

An Appealing, Robust Access to Furo-Fused Heteropolycycles

 , ,
, ,  ,
,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
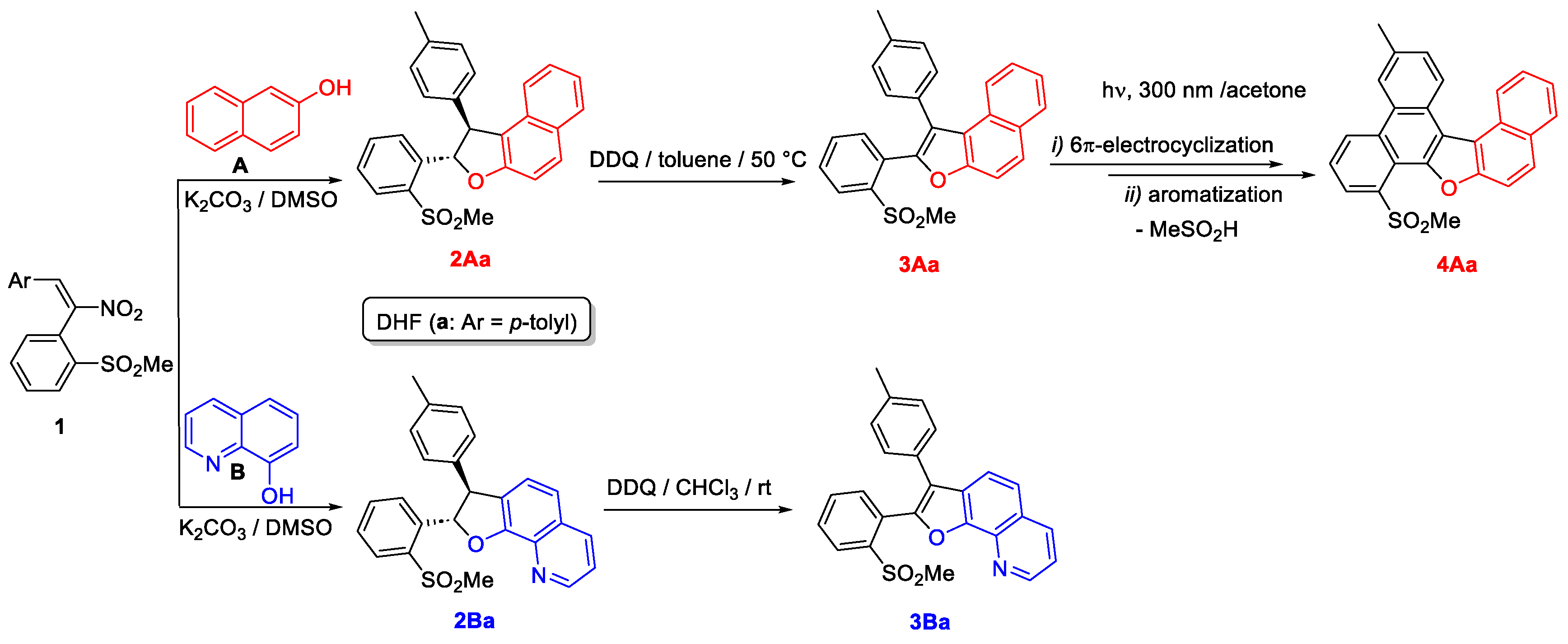
2.1. Reactions of Nitrostilbenes 1 with 8-Hydroxyquinoline (B)
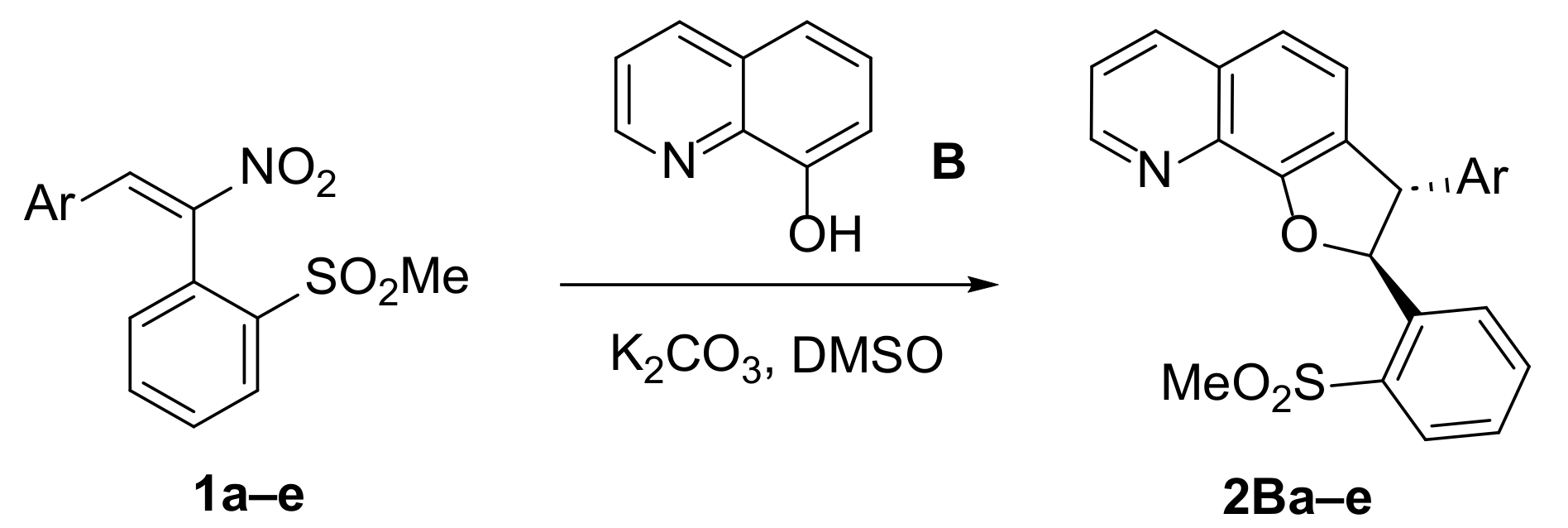
2.1.1. Synthesis of Dihydrofurans 2B
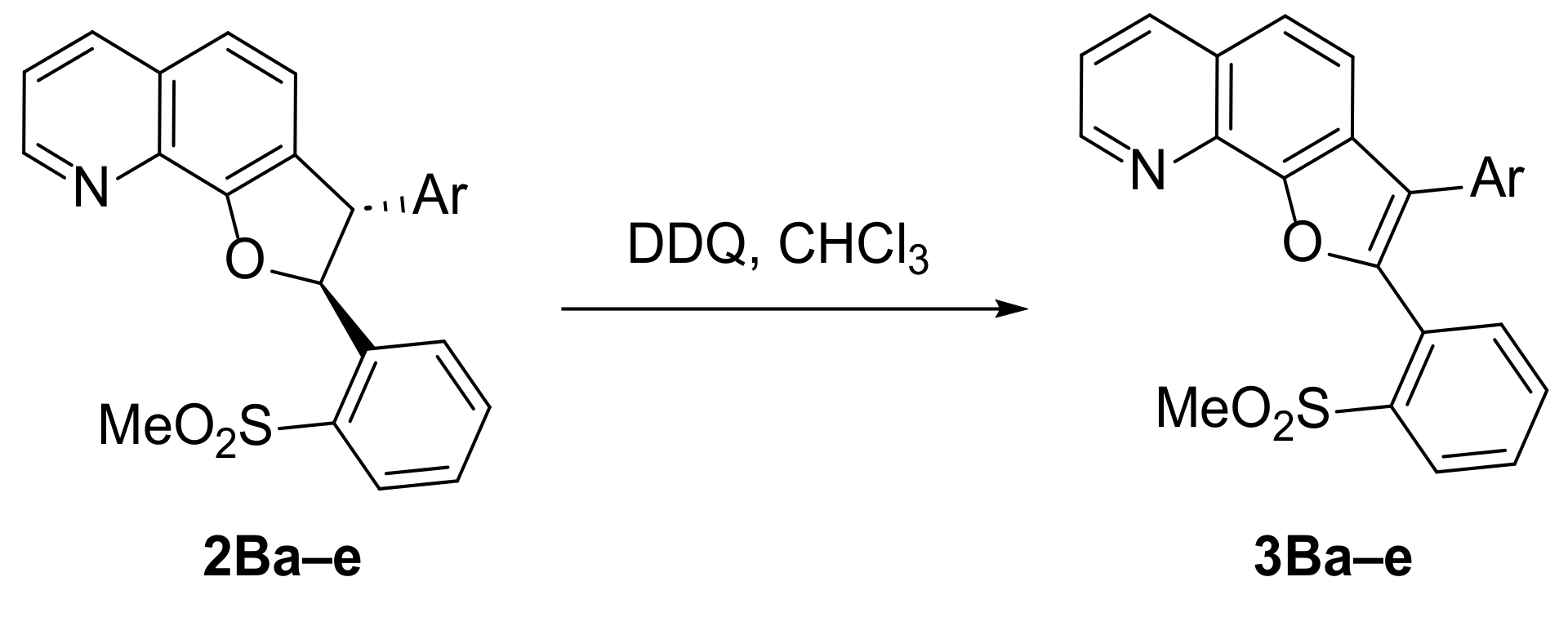
2.1.2. Aromatization of the Dihydrofuroquinolines 2B to 3B
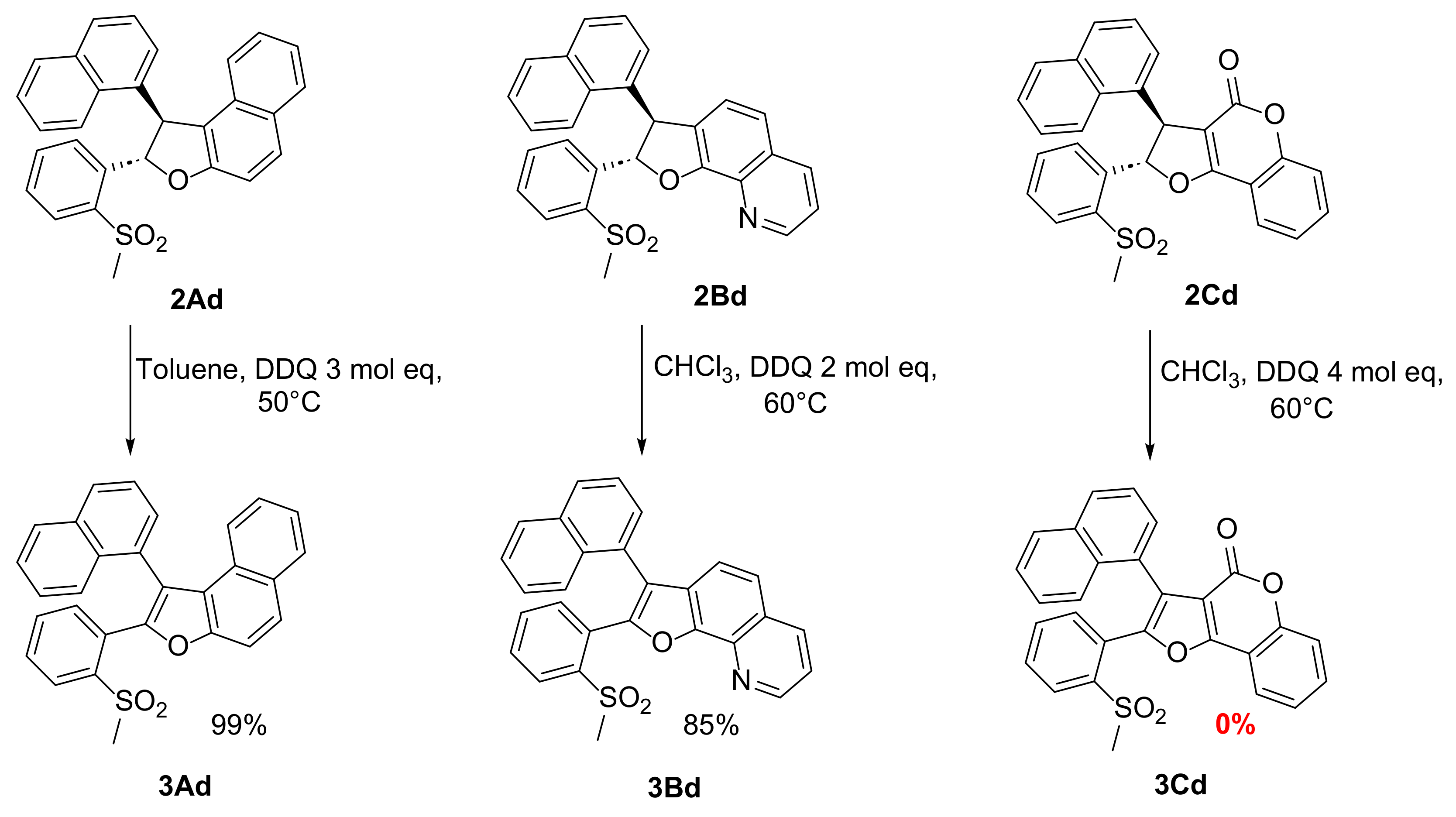
2.2. Reactions of Nitrostilbenes 1 with 4-Hydroxycoumarin (C)
2.2.1. Synthesis of Dihydrofurans 2C
2.2.2. Aromatization of the Dihydrofurocoumarins 2C to 3C
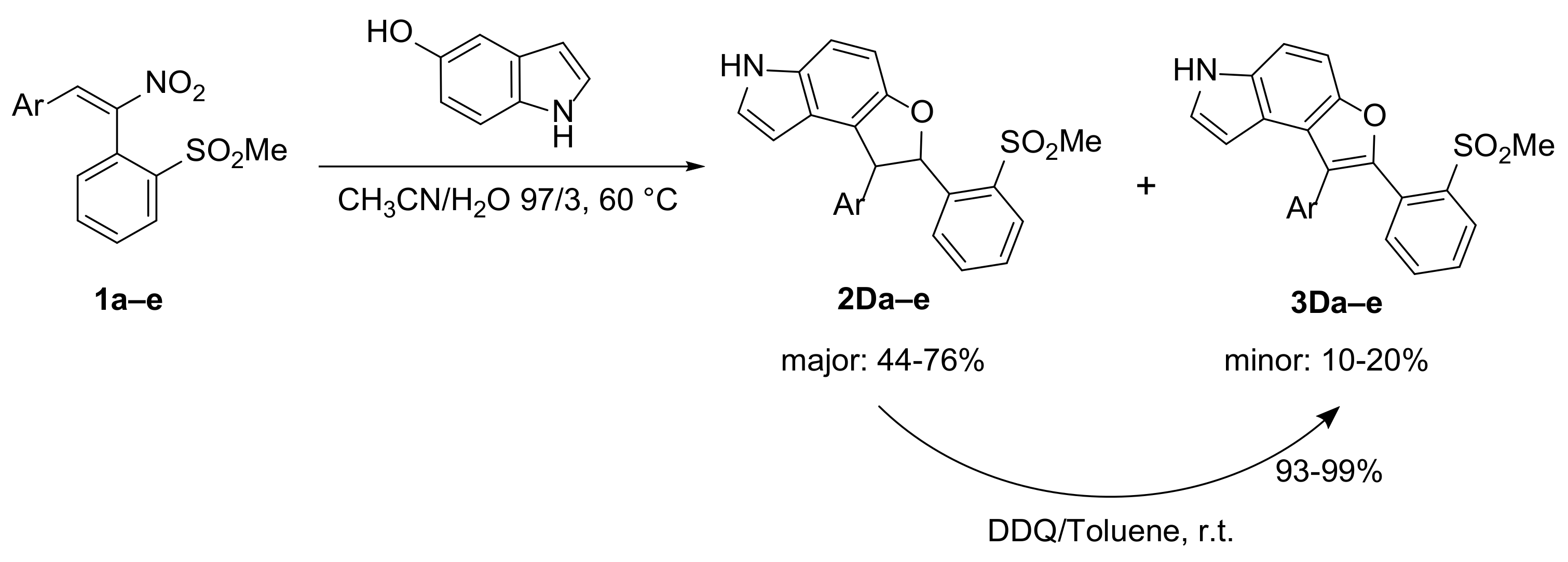
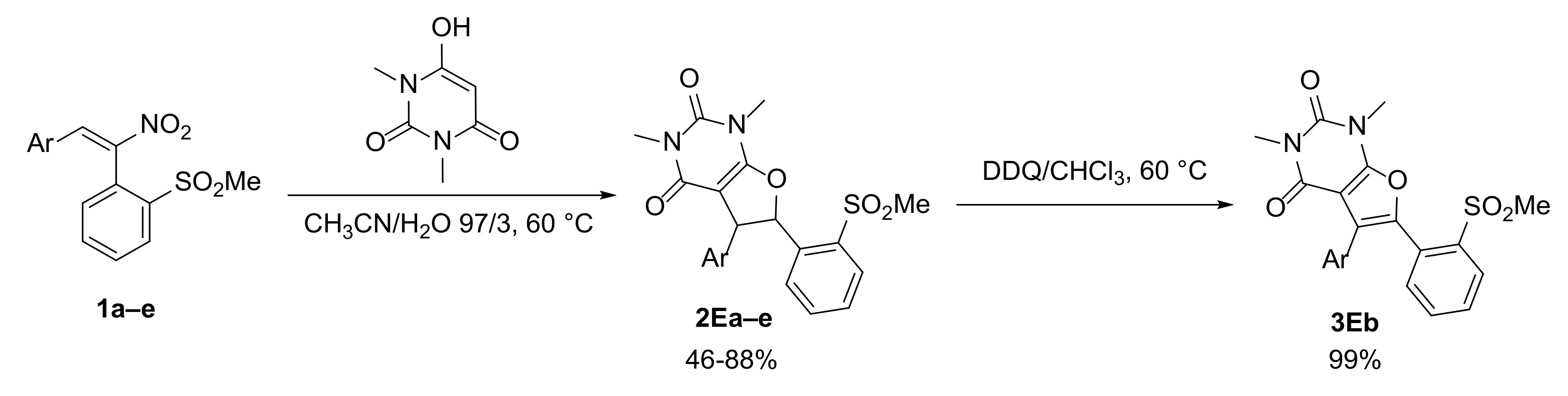
2.3. Extension of the Coupling Methodology with Nitrostilbenes 1 to Other Heterocyclic Phenols: 5-Hydroxyindole (D) and N,N′-Dimethylbarbituric Acid (E)
2.3.1. Synthesis of Dihydrofurans 2D and of the Relevant Aromatized Furans 3D
2.3.2. Synthesis of Dihydrofurans 2E and of the Relevant Aromatized Furans 3E
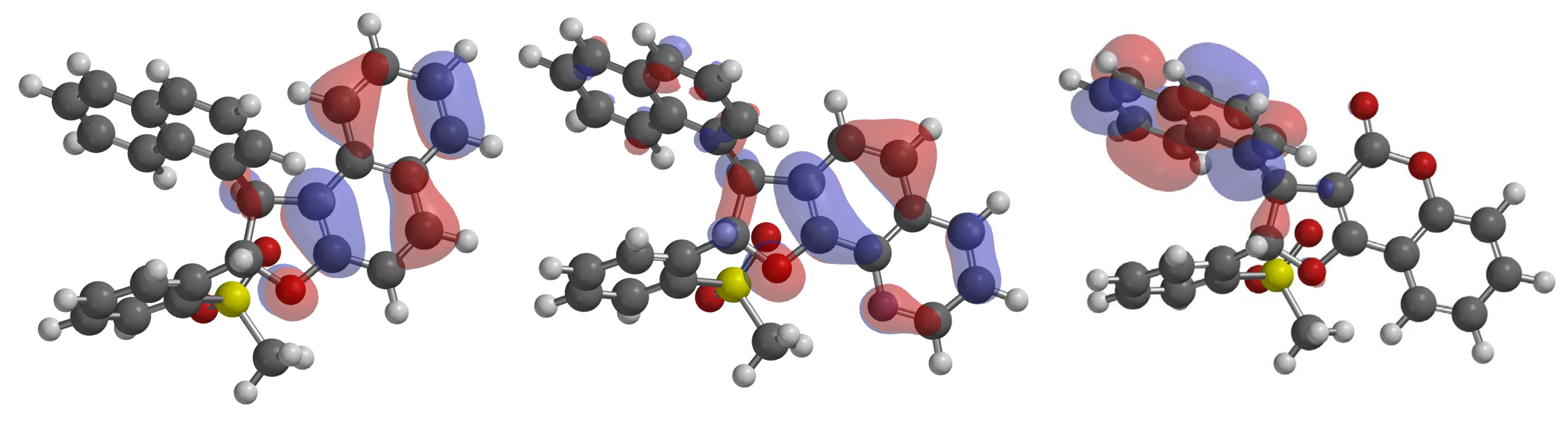
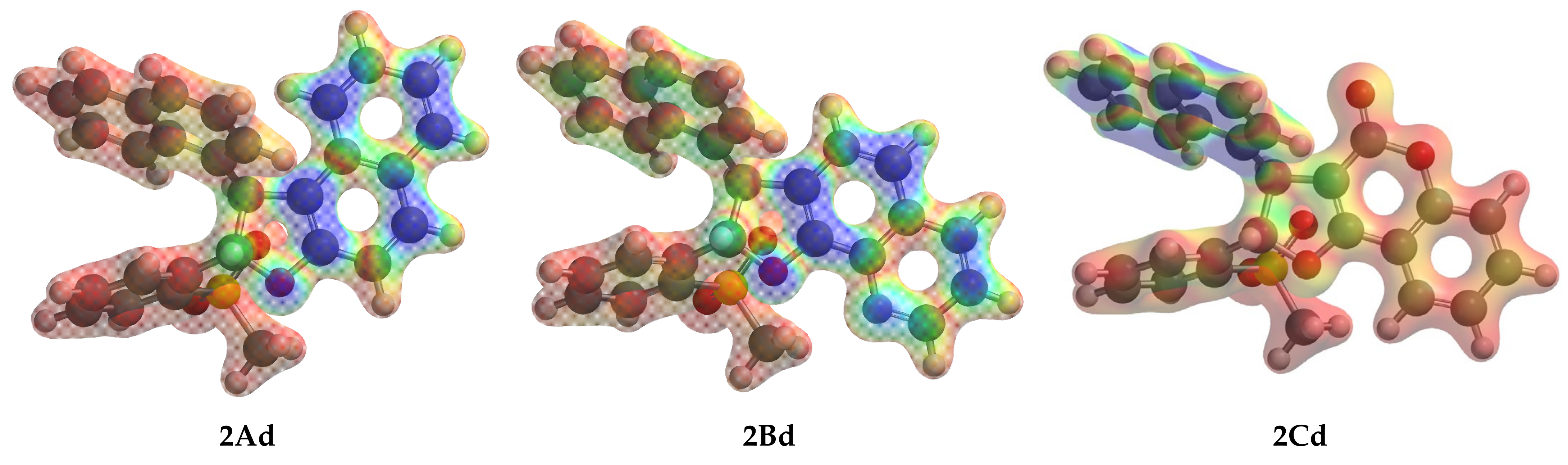
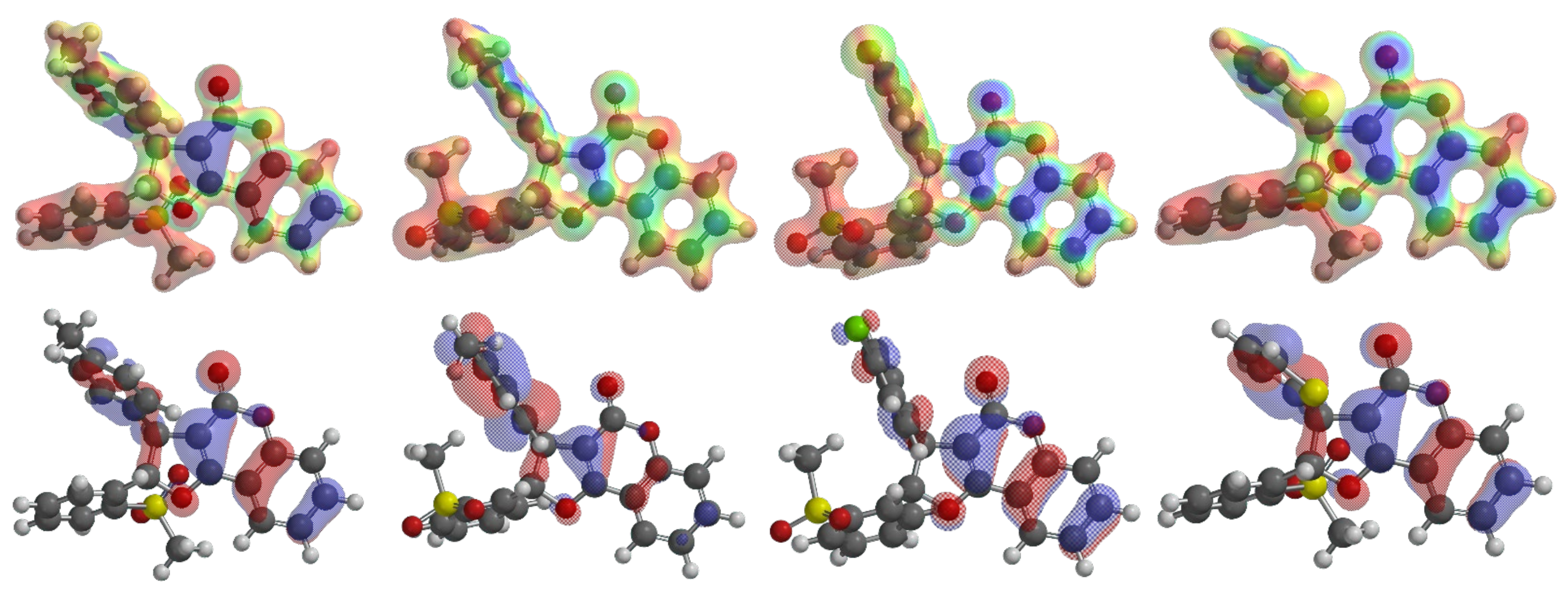
2.4. 6π-Electrocyclization, Followed by Aromatization, of Furoquinolines 3Ba–e and Furocoumarins 3Ca,b,e
2.5. Fluorescence Analysis
3. Experimental Section
3.1. Materials and Methods
3.2. Quantum Mechanical Calculations
- (1)
- Up to 200 conformers within a 40 Kcal/mol energy window above the global minimum conformer were initially selected for further geometry optimization in the gas phase using ab initio Hartree–Fock calculations at the 3-21G level.
- (2)
- Up to 100 conformers in a 20 Kcal/mol energy window were selected and further optimized using density-functional theory (DFT) implemented with ωB97X-D density functional and 6-31G* basis set.
- (3)
- Up to 50 conformers in a 10 Kcal/mol energy window were selected and further refined by DFT calculations with the ωB97X-V functional and the 6-311+G(2df,2p)[6-311G*] basis set.
- (4)
- Up to 25 conformers in a 5 Kcal/mol window were selected and their optimized structures were confirmed as real minima by IR frequency calculation (no imaginary frequencies).
3.3. General Procedure for the Reactions of Substrates 1a–e with 8-Hydroxyquinoline B
