The Versatility of the Roskamp Homologation in Synthesis


Abstract
:1. Introduction
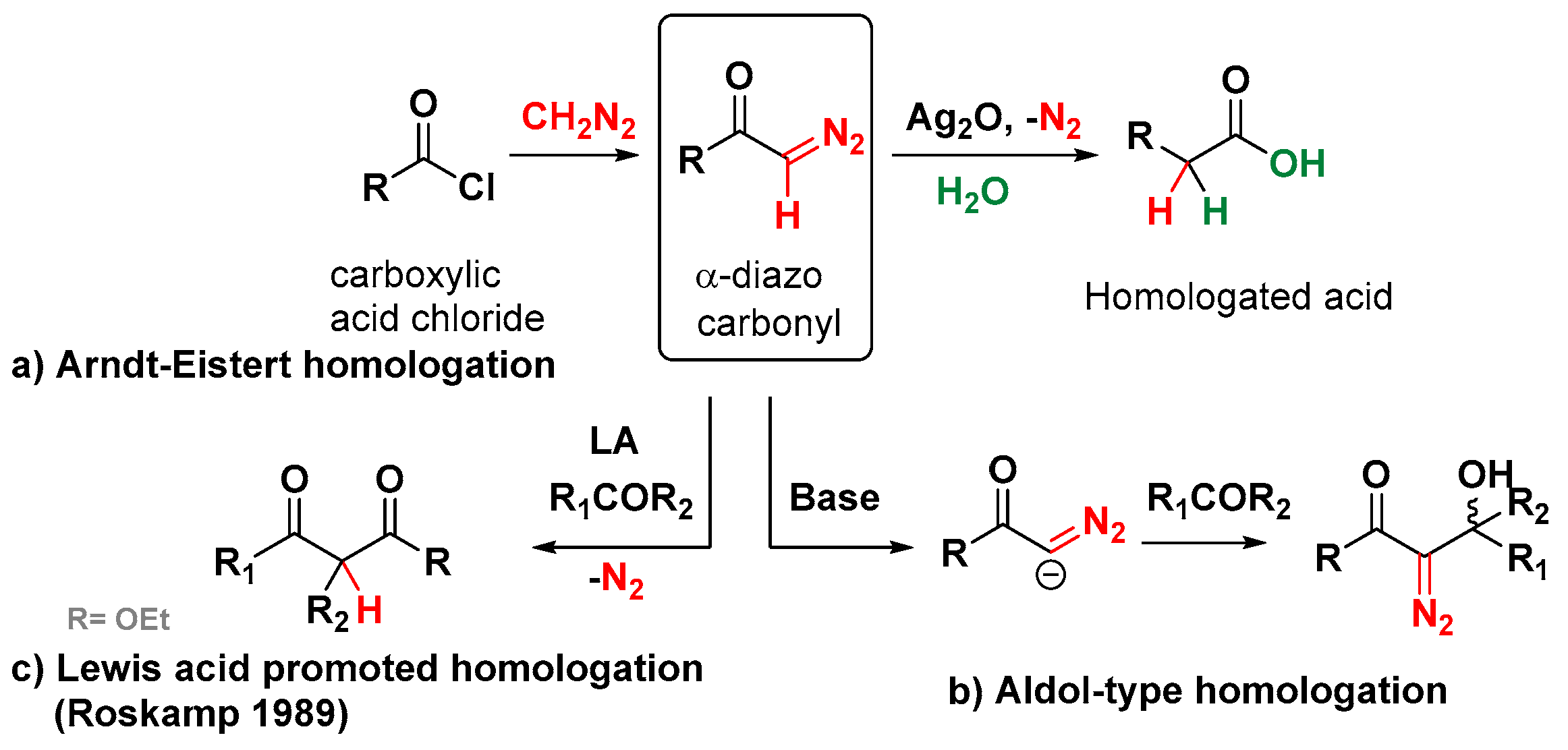
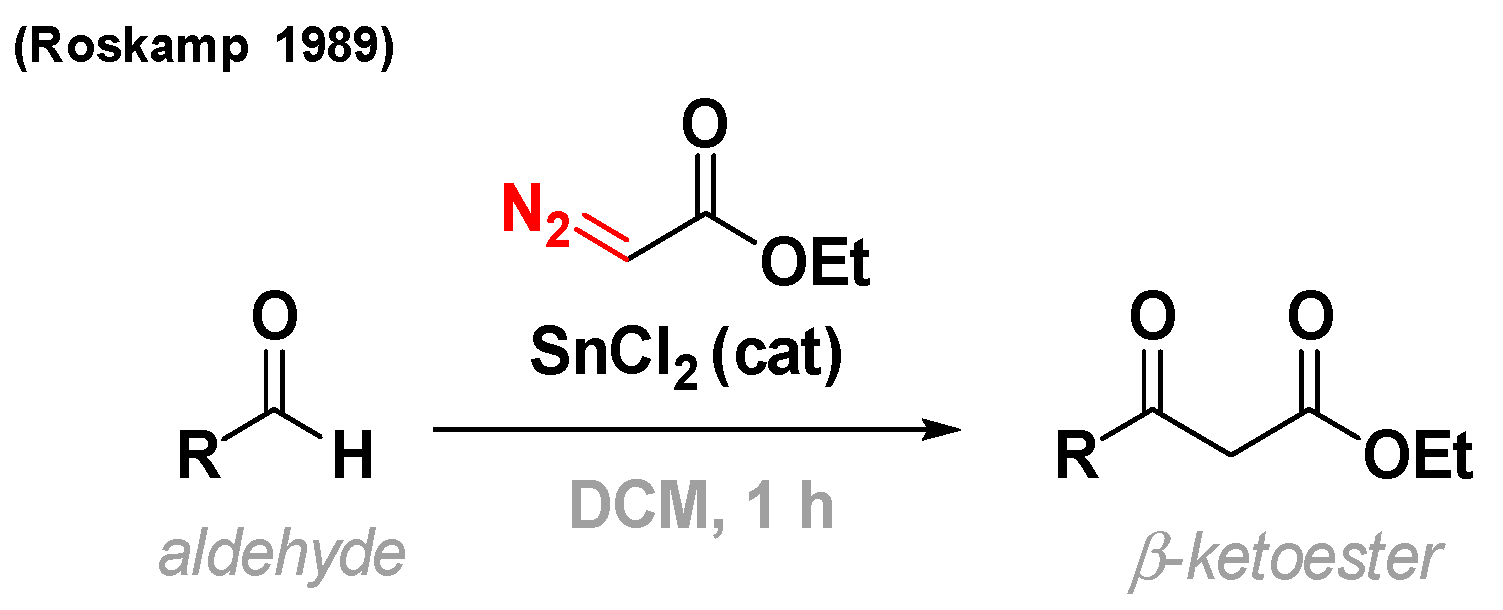
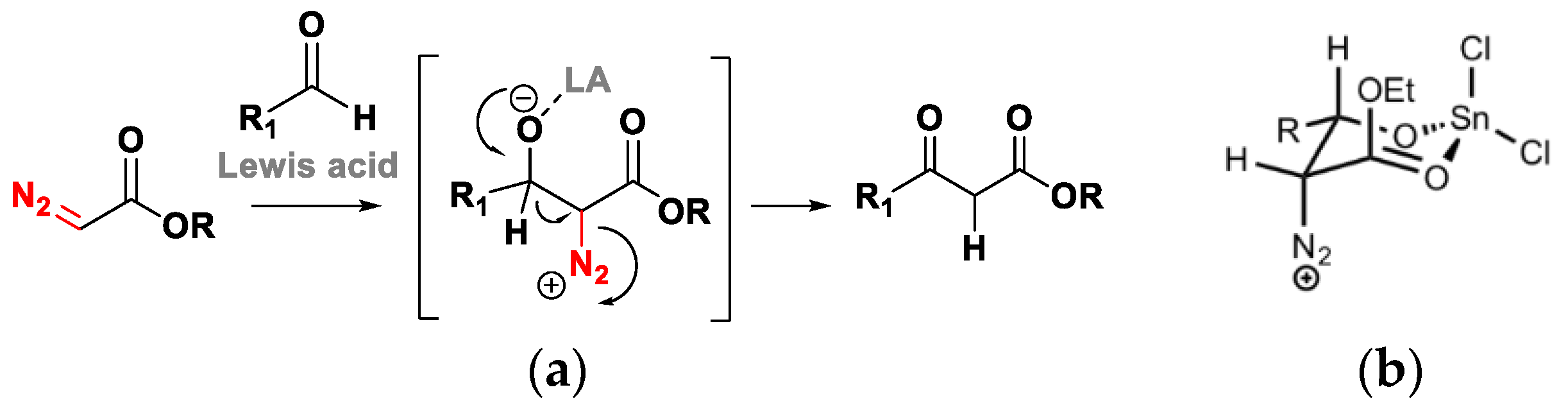
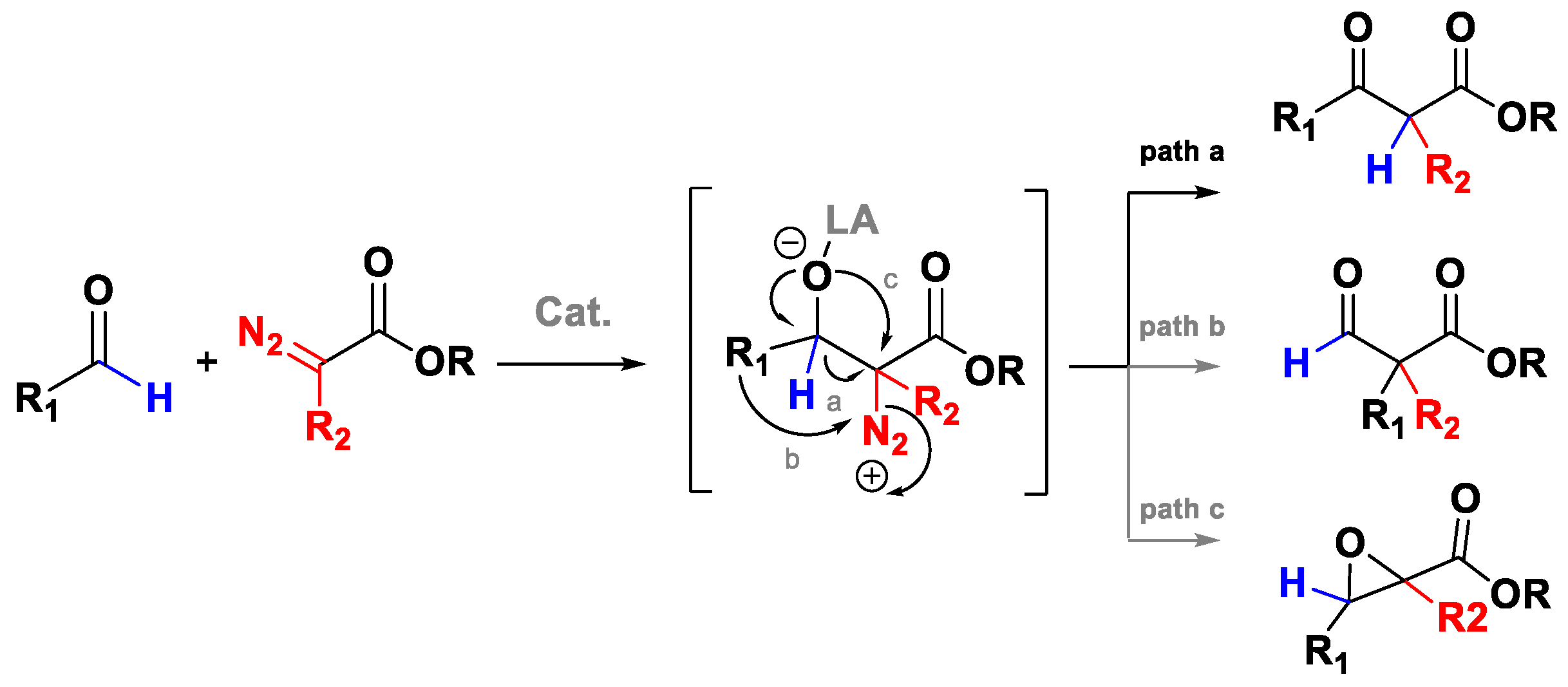
2. The Roskamp Reaction: A Powerful Method for the Synthesis of β-Keto Esters
3. A Strategy Toward Asymmetric Variants
3.1. Roskamp Reaction and Chiral Auxiliaries
3.2. New Directions in Asymmetric Roskamp Reactions
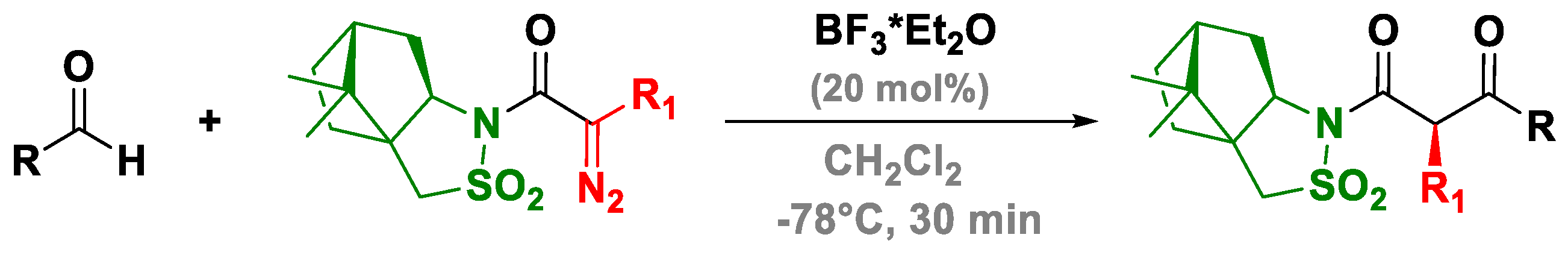
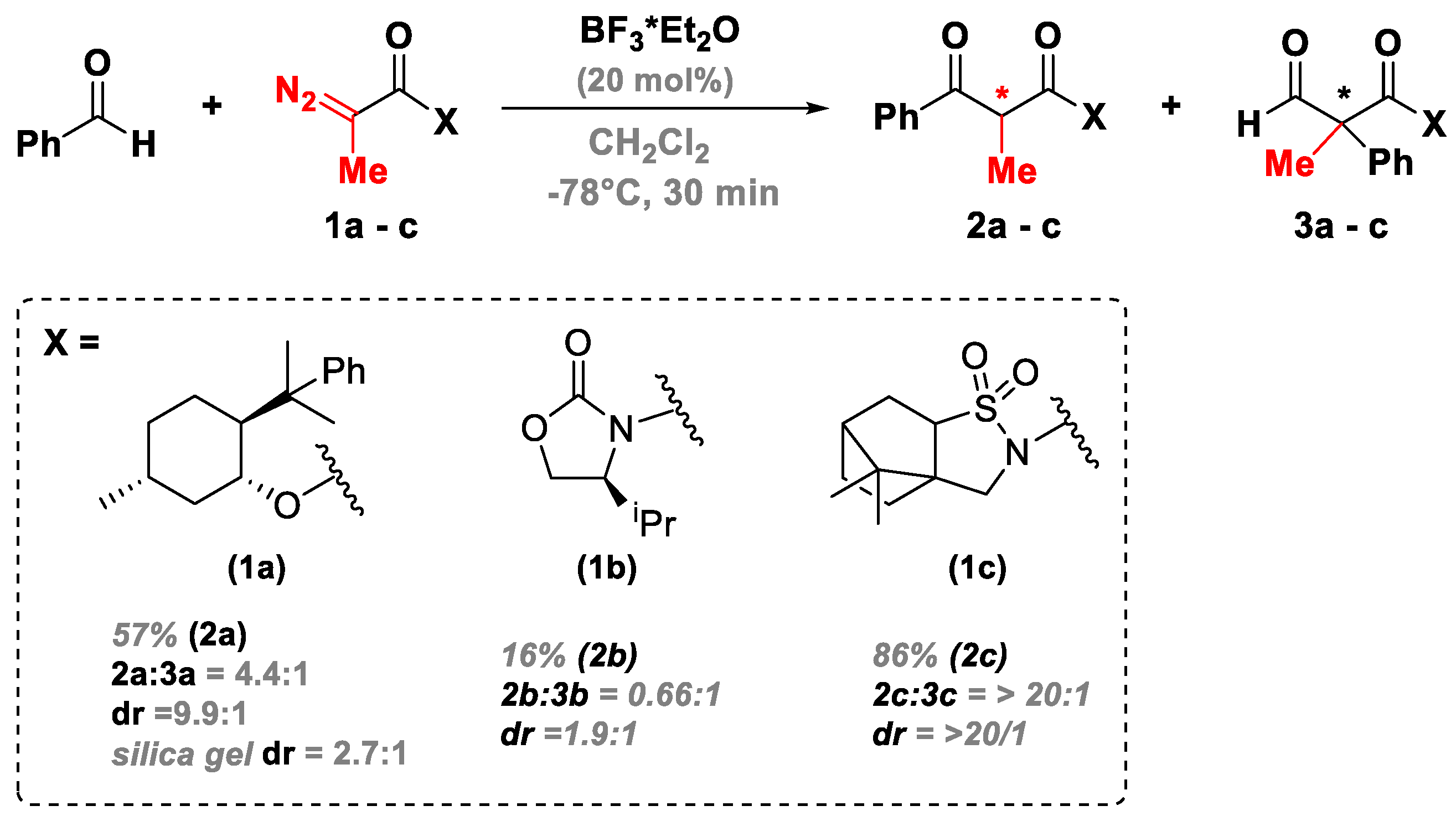
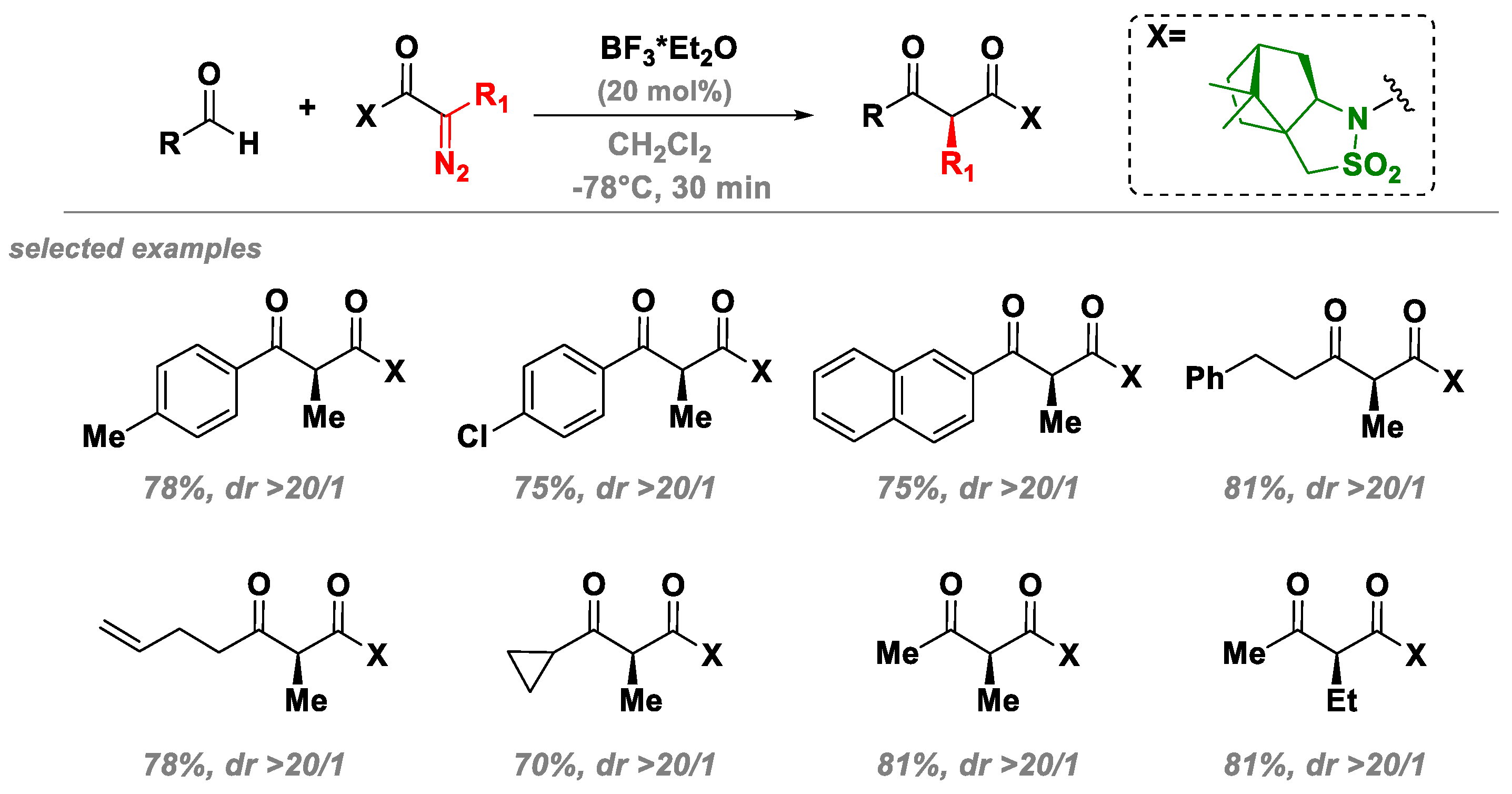
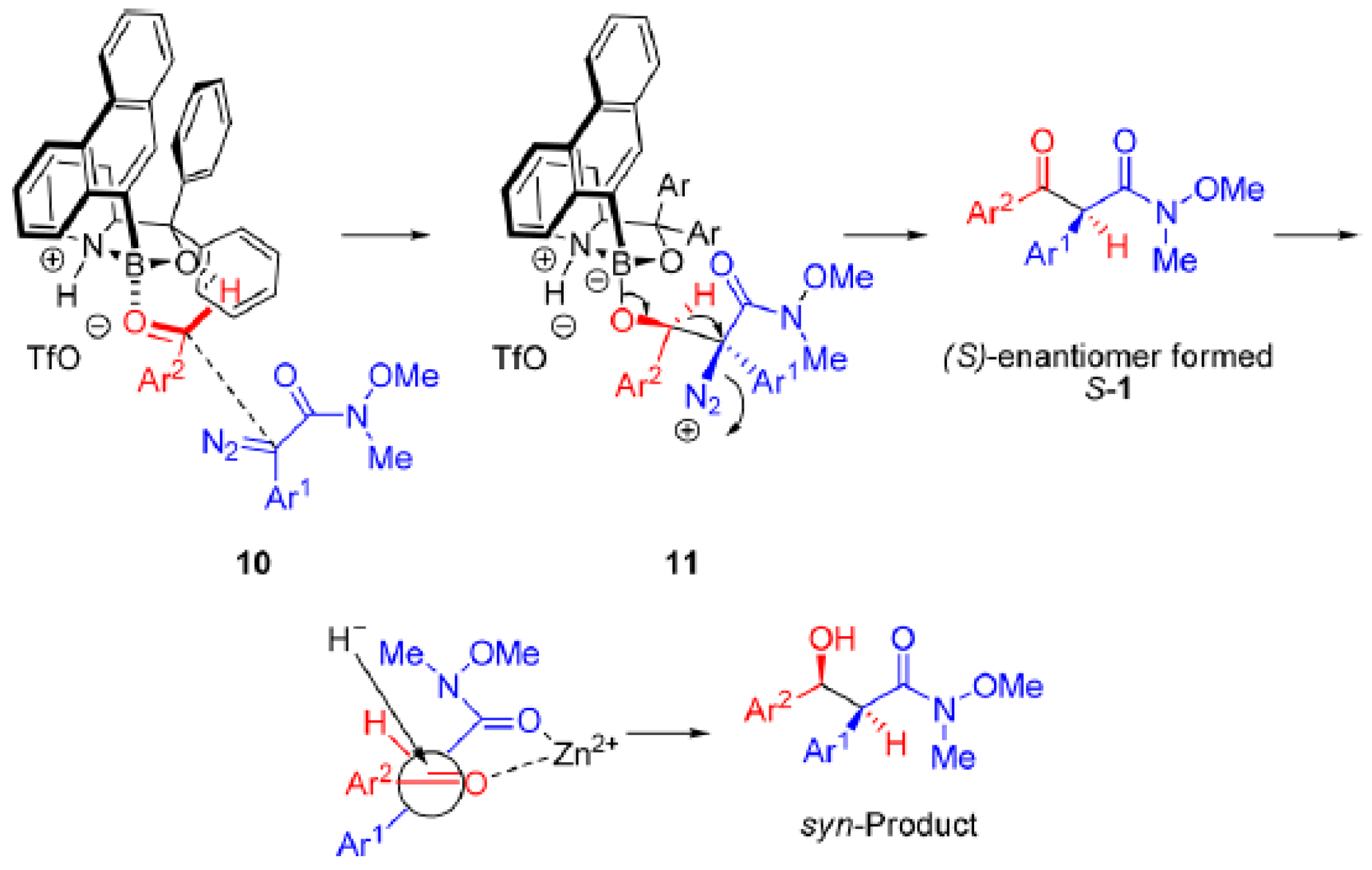
3.3. A Novel Lewis Acid Catalyst
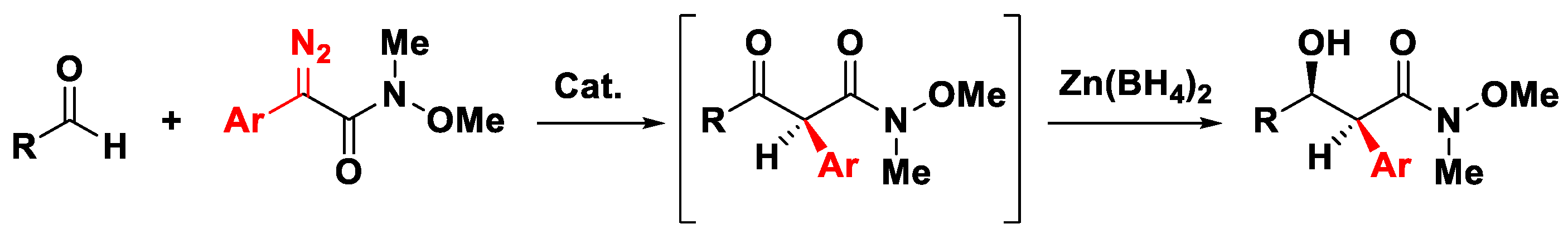
3.4. The Roskamp Reaction for Enantioriched Weinreb Amides
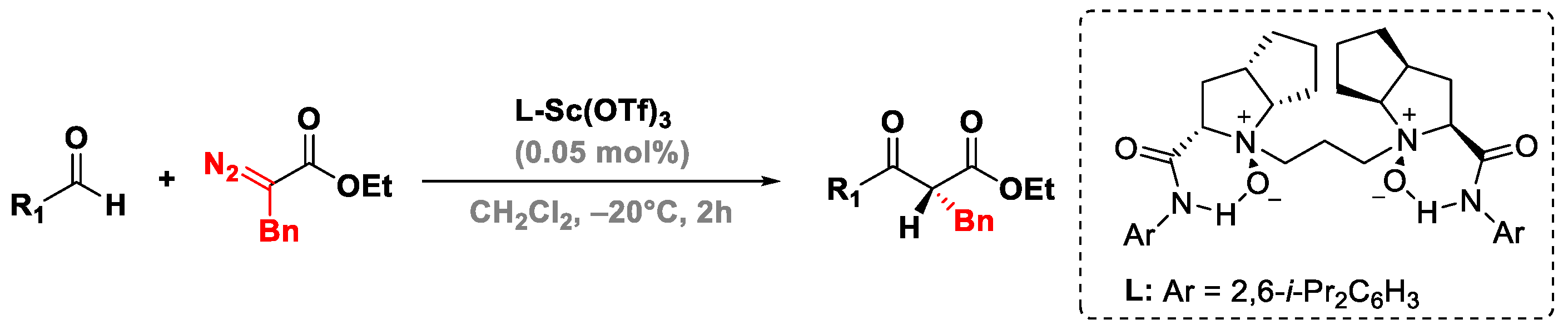
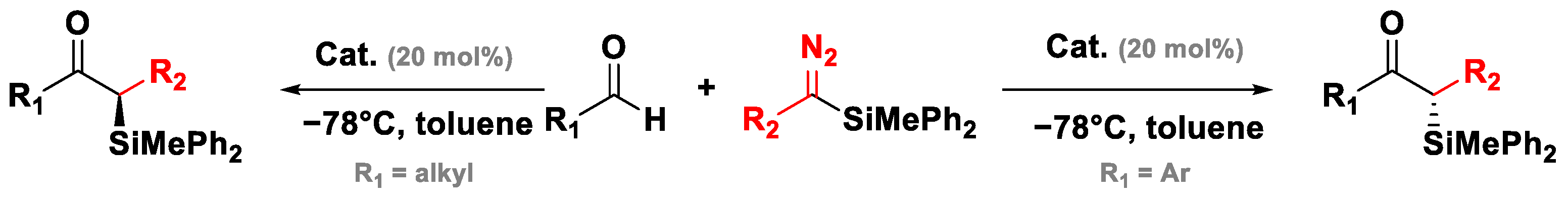
3.5. Roskamp Reaction of Silyl Diazoalkane
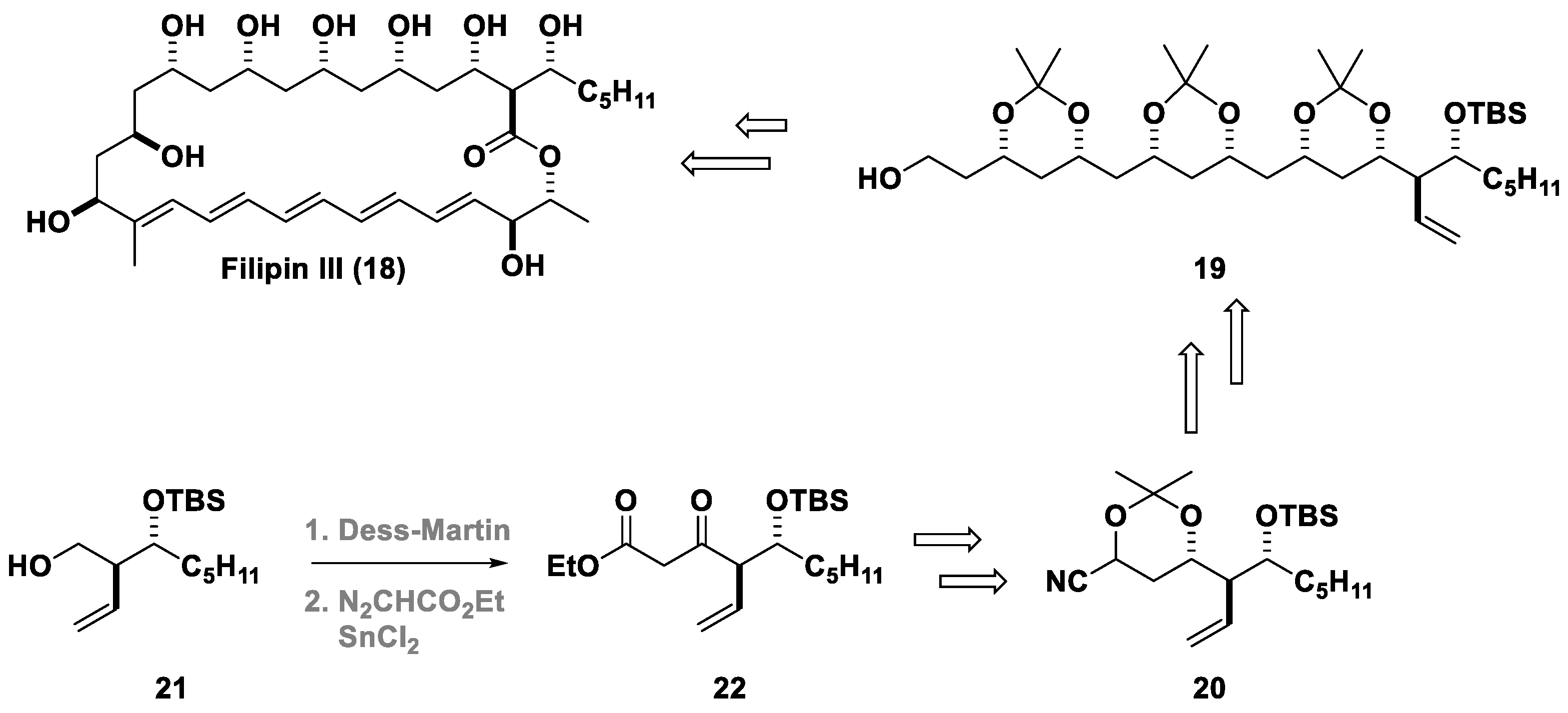
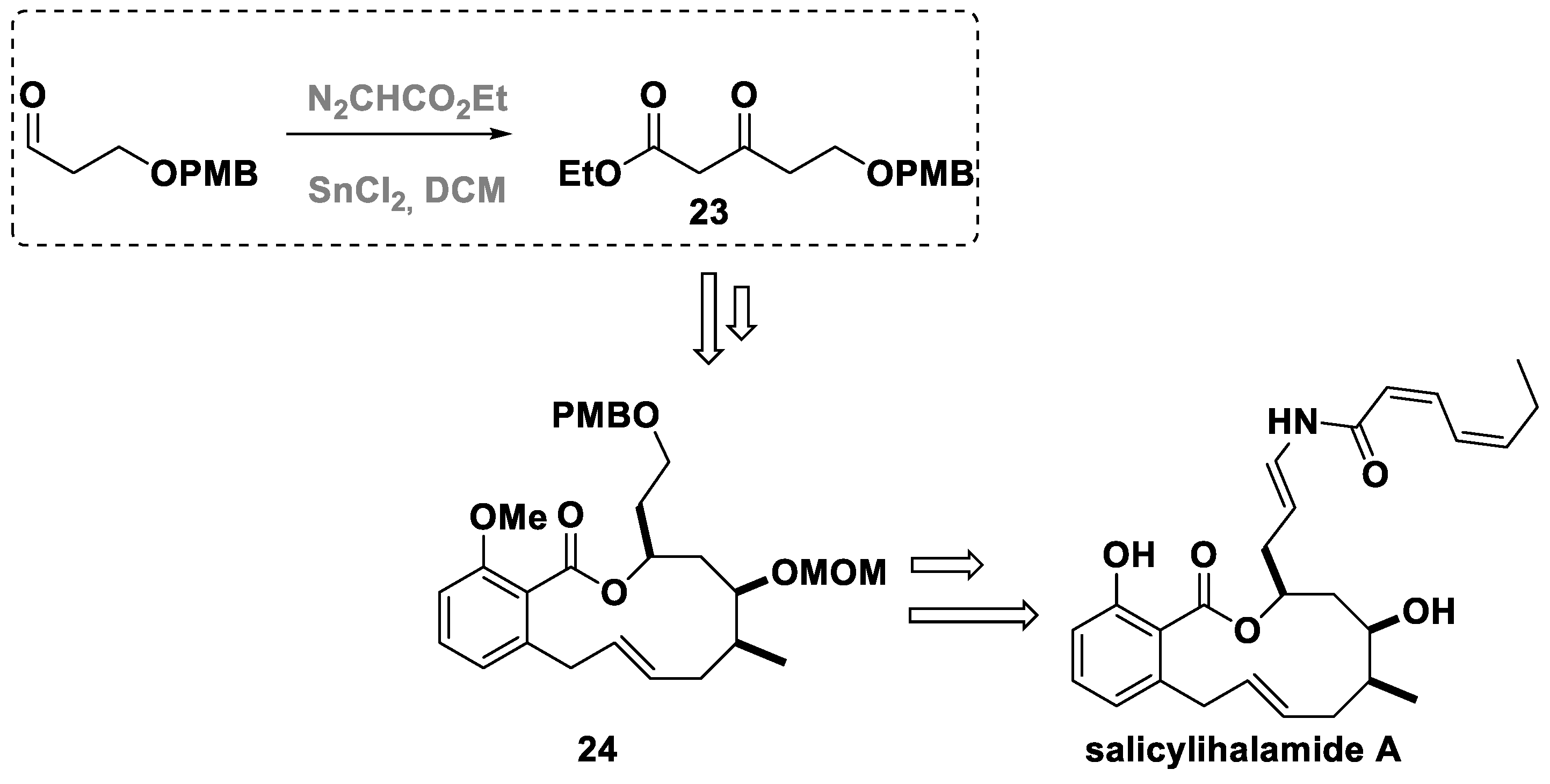
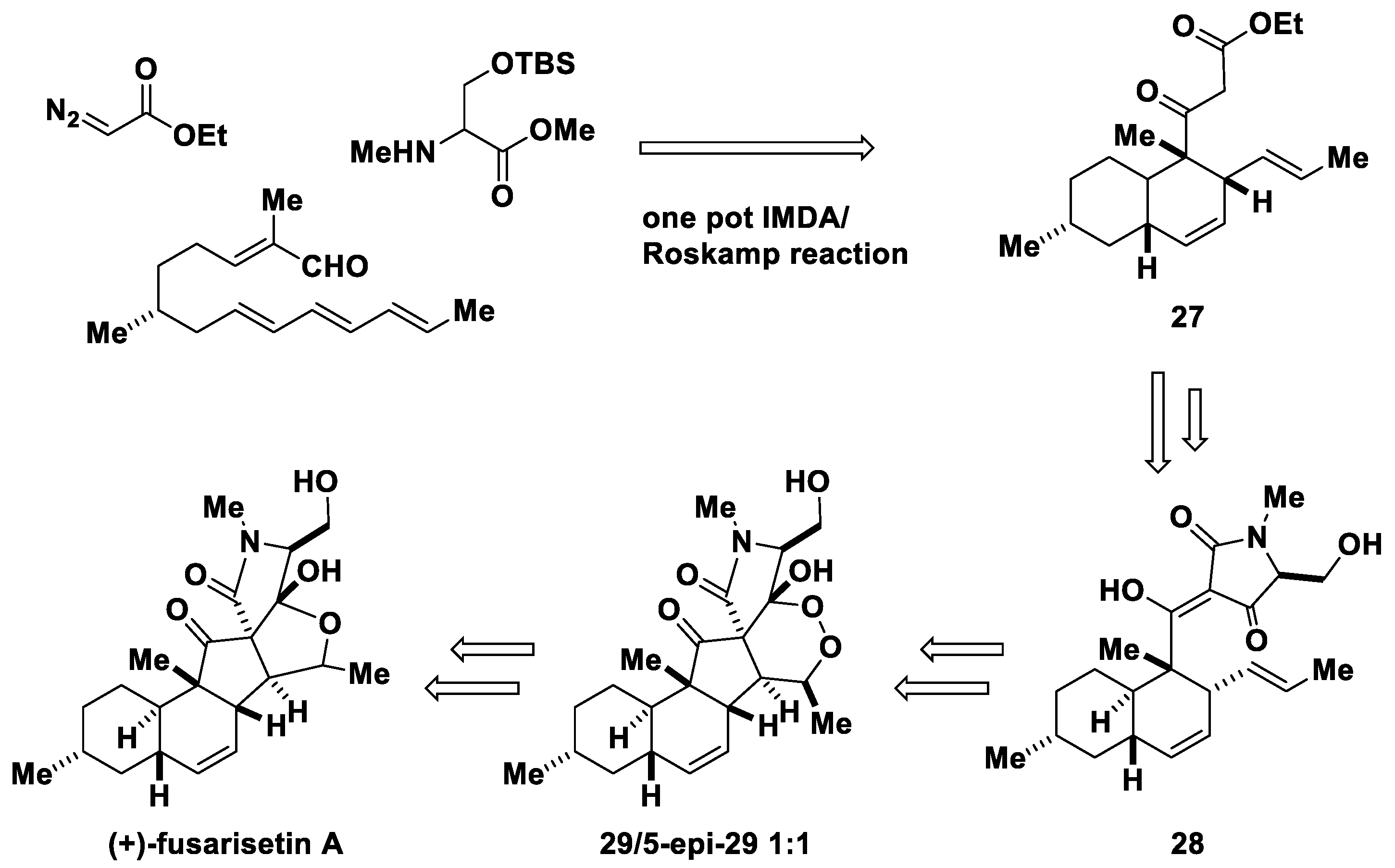
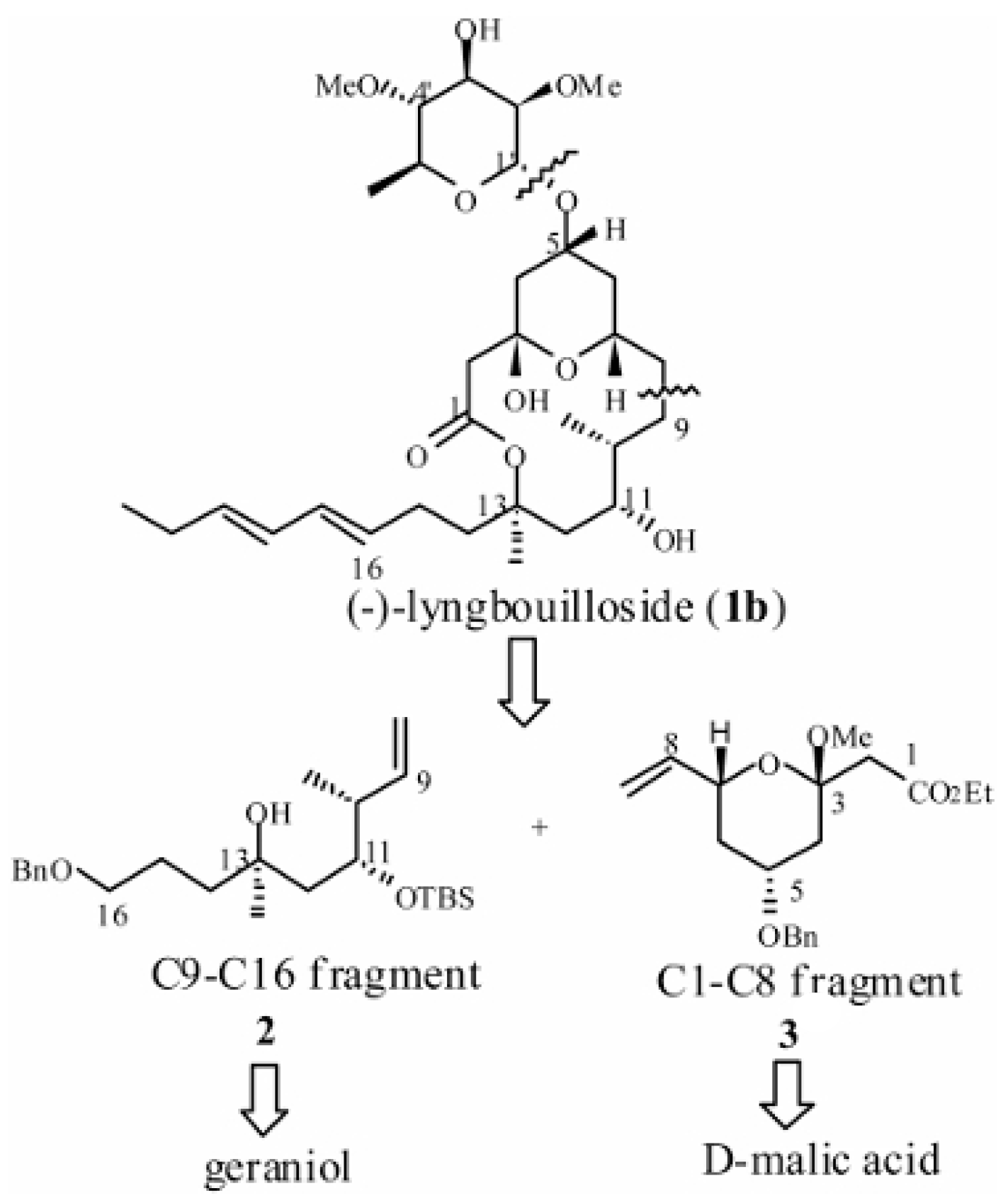
4. The Roskamp Reaction in Total Synthesis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Monticelli, S.; Castoldi, L.; Touqeer, S.; Miele, M.; Urban, E.; Pace, V. Recent advances in the synthesis and reactivity of spiro-epoxyoxindoles. Chem. Heterocycl. Compd. 2018, 54, 389–393. [Google Scholar] [CrossRef]
- Ielo, L.; Pillari, V.; Miele, M.; Castiglione, D.; Pace, V. Carbenoid-Mediated Homologation Tactics for Assembling (Fluorinated) Epoxides and Aziridines. Synlett 2021, 32, 551–560. [Google Scholar] [CrossRef]
- Ielo, L.; Miele, M.; Pillari, V.; Castoldi, L.; Pace, V. Lithium Carbenoids in Homologation Chemistry. In Homologation Reactions; Wiley: Hoboken, NJ, USA, 2023; pp. 79–142. [Google Scholar] [CrossRef]
