broussonetia papyrifera | 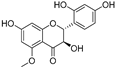 | - | - | 9.3 μM | [86] |
| Flavonoids—isoflavones | ||||||
| Formononetin (47) | Sophora flavescens |  | Non-competitive | 19.9 μM | - | [89] |
| (48) | Pichia pastoris | 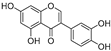 | Competitive | - | 15.9 μM | [95] |
| Daidzein (49) | Pueraria lobata |  | - | - | 17.5 μM | [96] |
| Lupinalbin A (50) | Apios americana | 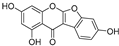 | Competitive | - | 10.3 μM | [97] |
| Calycosin (51) | Pueraria lobata | 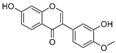 | Competitive | 1.5 μM | 7.0 μM | [98] |
| Semilicoisoflavone B (52) | Glycyrrhiza inflata | 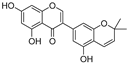 | - | - | 0.3 μM | [99] |
| Allolicoisoflavone B (53) | Glycyrrhiza inflata | 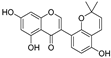 | - | - | 0.8 μM | [99] |
| Flavonoids—aurones | ||||||
| (54) | Morus notabilis | 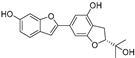 | Competitive | - | 14.8 μM | [100] |
| Moracin M (55) | Morus alba L. | 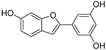 | - | 8.0 μM | - | [101] |
| Moracin B (56) | Morus alba L. | 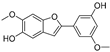 | -- | 34.4 μM | - | [101] |
| Moracin VN (57) | Artocarpus heterophyllus | 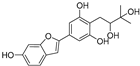 | Non-competitive | - | 0.8 μM | [102] |
| Flavonoids—chalcones | ||||||
| Isoliquiritigenin (58) | Pueraria lobata |  | - | - | 4.9 μM | [96] |
| Xanthohumol (59) | Humulus lupulus | 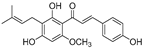 | Competitive | 15.4 μM | 31.1 μM | [91] |
| (60) | Humulus lupulus |  | Competitive | 34.3 μM | >50 μM | [91] |
| Xanthohumol C (61) | Humulus lupulus | 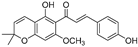 | Competitive | 20.6 μM | 41.3 μM | [91] |
| Xanthoumol B (62) | Humulus lupulus | 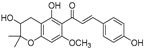 | Competitive | 22.1 μM | 46.7 μM | [91] |
| (63) | Morus alba L. | 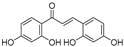 | - | 0.07 μM | - | [101] |
| Flavonoids—anthocyanidins | ||||||
| Cyanidin (64) | Diospyros kaki | 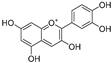 | Competitive | - | 9.1 μM | [103] |
| Luteolinidin (65) | Sorghum bicolor | 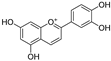 | Competitive | - | 3.7 μM | [104] |
| Stilbenes | ||||||
| Oxyresveratrol (66) | Morus alba |  | - | - | 1.7 μM | [94] |
| (67) | Morus alba | 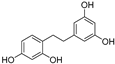 | - | - | 0.3 μM | [94] |
| (68) | Morus alba | 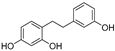 | - | - | 0.8 μM | [94] |
| Caricapapayol (69) | Carica papaya | 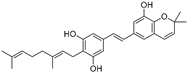 | - | 14.3 μM | - | [77] |
| Flavonolignans | ||||||
| Isosilybin A (70) | Silybum marianum | 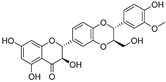 | Mixed | 2.1 μM | 16.7 μM | [105] |
| Isosilybin B (71) | Silybum marianum | 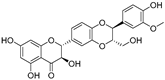 | Mixed | 4.9 μM | 19.8 μM | [105] |
| (72) | Silybum marianum | 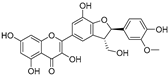 | Mixed | 7.6 μM | 35.9 μM | [105] |
| Silychristin A (73) | Silybum marianum |  | Mixed | 3.2 μM | 28.8 μM | [105] |
| Silychristin B (74) | Silybum marianum | 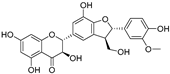 | Mixed | 4.5 μM | 44.9 μM | [105] |
| Other natural products—polyphenols | ||||||
| Neorauflavane (75) | Campylotropis hirtella |  | Competitive | 0.03 μM | 0.5 μM | [106] |
| trans-N-Coumaroyltyramine (76) | Humulus japonicus | 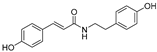 | - | - | 40.6 μM | [107] |
| Caffeine (77) | Camellia pollen | 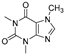 | Non-competitive | 18.6 μg/mL | - | [108] |
| Arichostatin A (78) | Streptomyces sp. | 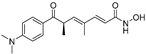 | Mixed | - | 2.3 μM | [109] |
| Deoxytrichostatin A (79) | Streptomyces sp. | 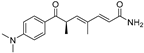 | - | - | 19.2 μM | [109] |
| 10-Methoxy-leonurine (80) | Leonurus japonicas | 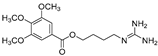 | Competitive | 7.4 μM | - | [110] |
| Leonurine (81) | Leonurus japonicas | 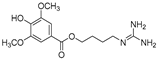 | Competitive | 12.4 μM | - | [110] |
| Emodin (82) | Leonurus japonicas | 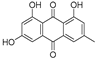 | - | - | 29.0 μM | [110] |
| Physcion (83) | Leonurus japonicas |  | - | - | 32.0μM | [111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, X.; Luo, X.; Jiang, X.; Chen, W.; Bai, R. Small-Molecule Tyrosinase Inhibitors for Treatment of Hyperpigmentation. Molecules 2025, 30, 788. https://doi.org/10.3390/molecules30040788
Ni X, Luo X, Jiang X, Chen W, Bai R. Small-Molecule Tyrosinase Inhibitors for Treatment of Hyperpigmentation. Molecules. 2025; 30(4):788. https://doi.org/10.3390/molecules30040788
Chicago/Turabian StyleNi, Xinhua, Xinyu Luo, Xiaoying Jiang, Wenchao Chen, and Renren Bai. 2025. "Small-Molecule Tyrosinase Inhibitors for Treatment of Hyperpigmentation" Molecules 30, no. 4: 788. https://doi.org/10.3390/molecules30040788
APA StyleNi, X., Luo, X., Jiang, X., Chen, W., & Bai, R. (2025). Small-Molecule Tyrosinase Inhibitors for Treatment of Hyperpigmentation. Molecules, 30(4), 788. https://doi.org/10.3390/molecules30040788