- 2-(2-(Methylsulfonyl)phenyl)-3-(1-naphthyl)-2,3-dihydrofuro[3,2-h]quinoline (2Bd). White solid. M.p. 151.0–152.7 °C (taken-up with E.P/DCM). 1H NMR (CDCl3, 400 MHz) δ 8.91 (dd, J = 4.5, 1.7 Hz, 1H), 8.19 (dd, J = 8.4, 1.7 Hz, 1H), 8.10 (dd, J = 7.7, 1.7 Hz, 1H), 7.86 (dd, J = 8.3, 1.3 Hz, 1H), 7.83–7.76 (m, 1H), 7.68 (s, 1H), 7.65–7.50 (m, 4H), 7.50–7.37 (m, 5H), 7.32–7.19 (m, 1H), 6.78 (br s, 1H), 6.03 (br s, 1H), 2.87 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 150.22, 140.38, 138.84, 138.04, 136.34, 136.03, 134.28, 133.85, 132.36, 131.93, 130.19, 129.55, 129.43, 129.02, 128.14, 126.35, 125.93, 125.66, 123.72, 122.74, 121.53, 121.38, 90.16, 53.53, 45.52 (three signals not visible because of isochrony). HRMS (ESI) m/z calculated [M + H]+ C28H22NO3S+ 452.1242, found 452.1244.
- 2-(2-(Methylsulfonyl)phenyl)-3-(2-thienyl)-2,3-dihydrofuro[3,2-h]quinoline (2Be). White solid. M.p. 174.0–175.6 °C (taken-up with E.P/DCM). 1H NMR (CDCl3, 400 MHz) δ 8.88 (dd, J = 4.2, 1.7 Hz, 1H), 8.20 (dd, J = 8.4, 1.7 Hz, 1H), 8.16 (dt, J = 7.6, 1.1 Hz, 1H), 7.62–7.58 (m, 2H), 7.54 (ddd, J = 7.6, 5.4, 3.5 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.44 (dd, J = 8.4, 4.2 Hz, 1H), 7.35 (dd, J = 8.2, 0.8 Hz, 1H), 7.25 (dd, J = 5.1, 1.3 Hz, 1H), 6.98 (dd, J = 5.1, 3.5 Hz, 1H), 6.95 (dd, J = 3.7, 1.3 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 5.38 (d, J = 8.1 Hz, 1H), 3.14 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 154.33, 150.25, 144.22, 139.54, 138.94, 136.35, 135.91, 134.16, 130.32, 129.73, 129.51, 129.47, 127.51, 127.24, 126.27, 125.30, 123.50, 121.65, 121.42, 90.23, 53.94, 45.78. HRMS (ESI) m/z calculated [M + H]+ C22H18NO3S2+ 408.0650, found 408.0658.
3.4. General Procedure for the Reactions of Substrates 1a–e with 4-Hydroxycoumarin C
- 2-(2-(Methylsulfonyl)phenyl)-3-(p-tolyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (2Ca). White solid. M.p. 219.9–220.8 °C. 1H NMR (CDCl3, 400 MHz) δ 8.12 (dd, J = 8.0, 1.4 Hz, 1H), 7.75 (dd, J = 7.8, 1.7 Hz, 1H), 7.71 (td, J = 7.5, 1.4 Hz, 1H), 7.67–7.57 (m, 3H), 7.42 (dd, J = 8.5, 1.0 Hz, 1H), 7.33 (td, J = 7.6, 1.1 Hz, 1H), 7.20–7.10 (m, 4H), 6.98 (d, J = 6.4 Hz, 1H), 4.65 (d, J = 6.4 Hz, 1H), 2.73 (s, 3H), 2.30 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.11, 159.53, 155.57, 138.94, 138.48, 137.93, 136.44, 134.83, 133.09, 130.04, 129.97, 129.90, 128.52, 127.63, 124.29, 123.23, 117.26, 112.28, 105.67, 90.12, 55.37, 45.57, 21.28. HRMS (ESI) m/z calculated [M + H]+ C25H21O5S+ 433.1031, found 433.1027.