- Miele, M.; Castoldi, L.; Simeone, X.; Holzer, W.; Pace, V. Straightforward synthesis of bench-stable heteroatom-centered difluoromethylated entities via controlled nucleophilic transfer from activated TMSCHF2. Chem. Commun. 2022, 58, 5761–5764. [Google Scholar] [CrossRef]
- Miele, M.; Pace, V. (Difluoromethyl)trimethylsilane (TMSCHF2): A Useful Difluoromethylating Nucleophilic Source. Aust. J. Chem. 2021, 74, 623–625. [Google Scholar] [CrossRef]
- Touqeer, S.; Ielo, L.; Miele, M.; Urban, E.; Holzer, W.; Pace, V. Direct and straightforward transfer of C1 functionalized synthons to phosphorous electrophiles for accessing gem-P-containing methanes. Org. Biomol. Chem. 2021, 19, 2425–2429. [Google Scholar] [CrossRef]
- Ielo, L.; Pillari, V.; Miele, M.; Holzer, W.; Pace, V. Consecutive C1-Homologation / Displacement Strategy for Converting Thiosulfonates into O,S-Oxothioacetals. Adv. Synth. Catal. 2020, 362, 5444–5449. [Google Scholar] [CrossRef]
- Miele, M.; Citarella, A.; Langer, T.; Urban, E.; Zehl, M.; Holzer, W.; Ielo, L.; Pace, V. Chemoselective Homologation–Deoxygenation Strategy Enabling the Direct Conversion of Carbonyls into (n+1)-Halomethyl-Alkanes. Org. Lett. 2020, 22, 7629–7634. [Google Scholar] [CrossRef]
- Castoldi, L.; Pace, V. Homologation reactions for olefin synthesis. Nat. Synth. 2024, 3, 288–290. [Google Scholar] [CrossRef]
- Malik, M.; Senatore, R.; Castiglione, D.; Roller-Prado, A.; Pace, V. Highly chemoselective homologative assembly of the α-substituted methylsulfinamide motif from N-sulfinylamines. Chem. Commun. 2023, 59, 11065–11068. [Google Scholar] [CrossRef]
- Castoldi, L.; Pace, V. Easy as one, two, three. Nat. Chem. 2018, 10, 1081–1082. [Google Scholar] [CrossRef]
- Miele, M.; Castoldi, L.; Beccalli, E.; Pace, V. Ruppert-Prakash Reagent (TMSCF3)-Catalyzed Chemoselective Esterification of Weinreb Amides. Adv. Synth. Catal. 2024, 366, 2277–2284. [Google Scholar] [CrossRef]
- Blangetti, M.; de la Vega-Hernández, K.; Miele, M.; Pace, V. Transition-Metal-Free Reactions of Amides by Tetrahedral Intermediates. In Amide Bond Activation; Wiley: Hoboken, NJ, USA, 2022; pp. 101–156. [Google Scholar] [CrossRef]
- Pace, V.; Murgia, I.; Westermayer, S.; Langer, T.; Holzer, W. Highly efficient synthesis of functionalized α-oxyketones via Weinreb amides homologation with α-oxygenated organolithiums. Chem. Commun. 2016, 52, 7584–7587. [Google Scholar] [CrossRef] [PubMed]
- Senatore, R.; Malik, M.; Touqeer, S.; Listro, R.; Collina, S.; Holzer, W.; Pace, V. Straightforward and direct access to β-seleno- amines and sulfonylamides via the controlled addition of phenylselenomethyllithium (LiCH2SePh) to imines. Tetrahedron 2020, 76, 131220. [Google Scholar] [CrossRef]
- de la Vega-Hernández, K.; Senatore, R.; Miele, M.; Urban, E.; Holzer, W.; Pace, V. Chemoselective reduction of isothiocyanates to thioformamides mediated by the Schwartz reagent. Org. Biomol. Chem. 2019, 17, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Miele, M.; D’Orsi, R.; Sridharan, V.; Holzer, W.; Pace, V. Highly chemoselective difluoromethylative homologation of iso(thio)cyanates: Expeditious access to unprecedented α,α-difluoro(thio)amides. Chem. Commun. 2019, 55, 12960–12963. [Google Scholar] [CrossRef]
- Miele, M.; Castoldi, L.; Roller-Prado, A.; Pisano, L.; Pace, V. Chemoselective Synthesis of Cyanoformamides from Isocyanates and a Highly Reactive Nitrile Anion Reservoir. Eur. J. Org. Chem. 2024, 27, e202400619. [Google Scholar] [CrossRef]