- 3-(4-Methoxyphenyl)-2-(2-(methylsulfonyl)phenyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (2Cb). White solid. M.p. 214.9–216.1 °C. 1H NMR (CDCl3, 400 MHz) δ 8.12 (dd, J = 7.9, 1.4 Hz, 1H), 7.75 (dd, J = 7.8, 1.6 Hz, 1H), 7.71 (td, J = 7.6, 1.4 Hz, 1H), 7.65–7.56 (m, 3H), 7.42 (dd, J = 8.5, 1.1 Hz, 1H), 7.33 (td, J = 7.6, 1.1 Hz, 1H), 7.25–7.16 (m, 2H), 6.97 (d, J = 6.4 Hz, 1H), 6.90–6.82 (m, 2H), 4.65 (d, J = 6.4 Hz, 1H), 3.77 (s, 3H), 2.76 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.01, 159.52, 159.43, 155.58, 138.96, 138.52, 134.80, 133.06, 131.47, 130.02, 129.90, 128.86, 128.49, 124.27, 123.21, 117.26, 114.64, 112.29, 105.70, 90.10, 55.35, 55.01, 45.60. HRMS (ESI) m/z calculated [M + H]+ C25H21O6S+ 449.0981, found 449.0990.
- 3-(4-Chlorophenyl)-2-(2-(methylsulfonyl)phenyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (2Cc). White solid. M.p. 253.0–253.7 °C. 1H NMR (CDCl3, 400 MHz) δ 8.13 (dd, J = 7.9, 1.4 Hz, 1H), 7.75 (dd, J = 7.8, 1.6 Hz, 1H), 7.70 (td, J = 7.6, 1.5 Hz, 1H), 767–7.60 (m, 2H), 7.60–7.55 (m, 1H), 7.44 (dd, J = 8.5, 1.0 Hz, 1H), 7.35 (dd, J = 7.6, 1.1 Hz, 1H), 7.34–7.28 (m, 2H), 7.26–7.19 (m, 2H), 6.94 (d, J = 5.9 Hz, 1H), 4.65 (d, J = 5.8 Hz, 1H), 2.88 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.23, 159.37, 155.62, 138.57, 138.33, 138.14, 134.85, 134.01, 133.31, 130.20, 130.07, 129.36 129.10, 128.16, 124.40, 123.18, 117.37, 112.10, 105.43, 89.92, 54.91, 45.65. HRMS (ESI) m/z calculated [M + H]+ C24H18ClO5S+ 453.0485, found 453.0492.
- 2-(2-(Methylsulfonyl)phenyl)-3-(1-naphthyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (2Cd). White solid. M.p. 216.0–217.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.11 (dd, J = 8.1, 1.4 Hz, 1H), 7.91 (d, J = 7.9 Hz, 1H), 7.87–7.78 (m, 3H), 7.76 (dd, J = 7.8, 1.6 Hz, 1H), 7.70–7.61 (m, 2H), 7.54–7.43 (m, 4H), 7.40 (ddd, J = 8.1, 6.8, 1.1 Hz, 1H), 7.34 (td, J = 7.6, 1.1 Hz, 1H), 7.26–7.23 (m, 1H), 6.89 (br s, 1H), 5.69 (br s, 1H), 2.29 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.98, 159.53, 155.70, 139.17, 138.87, 135.08, 134.20, 133.24, 131.46, 130.41, 130.11, 129.30, 128.90, 128.80, 126.54, 126.08, 126.00, 124.33, 123.30, 122.65, 117.29, 112.35, 104.60, 100.02, 90.28, 45.02 (two isochronous carbon). HRMS (ESI) m/z calculated [M + H]+ C28H21O5S+ 469.1031, found 469.1023.