- Monticelli, S.; Rui, M.; Castoldi, L.; Missere, G.; Pace, V. A practical guide for using lithium halocarbenoids in homologation reactions. Monatshefte Für Chem.-Chem. Mon. 2018, 149, 1285–1291. [Google Scholar] [CrossRef]
- Candeias, N.R.; Paterna, R.; Gois, P.M.P. Homologation Reaction of Ketones with Diazo Compounds. Chem. Rev. 2016, 116, 2937–2981. [Google Scholar] [CrossRef]
- Ye, T.; McKervey, M.A. Organic Synthesis with .alpha.-Diazo Carbonyl Compounds. Chem. Rev. 1994, 94, 1091–1160. [Google Scholar] [CrossRef]
- Wenkert, E.; McPherson, C.A. Condensations of acyldiazomethanes with aldehydes, ketones, and their derivatives. J. Am. Chem. Soc. 1972, 94, 8084–8090. [Google Scholar] [CrossRef]
- Pelicciari, R.; Castagnino, E.; Fringuelli, R.; Corsano, S. The preparation of acylacetylenic derivatives of α-cyclocitral on route to physiologically active terpenes. Tetrahedron Lett. 1979, 20, 481–484. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Mickus, D.E. Synthesis of ent-cholesterol, the unnatural enantiomer. J. Org. Chem. 1992, 57, 2732–2736. [Google Scholar] [CrossRef]
- Holmquist, C.R.; Roskamp, E.J. The conversion of olefins to β-keto esters: Ozonolysis of olefins followed by in situ reduction with tin(II) chloride in the presence of ethyl diazoacetate. Tetrahedron Lett. 1990, 31, 4991–4994. [Google Scholar] [CrossRef]
- Holmquist, C.R.; Roskamp, E.J. A selective method for the direct conversion of aldehydes into .beta.-keto esters with ethyl diazoacetate catalyzed by tin(II) chloride. J. Org. Chem. 1989, 54, 3258–3260. [Google Scholar] [CrossRef]