3.5. Alternative Procedure for the Reaction of Substrates 1c–e with 4-Hydroxycoumarin C
- 2-(2-(Methylsulfonyl)phenyl)-3-(thien-2-yl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (2Ce). Pale yellow solid. M.p. 213.0–214.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.15 (dd, J = 8.0, 1.4 Hz, 1H), 7.74 (dd, J = 7.8, 1.6 Hz, 1H), 7.70 (dd, J = 7.6, 1.5 Hz, 1H), 7.67–7.57 (m, 3H), 7.44 (d, J = 8.4 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.27–7.23 (m, 1H), 7.05–7.00 (m, 2H), 6.96 (dd, J = 5.1, 3.5 Hz, 1H), 5.04 (d, J = 6.4 Hz, 1H), 2.87 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.16, 159.34, 155.62, 142.45, 138.74, 138.21, 134.85, 133.36, 130.28, 130.09, 128.48, 127.47, 126.31, 125.63, 124.37, 123.33, 117.33, 112.15, 105.00, 90.07, 50.58, 45.63. HRMS (ESI) m/z calculated [M + H]+ C22H17O5S2+ 425.0439, found 425.0442.
3.6. Extending the Coupling Methodology to Other Heterocyclic Phenols: 5-Hydroxyindole D and N,N′-Dimethylbarbituric Acid E
- 2-(2-(Methylsulfonyl)phenyl)-1-(p-tolyl)-1,6-dihydro-2H-furo[3,2-e]indole (2Da). 1H NMR (CDCl3, 400 MHz) δ 8.11 (s, 1H), 8.08 (dd, J = 8.0, 1.4 Hz, 1H), 7.79 (dd, J = 7.9, 1.3 Hz, 1H), 7.62 (td, J = 7.6, 1.4 Hz, 1H), 7.50 (ddd, J = 8.0, 7.4, 1.4 Hz, 1H), 7.28 (dt, J = 8.7, 0.9 Hz, 1H), 7.14 (d, J = 8.1 Hz, 2H), 7.10–7.06 (m, 3H), 6.87 (d, J = 8.6 Hz, 1H), 6.60 (d, J = 7.2 Hz, 1H,), 5.93 (ddd, J = 3.1, 2.0, 0.9 Hz, 1H), 4.94 (d, J = 7.2 Hz, 1H), 2.76 (s, 3H,), 2.29 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 153.51, 141.49, 138.62, 138.47, 136.94, 134.29, 132.61, 129.53, 129.44, 129.15, 128.92, 128.19, 125.78, 124.70, 118.42, 111.25, 105.42, 99.86, 87.23, 58.17, 45.37, 21.23. HRMS (ESI) m/z calculated [M + H]+ C24H22NO3S+ 404.1242, found 404.1244.
- 8-(4-Methoxyphenyl)-7-(2-(methylsulfonyl)phenyl)-7,8-dihydro-1H-furo[2,3-g]indole (2Db). White solid. M.p. 119.0–120.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.14 (s, 1H), 8.08 (dd, J = 8.0, 1.4 Hz, 1H), 7.79 (dd, J = 7.9, 1.3 Hz, 1H), 7.62 (td, J = 7.6, 1.5 Hz, 1H), 7.55–7.45 (m, 1H), 7.28 (dt, J = 8.6, 0.9 Hz, 1H), 7.19–7.13 (m, 2H), 7.09 (t, J = 2.9 Hz, 1H), 6.87 (d, J = 8.7 Hz, 1H), 6.84–6.78 (m, 2H), 6.58 (d, J = 7.4 Hz, 1H), 5.92 (ddd, J = 3.1, 2.0, 0.9 Hz, 1H), 4.94 (d, J = 7.4 Hz, 1H), 3.75 (s, 3H), 2.78 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 158.86, 153.46, 141.45, 138.67, 134.28, 133.54, 132.62, 129.46, 129.39, 129.18, 128.92, 125.76, 124.67, 118.49, 114.18, 111.24, 105.44, 99.88, 87.28, 57.78, 55.27, 45.41. HRMS (ESI) m/z calculated [M + H]+ C24H22NO4S+ 420.1191, found 420.1188.
- 8-(4-Chlorophenyl)-7-(2-(methylsulfonyl)phenyl)-7,8-dihydro-1H-furo[2,3-g]indole (2Dc). Always in mixture with 3Dc: 1H-NMR peaks evaluated by subtraction. 1H NMR (CDCl3, 400 MHz) δ 8.23 (s, 1H), 8.08 (dd, J = 7.9, 1.5 Hz, 1H), 7.72 (dd, J = 7.9, 1.4 Hz, 1H), 7.64–7.56 (m, 1H), 7.56–7.45 (m, 1H), 7.29 (dt, J = 8.7, 1.0 Hz, 1H), 7.26–7.22 (m, 2H), 7.20–7.16 (m, 2H), 7.10 (t, J = 2.9 Hz, 1H), 6.88 (d, J = 8.6 Hz, 1H), 6.52 (d, J = 6.4 Hz, 1H), 5.92 (ddd, J = 3.1, 2.0, 1.0 Hz, 1H), 4.91 (d, J = 6.4 Hz, 1H), 2.93 (s, 3H).
- 7-(2-(Methylsulfonyl)phenyl)-8-(naphthalen-1-yl)-7,8-dihydro-1H-furo[2,3-g]indole (2Dd). White solid. M.p. 155.0–156.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.17 (s, 1H), 8.04 (dd, J = 8.0, 1.4 Hz, 1H), 7.96 (d, J = 7.5 Hz, 1H), 7.84 (d, J = 8.2 Hz, 1H), 7.80–7.73 (m, 1H), 7.69 (td, J = 7.7, 1.4 Hz, 1H), 7.63–7.49 (m, 2H), 7.44–7.36 (m, 3H), 7.32 (d, J = 8.6 Hz, 1H), 7.29–7.21 (m, 1H), 7.07 (s, 1H), 6.92 (d, J = 8.7 Hz, 1H), 6.61 (d, J = 6.9 Hz, 1H), 5.96 (s, 1H), 5.79 (t, J = 2.6 Hz, 1H), 2.38 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 164.31, 153.88, 141.39, 139.00, 137.44, 134.47, 133.88, 133.83, 132.60, 131.91, 129.61, 129.18, 129.06, 127.80, 126.16, 125.76, 125.72, 124.68, 122.90, 117.88, 111.42, 105.79, 100.45, 87.52, 52.91, 44.94 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C27H22NO3S+ 440.1242, found 440.1235.
- 7-(2-(Methylsulfonyl)phenyl)-8-(thiophen-2-yl)-7,8-dihydro-1H-furo[2,3-g]indole (2De). White solid. M.p. 138.0–139.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.20 (s, 1H), 8.11 (dd, J = 7.9, 1.4 Hz, 1H), 7.74 (dd, J = 7.8, 1.4 Hz, 1H), 7.61 (td, J = 7.6, 1.5 Hz, 1H), 7.52 (td, J = 7.7, 1.4 Hz, 1H), 7.30 (d, J = 8.9 Hz, 1H), 7.19 (dd, J = 5.0, 1.4 Hz, 1H), 7.13 (t, J = 2.9 Hz, 1H), 6.97–6.90 (m, 2H), 6.85 (d, J = 8.7 Hz, 1H), 6.63 (d, J = 7.4 Hz, 1H), 6.11–6.03 (m, 1H), 5.32 (d, J = 7.4 Hz, 1H), 2.89 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 153.23, 144.70, 140.67, 138.91, 134.30, 132.71, 129.67, 129.16, 127.03, 126.00, 125.80, 124.91, 124.65, 117.70, 111.78, 105.46, 99.80, 87.49, 53.18, 45.42 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C21H18NO3S2+ 396.0650, found 396.0659.
- 1,3-Dimethyl-6-(2-(methylsulfonyl)phenyl)-5-(p-tolyl)-5,6-dihydrofuro[2,3-d]pyrimidine-2,4(1H,3H)-dione (2Ea). White solid. M.p. 284.0–285.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.03 (dd, J = 7.8, 1.2 Hz, 1H), 7.72–7.61 (m, 2H), 7.55 (ddd, J = 8.6, 6.4, 2.2 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 7.9 Hz, 2H), 4.22–4.11 (m, 2H), 3.23 (s, 3H), 3.18 (s, 3H), 3.07 (s, 3H), 2.35 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.39, 165.69, 152.06, 139.56, 138.27, 133.97, 133.87, 132.12, 129.89, 129.59, 129.30, 129.11, 46.40, 43.78, 40.81, 28.93, 28.76, 21.37 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C22H23N2O5S+ 427.1249, found 427.1238.
- 5-(4-Methoxyphenyl)-1,3-dimethyl-6-(2-(methylsulfonyl)phenyl)-5,6-dihydrofuro[2,3-d]pyrimidine-2,4(1H,3H)-dione (2Eb). White solid. M.p. 108.0–109.0 °C. Mix stereoisomers. 1H NMR (CDCl3, 400 MHz) δ 8.08 (dd, J = 8.0, 1.4 Hz, 1H), 8.03 (dd, J = 8.1, 1.5 Hz, 1H), 7.70–7.61 (m, 3H), 7.58–7.51 (m, 1H), 7.51–7.43 (m, 2H), 7.33–7.26 (m, 4H), 6.95–6.85 (m, 4H), 5.74 (s, 2H), 4.19 (d, J = 9.4 Hz, 1H), 4.14 (d, J = 9.4 Hz, 1H), 3.81 (s, 3H), 3.80 (s, 3H), 3.47 (s, 3H), 3.32 (s, 3H), 3.25 (s, 3H), 3.22 (s, 3H), 3.18 (s, 3H), 3.07 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.37, 165.65, 162.27, 160.53, 159.98, 159.62, 152.06, 151.74, 142.42, 139.54, 139.05, 134.68, 133.99, 133.86, 132.10, 130.62, 129.65, 129.58, 129.46, 129.37, 129.10, 128.27, 127.69, 124.69, 114.34, 113.93, 97.30, 91.56, 55.38, 55.36, 47.04, 46.49, 45.49, 43.78, 43.31, 40.77, 29.98, 28.92, 28.75, 28.10. HRMS (ESI) m/z calculated [M + H]+ C22H23N2O5S+ 443.1199, found 443.1190.
- 5-(4-Chlorophenyl)-1,3-dimethyl-6-(2-(methylsulfonyl)phenyl)-5,6-dihydrofuro[2,3-d]pyrimidine-2,4(1H,3H)-dione (2Ec). White solid. M.p. 115.0–116.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.02 (d, J = 7.9 Hz, 1H), 7.68 (t, J = 7.5 Hz, 2H), 7.63 (d, J = 7.4 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 8.6 Hz, 2H), 4.15 (s, 2H), 3.23 (s, 3H), 3.18 (s, 3H), 3.07 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.06, 165.65, 151.89, 139.59, 134.33, 133.88, 133.45, 132.06, 131.54, 130.77, 129.62, 129.30, 128.78, 45.20, 43.85, 41.09, 28.95, 28.81 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C21H20ClN2O5S+ 447.0703, found 447.0709.
- 1,3-Dimethyl-6-(2-(methylsulfonyl)phenyl)-5-(naphthalen-1-yl)-5,6-dihydrofuro[2,3-d]pyrimidine-2,4(1H,3H)-dione (2Ed). White solid. M.p. 245.0–246.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.08 (dd, J = 7.8, 1.3 Hz, 1H), 7.93–7.86 (m, 2H), 7.77–7.68 (m, 2H), 7.66–7.57 (m, 3H), 7.54–7.46 (m, 3H), 4.48 (d, J = 9.3 Hz, 1H), 4.34 (d, J = 9.3 Hz, 1H), 3.29 (s, 3H), 3.10 (s, 3H), 3.02 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 166.53, 165.38, 152.07, 139.71, 133.99, 133.92, 133.60, 132.37, 132.17, 129.69, 129.61, 129.35, 129.32, 129.29, 127.34, 127.11, 126.18, 125.29, 122.34, 44.16, 43.86, 41.14, 28.97, 28.80 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C25H23N2O5S+ 463.1249, found 463.1240.
- 1,3-Dimethyl-6-(2-(methylsulfonyl)phenyl)-5-(thiophen-2-yl)-5,6-dihydrofuro[2,3-d]pyrimidine-2,4(1H,3H)-dione (2Ee). Always in mixture with a minor unknown compound after chromatography. 1H NMR (CDCl3, 400 MHz) δ 8.02 (dd, J = 7.9, 1.3 Hz, 1H), 7.72–7.62 (m, 2H), 7.55 (ddd, J = 8.7, 6.8, 1.9 Hz, 1H), 7.29 (dd, J = 5.2, 1.2 Hz, 1H), 7.13 (d, J = 3.5 Hz, 1H), 7.01 (dd, J = 5.1, 3.6 Hz, 1H), 4.20 (s, 2H), 3.26 (s, 3H), 3.17 (s, 3H), 3.07 (s, 3H).
3.7. General Procedure for the Aromatization of 2Ba–e with DDQ
- 3-(4-Methyphenyl)-2-(2-(methylsulfonyl)phenyl)furo[3,2-h]quinoline (3Ba). The compound was already characterized in [8].
- 3-(4-Methoxyphenyl)-2-(2-(methylsulfonyl)phenyl)furo[3,2-h]quinoline (3Bb). White solid. M.p. 146.5–147.7 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 8.92 (dd, J = 4.3, 1.7 Hz, 1H), 8.30–8.24 (m, 2H), 7.83 (d, J = 8.6 Hz, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.60 (td, J = 7.7, 1.5 Hz, 1H), 7.53 (td, J = 7.6, 1.5 Hz, 1H), 7.45 (dd, J = 8.3, 4.3 Hz, 1H), 7.43–7.37 (m, 3H), 6.96–6.88 (m, 2H), 3.81 (s, 3H), 3.62 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 159.23, 150.28, 149.35, 149.13, 141.19, 137.08, 136.42, 133.43, 133.34, 130.83, 130.66, 129.93, 128.04, 126.83, 123.66, 123.51, 120.75, 120.64, 120.03, 114.44, 55.35, 46.10 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C25H20NO4S+ 430.1235, found 430.1241.
- 3-(4-Chlorophenyl)-2-(2-(methylsulfonyl)phenyl)furo[3,2-h]quinoline (3Bc). The compound has been already characterized [8].
- 2-(2-(Methylsulfonyl)phenyl)-3-(naphth-1-yl)furo[3,2-h]quinoline (3Bd). White solid. M.p. 167.2–169.3 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 8.96 (dd, J = 4.4, 1.6 Hz, 1H), 8.27 (dd, J = 8.3, 1.7 Hz, 1H), 8.23 (dd, J = 8.0, 1.3 Hz, 1H), 7.97 (dd, J = 8.4, 1.1 Hz, 1H), 7.93–7.86 (m, 2H), 7.61 (d, J = 8.6 Hz, 1H), 7.57 (dd, J = 7.0, 1.3 Hz, 1H), 7.51–7.43 (m, 4H), 7.47–7.35 (m, 2H), 7.31 (td, J = 7.6, 1.3 Hz, 1H), 7.20 (dd, J = 7.7, 1.4 Hz, 1H), 3.77 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 151.00, 150.35, 149.27, 140.64, 137.16, 136.51, 133.89, 133.20, 132.62, 132.46, 130.72, 130.37, 129.82, 129.46, 129.37, 128.77, 128.72, 128.45, 126.99, 126.57, 126.19, 126.11, 125.82, 123.57, 120.83, 120.52, 119.69, 46.38. HRMS (ESI) m/z calculated [M + H]+ C28H20NO3S+ 450.1086, found 450.1095.
- 2-(2-(Methylsulfonyl)phenyl)-3-(thien-2-yl)furo[3,2-h]quinoline (3Be). White solid. M.p. 146.5–147.7 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 8.93 (dd, J = 4.3, 1.7 Hz, 1H),8.32–8.26 (m, 2H), 8.02 (d, J = 8.6 Hz, 1H), 7.74 (d, J = 8.6 Hz, 1H), 7.71–7.63 (m, 2H), 7.62 (td, J = 7.3, 1.8 Hz, 1H), 7.47 (dd, J = 8.3, 4.3 Hz, 1H), 7.32 (dd, J = 5.1, 1.2 Hz, 1H), 7.15 (dd, J = 3.6, 1.2 Hz, 1H), 7.05 (dd, J = 5.1, 3.6 Hz, 1H), 3.44 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ150.44, 149.39, 149.32, 141.39, 136.91, 136.49, 133.72, 133.43, 132.47, 130.63, 130.56, 130.20, 127.65, 127.30, 127.08, 126.94, 125.91, 123.81, 120.99, 120.07, 115.16, 45.83. HRMS (ESI) m/z calculated [M + H]+ C22H16NO3S2+ 406.0493, found 406.0493.
3.8. General Procedure for Aromatization of 2Ca,b,c,e and 2Eb with DDQ
- 2-(2-(Methylsulfonyl)phenyl)-3-(p-tolyl)-4H-furo[3,2-c]chromen-4-one (3Ca). White solid. M.p. 135.0–136.0 °C (XX EtOH). 1H NMR (CDCl3, 400 MHz) δ 8.22 (dd, J = 7.9, 1.5 Hz, 1H), 7.78 (dd, J = 7.8, 1.6 Hz, 1H), 7.65 (td, J = 7.7, 1.5 Hz, 1H), 7.61–7.50 (m, 2H), 7.47 (d, J = 8.3 Hz, 1H), 7.40–7.33 (m, 2H), 7.31 (d, J = 7.9 Hz, 2H), 7.10 (d, J = 7.8 Hz, 2H), 3.16 (s, 3H), 2.31 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 157.58, 157.40, 152.92, 148.68, 140.83, 138.24, 134.20, 133.70, 131.13, 130.76, 130.68, 130.28, 129.14, 125.92, 124.61, 123.72, 120.61, 117.52, 112.73, 110.30, 45.24, 21.40 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C25H19O5S+ 431.0875, found 431.0868.
- 3-(4-Methoxyphenyl)-2-(2-(methylsulfonyl)phenyl)-4H-furo[3,2-c]chromen-4-one (3Cb). White solid. M.p. 113.0–114.0 °C (XX EtOH). 1H NMR (CDCl3, 400 MHz) δ 8.23 (dd, J = 7.9, 1.4 Hz, 1H), 7.78 (dd, J = 7.8, 1.5 Hz, 1H), 7.65 (td, J = 7.7, 1.5 Hz, 1H), 7.59 (td, J = 7.5, 1.5 Hz, 1H), 7.54 (ddd, J = 8.7, 7.2, 1.6 Hz, 1H), 7.48 (dd, J = 8.4, 1.2 Hz, 1H), 7.40–7.30 (m, 4H), 6.86–6.80 (m, 2H), 3.78 (s, 3H), 3.19 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 159.61, 157.70, 157.39, 152.90, 148.45, 140.81, 134.20, 133.75, 131.72, 131.14, 130.75, 130.72, 129.20, 124.63, 123.41, 121.10, 120.60, 117.54, 113.90, 112.74, 110.26, 55.31, 45.27. HRMS (ESI) m/z calculated [M + H]+ C25H19O6S+ 447.0824, found 447.0818.
- 2-(2-(Methylsulfonyl)phenyl)-3-(thiophen-2-yl)-4H-furo[3,2-c]chromen-4-one (3Ce). White solid. M.p. 241.0–242.0 °C (XX EtOH). 1H NMR (CDCl3, 400 MHz) δ 8.29–8.23 (m, 1H), 7.83 (dd, J = 7.9, 1.6 Hz, 1H), 7.81–7.73 (m, 3H), 7.67–7.62 (m, 1H), 7.55 (ddd, J = 8.7, 7.3, 1.6 Hz, 1H), 7.48 (dd, J = 8.5, 1.2 Hz, 1H), 7.34 (ddd, J = 8.2, 7.3, 1.2 Hz, 1H), 7.20 (dd, J = 5.1, 1.2 Hz, 1H), 7.02 (dd, J = 5.1, 3.7 Hz, 1H), 2.91 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 157.87, 157.65, 152.94, 147.79, 141.33, 134.95, 133.88, 131.73, 131.44, 130.70, 130.64, 129.69, 128.80, 127.62, 126.68, 124.72, 121.08, 117.88, 117.34, 112.47, 109.29, 44.67. HRMS (ESI) m/z calculated [M + H]+ C22H15O5S+ 423.0283, found 423.0290.
- 5-(4-Methoxyphenyl)-1,3-dimethyl-6-(2-(methylsulfonyl)phenyl)furo[2,3-d]pyrimidine-2,4(1H,3H)-dione (3Eb). White solid. M.p. 223.0–224.0 °C (XX EtOH). 1H NMR (CDCl3, 400 MHz) δ 8.25–8.19 (m, 1H), 7.71–7.63 (m, 2H), 7.43–7.37 (m, 1H), 7.21–7.14 (m, 2H), 6.78–6.72 (m, 2H), 3.75 (s, 3H), 3.67 (s, 3H), 3.32 (s, 3H), 2.96 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 159.64, 158.29, 154.20, 150.53, 146.24, 140.02, 134.17, 133.08, 131.34, 130.39, 129.82, 126.80, 121.37, 114.36, 99.71, 55.37, 44.42, 29.70, 28.33 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C22H21N2O6S+ 441.1042, found 441.1038.
3.9. General Procedure for Aromatization of 2Da–e with DDQ
- 7-(2-(Methylsulfonyl)phenyl)-8-(p-tolyl)-1H-furo[2,3-g]indole (3Da). White solid. M.p. 152.0–153.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.40 (s, 1H), 8.23 (dd, J = 7.9, 1.5 Hz, 1H), 7.54 (td, J = 7.7, 1.5 Hz, 1H), 7.47 (td, J = 7.5, 1.5 Hz, 1H), 7.44–7.40 (m, 2H), 7.35 (s, 2H), 7.32 (dd, J = 7.6, 1.5 Hz, 1H), 7.19–7.13 (m, 3H), 6.54 (dd, J = 3.2, 1.9 Hz, 1H), 3.37 (s, 3H), 2.37 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 150.05, 147.35, 140.64, 137.12, 133.95, 133.21, 132.66, 131.22, 130.30, 130.17, 129.73, 129.38, 129.20, 123.76, 121.23, 120.37, 120.33, 109.01, 105.96, 101.88, 45.15, 21.41. HRMS (ESI) m/z calculated [M + H]+ C24H20NO3S+ 402.1086, found 402.1080.