- Claisen, L.; Claparède, A. Condensationen von Ketonen mit Aldehyden. Berichte Der Dtsch. Chem. Ges. 1881, 14, 2460–2468. [Google Scholar] [CrossRef]
- Benetti, S.; Romagnoli, R.; Risi, C.D.; Spalluto, G.; Zanirato, V. MASTERING BETA -KETO ESTERS. Chem. Rev. 1995, 95, 1065–1114. [Google Scholar] [CrossRef]
- de Fátima, Â.; Braga, T.C.; Neto, L.d.S.; Terra, B.S.; Oliveira, B.G.F.; da Silva, D.L.; Modolo, L.V. A mini-review on Biginelli adducts with notable pharmacological properties. J. Adv. Res. 2015, 6, 363–373. [Google Scholar] [CrossRef]
- Attanasi, O.A.; Filippone, P.; Fiorucci, C.; Foresti, E.; Mantellini, F. Reaction of Some 1,2-Diaza-1,3-butadienes with Activated Methine Compounds. A Straightforward Entry to 1,4-Dihydropyridazine, Pyridazine, and 4,5(4H,5H)-Cyclopropylpyrazole Derivatives. J. Org. Chem. 1998, 63, 9880–9887. [Google Scholar] [CrossRef]
- Rutqvist, J.; Rinaldi, A.P.; Cappa, F.; Moridis, G.J. Modeling of fault activation and seismicity by injection directly into a fault zone associated with hydraulic fracturing of shale-gas reservoirs. J. Pet. Sci. Eng. 2015, 127, 377–386. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R.; Ley, S.V.; Nikbin, N. An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals. Beilstein J. Org. Chem. 2011, 7, 442–495. [Google Scholar] [CrossRef]
- LeBlanc, A.; Cuperlovic-Culf, M.; Morin, P.J.; Touaibia, M. Structurally Related Edaravone Analogues: Synthesis, Antiradical, Antioxidant, and Copper-Chelating Properties. CNS Neurol. Disord. Drug Targets 2019, 18, 779–790. [Google Scholar] [CrossRef]