- 8-(4-Methoxyphenyl)-7-(2-(methylsulfonyl)phenyl)-1H-furo[2,3-g]indole (3Db). White solid. M.p. 137.0–138.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.38 (s, 1H), 8.23 (dd, J = 7.9, 1.5 Hz, 1H), 7.54 (td, J = 7.7, 1.5 Hz, 1H), 7.51–7.43 (m, 3H), 7.34 (s, 2H), 7.32 (dd, J = 7.5, 1.5 Hz, 1H), 7.18–7.16 (m, 1H), 6.93–6.87 (m, 2H), 6.54 (dd, J = 3.2, 2.1 Hz, 1H), 3.82 (s, 3H), 3.38 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 159.04, 150.02, 147.25, 140.65, 133.94, 133.21, 132.65, 131.45, 131.24, 130.32, 129.36, 125.01, 123.81, 120.90, 120.41, 120.35, 113.96, 109.00, 105.98, 101.81, 55.31, 45.17. HRMS (ESI) m/z calculated [M + H]+ C24H20NO4S+ 418.1035, found 418.11040.
- 8-(4-Chlorophenyl)-7-(2-(methylsulfonyl)phenyl)-1H-furo[2,3-g]indole (3Dc). White solid. M.p. 162.0–163.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.43 (s, 1H), 8.24 (dd, J = 7.9, 1.4 Hz, 1H), 7.57 (td, J = 7.7, 1.4 Hz, 1H), 7.54–7.45 (m, 3H), 7.37–7.30 (m, 4H), 7.27 (dd, J = 7.6, 1.4 Hz, 1H), 7.17 (d, J = 3.0 Hz, 1H), 6.49 (t, J = 2.5 Hz, 1H), 3.41 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 150.09, 147.80, 140.75, 133.85, 133.42, 133.39, 132.71, 131.61, 131.42, 130.78, 130.46, 129.72, 128.78, 124.03, 120.12, 119.89, 109.26, 105.94, 101.60, 45.30 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C23H17ClNO3S+ 422.0539, found 422.0537.
- 7-(2-(Methylsulfonyl)phenyl)-8-(naphthalen-1-yl)-1H-furo[2,3-g]indole (3Dd). White solid. M.p. 276.0–277.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.30 (s, 1H), 8.18 (dd, J = 8.0, 1.3 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.93–7.84 (m, 2H), 7.58 (dd, J = 7.0, 1.3 Hz, 1H), 7.51–7.29 (m, 6H), 7.30–7.21 (m, 1H), 7.17 (dd, J = 7.7, 1.4 Hz, 1H), 6.95 (t, J = 2.9 Hz, 1H), 5.59 (t, J = 2.5 Hz, 1H), 3.53 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 149.94, 148.94, 140.02, 133.74, 133.15, 133.00, 132.70, 132.55, 130.89, 130.51, 130.47, 129.30, 129.24, 128.31, 128.22, 126.70, 126.41, 126.05, 125.75, 123.85, 121.85, 120.50, 119.39, 109.16, 105.87, 101.73, 45.52. HRMS (ESI) m/z calculated [M + H]+ C27H20NO3S+ 438.1086, found 438.1082.
- 7-(2-(Methylsulfonyl)phenyl)-8-(thiophen-2-yl)-1H-furo[2,3-g]indole (3De). White solid. M.p. 200.0–201.0 °C. 1H NMR (CDCl3, 400 MHz) δ 8.44 (1H, s), 8.25–8.20 (m, 1H), 7.64–7.54 (m, 2H), 7.52–7.47 (m, 1H), 7.37–7.33 (m, 2H), 7.30 (dd, J = 5.1, 1.2 Hz, 1H), 7.27–7.24 (m, 1H), 7.20 (t, J = 2.8 Hz, 1H), 7.06 (dd, J = 5.1, 3.5 Hz, 1H), 6.72 (dd, J = 3.2, 2.1 Hz, 1H), 3.24 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 150.00, 148.21, 140.87, 134.08, 133.41, 133.20, 132.79, 130.60, 130.16, 129.96, 128.31, 127.31, 126.04, 124.03, 120.09, 120.01, 114.81, 109.39, 106.02, 101.78, 44.91. HRMS (ESI) m/z calculated [M + H]+ C21H16NO3S2+ 394.0493, found 394.0497.
3.10. General Procedure for 6π-Electrocyclization of 3B and 3C
- 9-Methylphenanthro[9′,10′:4,5]furo[3,2-h]quinoline (5Ba). White solid. M.p. 209.2–210.8 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 9.11 (d, J = 3.4 Hz, 1H), 8.82 (dd, J = 8.0, 1.6 Hz, 1H), 8.75 (d, J = 7.4 Hz, 1H), 8.61–8.54 (m, 2H), 8.51 (d, J = 8.6 Hz, 1H), 8.33 (dt, J = 8.2, 1.2 Hz, 1H), 7.84 (d, J = 8.5 Hz, 1H), 7.78–7.67 (m, 2H), 7.60 (d, J = 9.0 Hz, 1H), 7.51 (dd, J = 8.4, 4.1 Hz, 1H), 2.67 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 151.50, 150.68, 150.42,137.06, 136.53, 135.11, 130.36, 130.28, 129.09, 128.99, 128.72, 127.25, 127.19, 126.84, 126.07, 124.98, 123.95, 123.89, 123.53, 123.41, 122.46, 122.27, 120.93, 22.16. HRMS (ESI) m/z calculated [M + H]+ C24H16NO+ 334.1154, found 334.1150.
- 9-Methoxyphenanthro[9′,10′:4,5]furo[3,2-h]quinoline (5Bb). White solid. M.p. 217.8–218.3 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 9.10 (dd, J = 4.3, 1.7 Hz, 1H), 8.80 (dd, J = 8.1, 1.5 Hz, 1H), 8.66 (dd, J = 8.1, 1.3 Hz, 1H), 8.58 (d, J = 8.8 Hz, 1H), 8.45 (d, J = 8.6 Hz, 1H), 8.32 (dd, J = 8.3, 1.7 Hz, 1H), 8.17 (d, J = 2.6 Hz, 1H), 7.82 (d, J = 8.6 Hz, 1H), 7.75 (ddd, J = 7.9, 7.0, 1.4 Hz, 1H), 7.70 (ddd, J = 8.5, 7.0, 1.7 Hz, 1H) 7.50 (dd, J = 8.2, 4.2 Hz, 1H), 7.40 (dd, J = 8.8, 2.5 Hz, 1H), 4.05 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 157.53, 150.69, 150.65, 150.39, 137.04, 136.50, 130.10, 129.97, 127.46, 127.00, 126.84, 125.34, 124.80, 123.47, 123.44, 122.64, 122.53, 122.32, 120.91, 120.80, 116.83, 115.23, 106.19, 55.63. HRMS (ESI) m/z calculated [M + H]+ C24H16NO2+ 350.1103, found 350.1110.
- 9-Chlorophenanthro[9′,10′:4,5]furo[3,2-h]quinoline (5Bc). White solid. M.p. 211.4–212.9 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 9.11 (d, J = 2.6 Hz, 1H), 8.81 (d, J = 7.8 Hz, 1H), 8.72 (d, J = 2.2 Hz, 1H), 8.66 (d, J = 8.1 Hz, 1H), 8.58 (d, J = 8.6 Hz, 1H), 8.44 (d, J = 8.6 Hz, 1H), 8.34 (dd, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.82–7.66 (m, 3H), 7.53 (dd, J = 8.3, 4.3 Hz, 1H). 13C NMR (CDCl3, 101 MHz) δ 151.83, 150.84, 150.53, 137.00, 136.55, 131.51, 129.93, 129.53, 128.05, 127.84, 127.64, 126.99, 126.59, 125.38, 124.51, 123.86, 123.76, 123.49, 122.61, 122.36, 121.16, 120.56, 114.85. HRMS (ESI) m/z calculated [M + H]+ C23H13ClNO+ 354.0607, found 354.0601.
- Chryseno[5′,6′:4,5]furo[3,2-h]quinoline (5Bd). White solid. M.p. 261.1–262.9 °C (XX MeOH). 1H NMR (CDCl3, 400 MHz) δ 9.18–9.09 (m, 2H), 8.99–8.91 (m, 1H), 8.82–8.76 (m, 1H), 8.74 (d, J = 9.0 Hz, 1H), 8.65 (d, J = 8.8 Hz, 1H), 8.33 (dd, J = 8.2, 1.7 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 8.03 (d, J = 8.9 Hz, 1H), 7.83–7.67 (m, 5H), 7.53 (dd, J = 8.2, 4.3 Hz, 1H). 13C NMR (CDCl3, 101 MHz) δ 153.35, 150.96, 150.70, 137.22, 136.38, 132.94, 130.47, 129.84, 128.10, 127.66, 127.22, 127.08, 126.89, 126.83, 126.27, 125.94, 125.39, 125.16, 123.74, 122.88, 122.50, 122.17, 121.71, 121.21, 115.75 (two signals not visible because of isochrony). HRMS (ESI) m/z calculated [M + H]+ C27H16NO+ 370.1154, found 370.1158.
- Thieno[2″,3″:3′,4′]naphtho [2′,1′:4,5]furo[3,2-h]quinoline (5Be). White solid. M.p. 232.5–233.7 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 9.12 (dd, J = 4.3, 1.7 Hz, 1H), 8.90–8.81 (m, 1H), 8.50–8.41 (m, 1H), 8.35 (dd, J = 8.3, 1.7 Hz, 1H), 8.26 (d, J = 8.5 Hz, 1H), 8.11 (d, J = 5.3 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H), 7.76–7.67 (m, 2H), 7.66 (d, J = 5.3 Hz, 1H), 7.53 (dd, J = 8.2, 4.3 Hz, 1H). 13C NMR (CDCl3, 101 MHz) δ 151.13, 150.76, 150.48, 136.96, 136.75, 133.58, 130.11, 128.79, 127.46, 127.04, 126.11, 124.44, 124.20, 123.79, 123.50, 122.50, 122.33, 121.08, 120.37, 120.01, 115.07. HRMS (ESI) m/z calculated [M + H]+ C21H12NOS+ 326.0561, found 326.0558.
- 9-Methyl-6H-phenanthro[9′,10′:4,5]furo[3,2-c]chromen-6-one (5Ca). White solid. M.p. 265.0–266.0 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 9.57 (d, J = 8.4 Hz, 1H), 8.64 (dd, J = 7.0, 2.3 Hz, 1H), 8.42 (s, 1H), 8.35 (dd, J = 7.5, 1.9 Hz, 1H), 8.09 (dd, J = 7.8, 1.6 Hz, 1H), 7.74–7.62 (m, 2H), 7.62–7.53 (m, 2H), 7.50 (dd, J = 8.5, 1.1 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 2.62 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 158.74, 158.61, 153.14, 150.02, 136.26, 131.34, 130.17, 129.38, 128.86, 128.07, 127.24, 124.67, 124.42, 123.51, 123.02, 121.57, 121.16, 120.83, 117.25, 117.19, 112.77, 108.51, 22.20 (two isochronous carbons). HRMS (ESI) m/z calculated [M + H]+ C24H15O3+ 351.0943, found 351.0950.