- Bretschneider, T.; Zocher, G.; Unger, M.; Scherlach, K.; Stehle, T.; Hertweck, C. A ketosynthase homolog uses malonyl units to form esters in cervimycin biosynthesis. Nat. Chem. Biol. 2011, 8, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Bingham, S.J.; Tyman, J.H.P. The synthesis of kermesic acid and isokermesic acid derivatives and of related dihydroxyanthraquinones. J. Chem. Soc.-Perkin Trans. 1 1997, 3637–3642. [Google Scholar] [CrossRef]
- Arndt, F.; Eistert, B. Ein Verfahren zur Überführung von Carbonsäuren in ihre höheren Homologen bzw. deren Derivate. Berichte Der Dtsch. Chem. Ges. (A B Ser.) 1935, 68, 200–208. [Google Scholar] [CrossRef]
- Pace, V.; Verniest, G.; Sinisterra, J.V.; Alcántara, A.R.; De Kimpe, N.J. Improved Arndt- Eistert synthesis of α-diazoketones requiring minimal diazomethane in the presence of calcium oxide as acid scavenger. Org. Chem. 2010, 75, 5760–5763. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, L.; Ielo, L.; Holzer, W.; Giester, G.; Roller, A.; Pace, V.J. α-Arylamino diazoketones: Diazomethane-loading controlled synthesis, spectroscopic investigations, and structural X-ray analysis. Org. Chem. 2018, 83, 4336–4347. [Google Scholar] [CrossRef] [PubMed]
- Buchner, E.; Curtius, T. Synthese von Ketonsäureäthern aus Aldehyden und Diazoessigäther. Berichte Der Dtsch. Chem. Ges. 1885, 18, 2371–2377. [Google Scholar] [CrossRef]
- Schlotterbeck, F. Umwandlung von Aldehyden in Ketone durch Diazomethan. Berichte Der Dtsch. Chem. Ges. 1907, 40, 479–483. [Google Scholar] [CrossRef]
- Meyer, H. Über die Einwirkung von Diazomethan auf Aldehydsäuren und Aldehyde. Monatshefte Für Chem. Und Verwandte Teile Anderer Wiss. 1905, 26, 1295–1301. [Google Scholar] [CrossRef]
- Smith, P.A.S.; Baer, D.R. The Demjanov and Tiffeneau-Demjanov Ring Expansions. In Organic Reactions; Wiley: Hoboken, NJ, USA, 1960; pp. 157–188. [Google Scholar] [CrossRef]