- 9-Methoxy-6H-phenanthro[9′,10′:4,5]furo[3,2-c]chromen-6-one (5Cb). White solid. M.p. 250.0–251.0 °C (taken up with EP/DCM). 1H NMR (CDCl3, 400 MHz) δ 9.59 (d, J = 9.0 Hz, 1H), 8.54 (d, J = 8.0 Hz, 1H), 8.32 (dd, J = 7.5, 1.8 Hz, 1H), 8.07 (dd, J = 7.9, 1.6 Hz, 1H), 7.99 (d, J = 2.6 Hz, 1H), 7.69–7.60 (m, 2H), 7.58 (td, J = 7.9, 7.2, 1.6 Hz, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 7.34 (dd, J = 9.0, 2.6 Hz, 1H), 4.01 (s, 3H). 13C NMR (CDCl3, 101 MHz) δ 158.70, 158.67, 158.21, 153.10, 149.24, 131.33, 130.39, 129.75, 127.46, 127.06, 124.68, 123.54, 121.55, 121.32, 120.86, 117.27, 117.18, 116.60, 112.77, 108.34, 105.55, 55.53 (two signals not visible because of isochrony). HRMS (ESI) m/z calculated [M + H]+ C24H15O4+ 367.0892, found 367.0898.
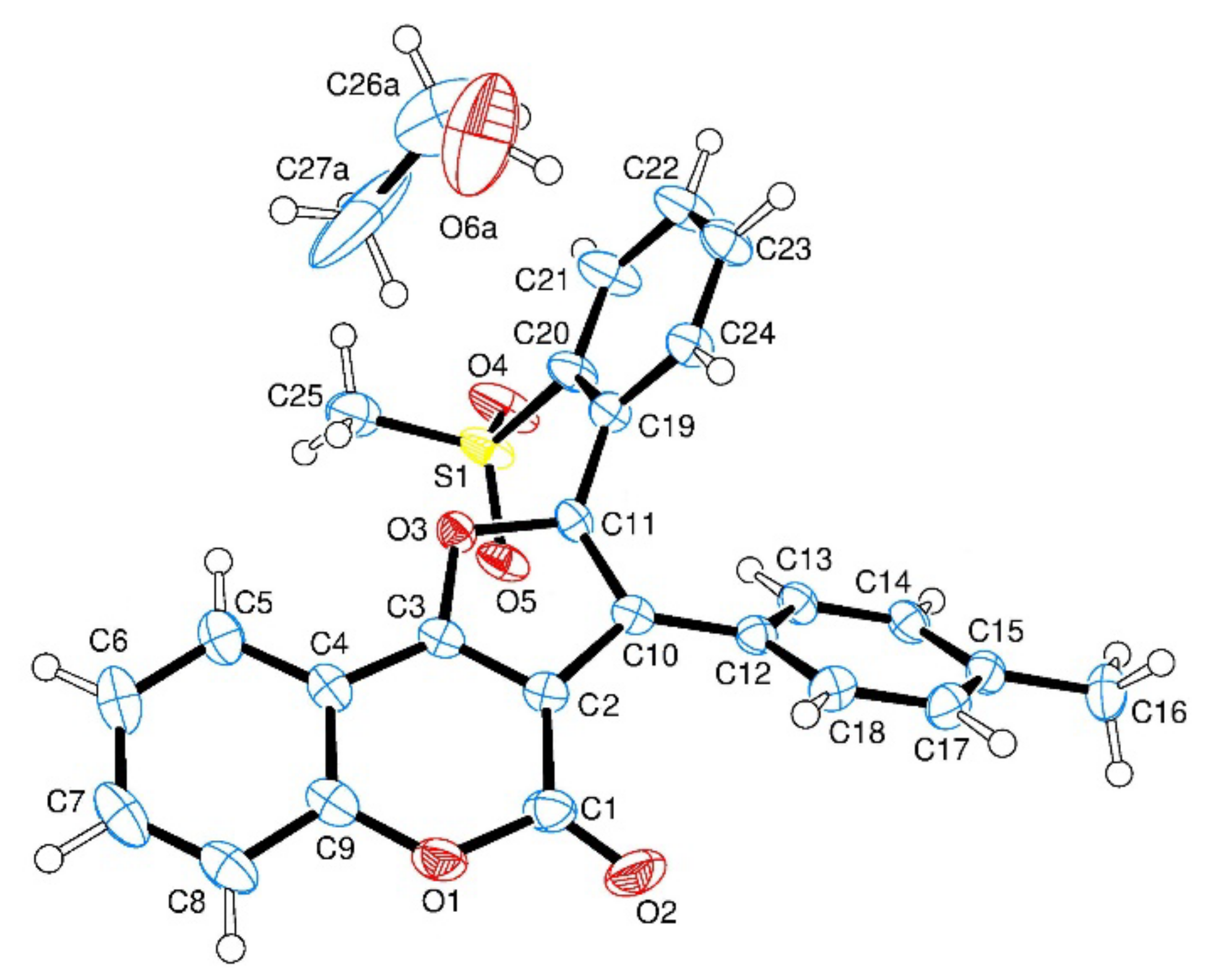
- Crystal structure of 3Ca
- A single crystal of compound 3Ca was submitted to X-ray data collection on a Bruker APEX-II CCD diffractometer (Billerica, MA, USA) with a graphite monochromated Cu-Kα radiation (λ = 1.5418 Å) at 100 K. The structure was solved by direct methods implemented in the SHELXS-97 program [39]. The refinement was carried out by full-matrix anisotropic least squares on F 2 for all reflections for non-H atoms using the SHELXL-97 program (Version 2019/2) [40]. 3Ca crystallizes with a molecule of ethanol. Crystallographic data for this structure have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2418973. Copies of the data can be obtained, free of charge, by application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; (fax: + 44-(0)-1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarivate Web of Science Database. Searched for “*polyheterocycl* OR *heteropolycycl*” over Topic Field and Displayed by Publication Year [A] and Then Refined for “Fused” and Displayed by WoS Category (Only Those with Minimum 3 Results Are Displayed) [B]. Available online: https://www.webofscience.com (accessed on 7 November 2024).
- Tsuji, H.; Nakamura, E. Design and Functions of Semiconducting Fused Polycyclic Furans for Optoelectronic Applications. Acc. Chem. Res. 2017, 50, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Sousa, C.M.; Coelho, P.J. Colour Switching with Photochromic Vinylidene-Naphthofurans. Tetrahedron 2018, 74, 7372–7379. [Google Scholar] [CrossRef]
- Duarte, C.D.; Barreiro, J.; Fraga, C.A.M. Privileged Structures: A Useful Oncept for the Rational Design of New Lead Drug Candidates. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.; Park, S.B. Privileged Structures: Efficient Chemical “Navigators” toward Unexplored Biologically Relevant Chemical Spaces. J. Am. Chem. Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, Y.T.; Gan, S.; Ren, A.; Milaneh, S.; Xiang, D.J.; Wang, W.L. Recent Progress of Organic Fluorescent Molecules for Bioimaging Applications: Cancer-Relevant Biomarkers. J. Mater. Chem. C. Mater. 2023, 11, 16859–16889. [Google Scholar] [CrossRef]
- Fylaktakidou, K.C.; Hadjipavlou-Litina, J.; Litinas, K.E.; Nicolaides, D.N. Natural and Synthetic Coumarin Derivatives with Anti-Inflammatory/Antioxidant Activities. Curr. Pharm. Des. 2004, 10, 3813–3833. [Google Scholar] [CrossRef]
- Benzi, A.; Bianchi, L.; Giorgi, G.; Maccagno, M.; Petrillo, G.; Spinelli, D.; Tavani, C. An Easy Access to Furan-Fused Polyheterocyclic Systems. Molecules 2022, 27, 3147. [Google Scholar] [CrossRef]
- Benzi, A. Synthesis of Fused N-and O-Heterocyclic Systems from Nitrodienic Building-Blocks. Ph.D. Thesis, Università di Genova, Genova, Italy, 2021. [Google Scholar]
- Zhang, Y.; Chen, F.; Yang, Y.; Tang, C.Z.; Tian, F.; Peng, L.; Wang, L.X. An Unexpected Metal-Free DMAP Catalyzed Michael Addition-Elimination Domino Reaction between 2-Naphthols and Bromomaleimides for the Effective Construction of 3-Arylmaleimides. Tetrahedron Lett. 2016, 57, 1261–1264. [Google Scholar] [CrossRef]
- Speranza, G.; Morelli, C.F.; Manitto, P. The Michael Reaction of N-Cinnamoylazoles with Phenols. A Simple Synthesis of 4-Arylchroman-2-Ones and 1-Arylbenzo[f]Chroman-3-Ones. Synthesis 2000, 2000, 123–126. [Google Scholar] [CrossRef]
- Faizullina, L.K.; Karamysheva, L.S.; Khalilova, Y.A.; Salikhov, S.M.; Valeev, F.A. Reactions of Phenol and Its Derivatives with Levoglucosenone. Chem. Heterocycl. Compd. 2023, 59, 254–259. [Google Scholar] [CrossRef]
- Vaughan, D.; Jha, A. Convenient Synthesis of Novel 2,2-Dialkyl-1,2-Dihydronaphtho[2,1-b]Furans. Tetrahedron Lett. 2009, 50, 5709–5712. [Google Scholar] [CrossRef]
- Bassetti, B.; Principi, M.; Ballini, R.; Petrini, M.; Palmieri, A. β-Nitroacrylates and Phenols as Key Precursors of Arenofuran-3-Carboxylates. Synthesis 2024, 56, 3167–3172. [Google Scholar]
- Abdou, M.M.; El-Saeed, R.A.; Bondock, S. Recent Advances in 4-Hydroxycoumarin Chemistry. Part 1: Synthesis and Reactions. Arab. J. Chem. 2019, 12, 88–121. [Google Scholar] [CrossRef]
- Abdou, M.M.; El-Saeed, R.A.; Bondock, S. Recent Advances in 4-Hydroxycoumarin Chemistry. Part 2: Scaffolds for Heterocycle Molecular Diversity. Arab. J. Chem. 2019, 12, 974–1003. [Google Scholar] [CrossRef]
- Ganguly, N.C.; Mondal, P.; Roy, S. A Mild Efficient Iodine-Catalyzed Synthesis of Novel Anticoagulants with 2,8-Dioxabicyclo[3.3.1]Nonane Core. Tetrahedron Lett. 2013, 54, 2386–2390. [Google Scholar] [CrossRef]
- Kasperkiewicz, K.; Ponczek, M.B.; Owczarek, J.; Guga, P.; Budzisz, E. Antagonists of Vitamin K-Popular Coumarin Drugs and New Synthetic and Natural Coumarin Derivatives. Molecules 2020, 25, 1465. [Google Scholar] [CrossRef]
- Cai, G.; Yu, W.; Song, D.; Zhang, W.; Guo, J.; Zhu, J.; Ren, Y.; Kong, L. Discovery of Fluorescent Coumarin-Benzo[b]Thiophene 1, 1-Dioxide Conjugates as Mitochondria-Targeting Antitumor STAT3 Inhibitors. Eur. J. Med. Chem. 2019, 174, 236–251. [Google Scholar] [CrossRef]
- Langer, P. Adventures in Coumarin Chemistry. Synlett 2024, 36, 29–43. [Google Scholar] [CrossRef]
- Kurti, L.; Chako, B. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier: San Diego, CA, USA, 2005. [Google Scholar]
- Noboro, O. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, NY, USA, 2001. [Google Scholar]
- Ghosh, M.; Hajra, A. DABCO-Promoted One-Pot Facile Synthesis of Angularly Fused Furoquinolinones and Furocoumarins. Eur. J. Org. Chem. 2015, 2015, 7836–7841. [Google Scholar] [CrossRef]
- Miao, C.B.; Liu, R.; Sun, Y.F.; Sun, X.Q.; Yang, H.T. Base-Controlled Selective Construction of Polysubstituted Dihydrofuran and Furan Derivatives through an I2-Mediated Cyclization. Tetrahedron Lett. 2017, 58, 541–545. [Google Scholar] [CrossRef]
- Yang, Z.; Fan, M.; Mu, R.; Liu, W.; Liang, Y. A Facile Synthesis of Highly Functionalized Dihydrofurans Based on 1,4-Diazabicyclo[2.2.2]Octane (DABCO) Catalyzed Reaction of Halides with Enones. Tetrahedron 2005, 61, 9140–9146. [Google Scholar] [CrossRef]
- Lyapustin, D.N.; Ulomsky, E.N.; Balyakin, I.A.; Shchepochkin, A.V.; Rusinov, V.L.; Chupakhin, O.N. Oxidative Aromatization of 4,7-Dihydro-6-Nitroazolo[1,5-a] Pyrimidines: Synthetic Possibilities and Limitations, Mechanism of Destruction, and the Theoretical and Experimental Substantiation. Molecules 2021, 26, 4719. [Google Scholar] [CrossRef]