- Kohlbacher, S.M.; Ionasz, V.-S.; Ielo, L.; Pace, V. The synthetic versatility of the Tiffeneau–Demjanov chemistry in homologation tactics. Monatsh. Chem. 2019, 150, 2011–2019. [Google Scholar] [CrossRef]
- Zeng, L.; Lai, Z.; Cui, S. One-Pot Reaction of Carboxylic Acids and Ynol Ethers for The Synthesis of β-Keto Esters. J. Org. Chem. 2018, 83, 14834–14841. [Google Scholar] [CrossRef]
- Li, W.; Wang, J.; Hu, X.; Shen, K.; Wang, W.; Chu, Y.; Lin, L.; Liu, X.; Feng, X. Catalytic Asymmetric Roskamp Reaction of α-Alkyl-α-diazoesters with Aromatic Aldehydes: Highly Enantioselective Synthesis of α-Alkyl-β-keto Esters. J. Am. Chem. Soc. 2010, 132, 8532–8533. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, K.; Chand, D.K. Molybdenum(VI) Dichloride Dioxide Catalyzed Synthesis of β-Keto Esters by C-H Insertion of Ethyl Diazoacetate into Aldehydes. Synthesis 2008, 2008, 1685–1687. [Google Scholar] [CrossRef]
- More, K.R.; Mali, R.S. Facile synthesis of naturally occurring nor-lignans (±)-Machicendiol and its analogs. Synth. Commun. 2017, 47, 788–792. [Google Scholar] [CrossRef]
- Booker, J.E.M.; Boto, A.; Churchill, G.H.; Green, C.P.; Ling, M.; Meek, G.; Prabhakaran, J.; Sinclair, D.; Blake, A.J.; Pattenden, G. Approaches to the quaternary stereocentre and to the heterocyclic core in diazonamide A using the Heck reaction and related coupling reactions. Org. Biomol. Chem. 2006, 4, 4193–4205. [Google Scholar] [CrossRef]
- Rahaman, M.; Ali, M.S.; Jahan, K.; Hinz, D.; Belayet, J.B.; Majinski, R.; Hossain, M.M. Synthetic Scope of Brønsted Acid-Catalyzed Reactions of Carbonyl Compounds and Ethyl Diazoacetate. J. Org. Chem. 2021, 86, 6138–6147. [Google Scholar] [CrossRef] [PubMed]