- Sun, Y.; Gan, J.; Fan, R. Facile Construction of Oxa-Aza Spirobicycles via a Tandem Carbon-Hydrogen Bond Oxidation. Adv. Synth. Catal. 2011, 353, 1735–1740. [Google Scholar] [CrossRef]
- Lou, B.-H.; Chen, S.-B.; Wang, J.; Chen, Y.; Li, J.-H. An Efficient Transition-Metal-Chloride/Sodium-Nitrite/TEMPO Catalytic System for Aerobic Oxidative Aromatisation of Hantzsch 1,4-Dihydropyridines. J. Chem. Res. 2013, 37, 409–412. [Google Scholar] [CrossRef]
- Hong, G.; Gan, X.; Leonhardt, C.; Zhang, Z.; Seibert, J.; Busch, J.M.; Bräse, S. A Brief History of OLEDs—Emitter Development and Industry Milestones. Adv. Mater. 2021, 33, 2005630. [Google Scholar] [CrossRef]
- Taddei, G.M. Sintesi Di Sistemi Polieterociclici Ossigenati. Master’s Thesis, Università di Genova, Genova, Italy, 2022. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publishers: Dordrecht, The Netherlands; New York, NY, USA, 1999. [Google Scholar]
- Bianchi, L.; Dell’Erba, C.; Maccagno, M.; Morganti, S.; Novi, M.; Petrillo, G.; Rizzato, E.; Sancassan, F.; Severi, E.; Spinelli, D.; et al. Easy Access to 4-Nitrothiochroman S,S-Dioxides via Ring-Enlargement from 3-Nitrobenzo[b]Thiophene. Tetrahedron 2004, 60, 4967–4973. [Google Scholar] [CrossRef]
- Bianchi, L.; Maccagno, M.; Petrillo, G.; Rizzato, E.; Sancassan, F.; Severi, E.; Spinelli, D.; Stenta, M.; Galatini, A.; Tavani, C. A New Route to Thiopyran S,S-Dioxide Derivatives via an Overall Ring-Enlargement Protocol from 3-Nitrothiophene. Tetrahedron 2009, 65, 336–343. [Google Scholar] [CrossRef]
- Tanemura, K.; Suzuki, T.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. A Mild and Efficient Method for the Mononitration of Aromatic Ompounds by Cerium (III) Ammonium Nitrate in Acetic Anhydride. J. Chem. Res. 2003, 2003, 497–499. [Google Scholar] [CrossRef]
- Tanemura, K.; Suzuki, T.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. Cleavage of 2-Methoxyethoxymethyl Ethers Catalyzed by Cerium(IV) Ammonium Nitrate (CAN) in Acetic Anhydride. Chem. Lett. 2001, 30, 1012–1013. [Google Scholar] [CrossRef]
- Dell’erba, C.; Gabellini, A.; Novi, M.; Petrillo, G.; Tavani, C.; Cosimelli, B.; Spinelli, D. Ring Opening of 2-Substituted 4-Nitrothiophenes with Pyrrolidine. Access to New Functionalized Nitro-Unsaturated Building Blocks. Tetahedron 2001, 57, 8159–8165. [Google Scholar] [CrossRef]
- J Armstrong, B.K.; Martin-Smith, M.; Brophy, G.C.; Sternhell, S.; Fries, K.; Heering, H.; Hemmecke, K.; Siebert, G.; Boswell, D.E.; Brennan, J.A.; et al. Benzo[b]Thiophen Derivatives. Part 1X.l Nitration of Benzo[b]Thiophen and the Isomeric Nitrobenzo[b]Thiophens. J. Chem. Soc. C 1969, 537, 1766–1775. [Google Scholar] [CrossRef]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction, Inc.: Irvine, CA, USA, 2003; ISBN 189066118X. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ar in 1 | t | Yields in 2Ba–e | t a | Yields in 2Ba–c a | |
|---|---|---|---|---|---|
| p-Tolyl | a | 24 h | 70% | 4 h | 90% |
| p-MeO-Phenyl | b | 24 h | 66% | 4 h | 58% |
| p-Cl-Phenyl | c | 24 h | 74% | 4 h | 81% |
| 1-Naphthyl | d | 45 h | 30% | ||
| 2-Thienyl | e | 24 h | 40% |
| Ar in 2B | t | T | Yields in 3Ba–e | |
|---|---|---|---|---|
| p-Tolyl | a | 24 h | r.t. | 91% |
| p-MeO-Phenyl | b | 24 h | r.t. | 90% |
| p-Cl-Phenyl | c | 27 h | r.t. | 73% |
| 1-Naphthyl | d | 30 h | 60 °C | 85% |
| 2-Thienyl | e | 24 h | r.t. | 99% |
| Ar in 1 | Yields in 2Ca–e | |
|---|---|---|
| p-Tolyl | a | 79% |
| p-MeO-Phenyl | b | 68% |
| p-Cl-Phenyl | c | 83% |
| 1-Naphthyl | d | 76% |
| 2-Thienyl | e | 10% a |
| Ar in 1 | t | Yields in 2Ce,c,d | |
|---|---|---|---|
| 2-Thienyl | e | 16 h | 74% |
| p-Cl-Phenyl | c | 4 h | 70% |
| 1-Naphthyl | d | 24 h | 76% |
| Solvent | DDQ mol equiv. | T | t | Yields in 3Ca |
|---|---|---|---|---|
| CHCl3 | 2 | r.t. | 24 h | 14% |
| CHCl3 | 3 | 60 °C | 48 h | 40% |
| Toluene | 8 | 110 °C | 28 h | 12% |
| CHCl3 | 4 | 60 °C | 5 days | 90% |
| Ar in 2C | t | Yields in 3Ca–e | |
|---|---|---|---|
| p-Tolyl | a | 96 h | 99% |
| p-MeO-Phenyl | b | 48 h | 99% |
| p-Cl-Phenyl | c | 11 days | 42% a |
| 1-Naphthyl | d | 28 h | 0% |
| 2-Thienyl | e | 24 h | 99% |
| CH3CN/H2O 97/3, 60 °C | DDQ/Tol, r.t. 10 min | ||||||
|---|---|---|---|---|---|---|---|
| Ar in 1 | t | 2D | 3D | Ar in 2D | 3D | ||
| p-Tolyl | a | 16 h | 76% | 12% | p-Tolyl | a | 93% |
| p-MeO-Phenyl | b | 40 h | 66% | 12% | p-MeO-Phenyl | b | 99% |
| p-Cl-Phenyl | c | 5 h | 44% | 20% | p-Cl-Phenyl | c | 98% |
| 1-Naphthyl | d | 48 h | 60% | 10% | 1-Naphthyl | d | 97% |
| 2-Thienyl | e | 52 h | 61% | 16% | 2-Thienyl | e | 99% |
| CH3CN/H2O 97/3, 60 °C, 24 h | DDQ/CHCl3, 60 °C, 29 h | ||||
|---|---|---|---|---|---|
| Ar in 1 | 2E | Ar in 2E | 3E | ||
| p-Tolyl | a | 68% | |||
| p-MeO-Phenyl | b | 48% | p-MeO-Phenyl | b | 99% |
| p-Cl-Phenyl | c | 71% | |||
| 1-Naphthyl | d | 88% | |||
| 2-Thienyl | e | 46% * |
| Ar in 3B | hν | t | 5B | hν a | t a | 5B | |
|---|---|---|---|---|---|---|---|
| p-Tolyl | a | 300 | 24 h | 52% | 350 | 16 h | 70% |
| p-MeO-Phenyl | b | 300 | 24 h | 29% | 350 | 16 h | 73% |
| p-Cl-Phenyl | c | 300 | 24 h | 57% | 350 | 17 h | 62% |
| 1-Naphthtyl | d | 300 | 24 h | 350 | 24 h | 68% | |
| 2-Thyenyl | e | 300 | 24 h | 350 | 16 h | 50% |
| Ar in 3C | 5C | |
|---|---|---|
| p-Tolyl | a | 70% |
| p-MeO-Phenyl | b | 73% |
| 2-Thienyl | e | 65% a |
| Compound | λmax (ab) [nm] | λmax (em) [nm] | [cm−1] | |
|---|---|---|---|---|
| 2Bb | 330 | 411 | 5972 | 0.037 |
| 3Bb | 329 | 433 | 7300 | 0.160 |
| 3Bc | 324 | 396 | 5612 | 0.200 |
| 3Bd | 297 | 407 | 9100 | 0.410 |
| 3Be | 325 | 434 | 7728 | 0.151 |
| 5Ba | 327 | 420 | 6772 | 0.694 |
| 5Bb | 328 | 442 | 7863 | 0.459 |
| 5Bc | 326 | 407 | 6105 | 0.258 |
| 5Bd | 305 | 407 | 8217 | 0.712 |
| 5Be | 290 | 423 | 10,842 | 0.478 |
| 3Ca | 334 | 405 | 5249 | 0.206 |
| 3Cb | 341 | 438 | 6494 | 0.109 |
| 3Ce | 332 | 442 | 7496 | 0.126 |
| 5Ca | 364 | 412 | 3201 | 0.427 |
| 5Cb | 371 | 441 | 4278 | 0.329 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benzi, A.; Bianchi, L.; Giorgi, G.; Lentini, G.; Maccagno, M.; Marcantoni Taddei, G.; Petrillo, G.; Tavani, C. An Appealing, Robust Access to Furo-Fused Heteropolycycles. Molecules 2025, 30, 948. https://doi.org/10.3390/molecules30040948
Benzi A, Bianchi L, Giorgi G, Lentini G, Maccagno M, Marcantoni Taddei G, Petrillo G, Tavani C. An Appealing, Robust Access to Furo-Fused Heteropolycycles. Molecules. 2025; 30(4):948. https://doi.org/10.3390/molecules30040948
Chicago/Turabian StyleBenzi, Alice, Lara Bianchi, Gianluca Giorgi, Giovanni Lentini, Massimo Maccagno, Guglielmo Marcantoni Taddei, Giovanni Petrillo, and Cinzia Tavani. 2025. "An Appealing, Robust Access to Furo-Fused Heteropolycycles" Molecules 30, no. 4: 948. https://doi.org/10.3390/molecules30040948
APA StyleBenzi, A., Bianchi, L., Giorgi, G., Lentini, G., Maccagno, M., Marcantoni Taddei, G., Petrillo, G., & Tavani, C. (2025). An Appealing, Robust Access to Furo-Fused Heteropolycycles. Molecules, 30(4), 948. https://doi.org/10.3390/molecules30040948