- Balaji, B.S.; Chanda, B.M. Simple and high yielding syntheses of β-keto esters catalysed by zeolites. Tetrahedron 1998, 54, 13237–13252. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Pandit, S.S.; Sadavarte, V.S. Montmorillonite K-10 catalyzed synthesis of β-keto esters: Condensation of ethyl diazoacetate with aldehydes under mild conditions. Green Chem. 2001, 3, 247–249. [Google Scholar] [CrossRef]
- Pandey, R.K.; Deshmukh, A.N.; Kumar, P. Synthesis of β-Keto Esters Promoted by Yttria-Zirconia Based Lewis Acid Catalyst. Synth. Commun. 2004, 34, 1117–1123. [Google Scholar] [CrossRef]
- Murata, H.; Ishitani, H.; Iwamoto, M. Selective synthesis of α-substituted β-keto esters from aldehydes and diazoesters on mesoporous silica catalysts. Tetrahedron Lett. 2008, 49, 4788–4791. [Google Scholar] [CrossRef]
- Baburajan, P.; Elango, K.P. One pot direct synthesis of β-ketoesters via carbonylation of aryl halides using cobalt carbonyl. Tetrahedron Lett. 2014, 55, 3525–3528. [Google Scholar] [CrossRef]
- Krishna Reddy, S.M.; Prasanna Kumari, S.; Selva Ganesan, S. Palladium catalysed hydrolysis-free arylation of aliphatic nitriles for the synthesis of 4-arylquinolin-2-one/pyrazolone derivatives. Tetrahedron Lett. 2021, 79, 153296. [Google Scholar] [CrossRef]
- Kandasamy, M.; Ishitani, H.; Kobayashi, S. Continuous-Flow Synthesis of β-Ketoesters and Successive Reactions in One-Flow using Heterogeneous Catalysis. Adv. Synth. Catal. 2022, 364, 3389–3395. [Google Scholar] [CrossRef]
- Yin, J.; Wang, C.; Kong, L.; Cai, S.; Gao, S. Asymmetric synthesis and biosynthetic implications of (+)-fusarisetin A. Angew. Chem. Int. Ed. Engl. 2012, 51, 7786–7789. [Google Scholar] [CrossRef]
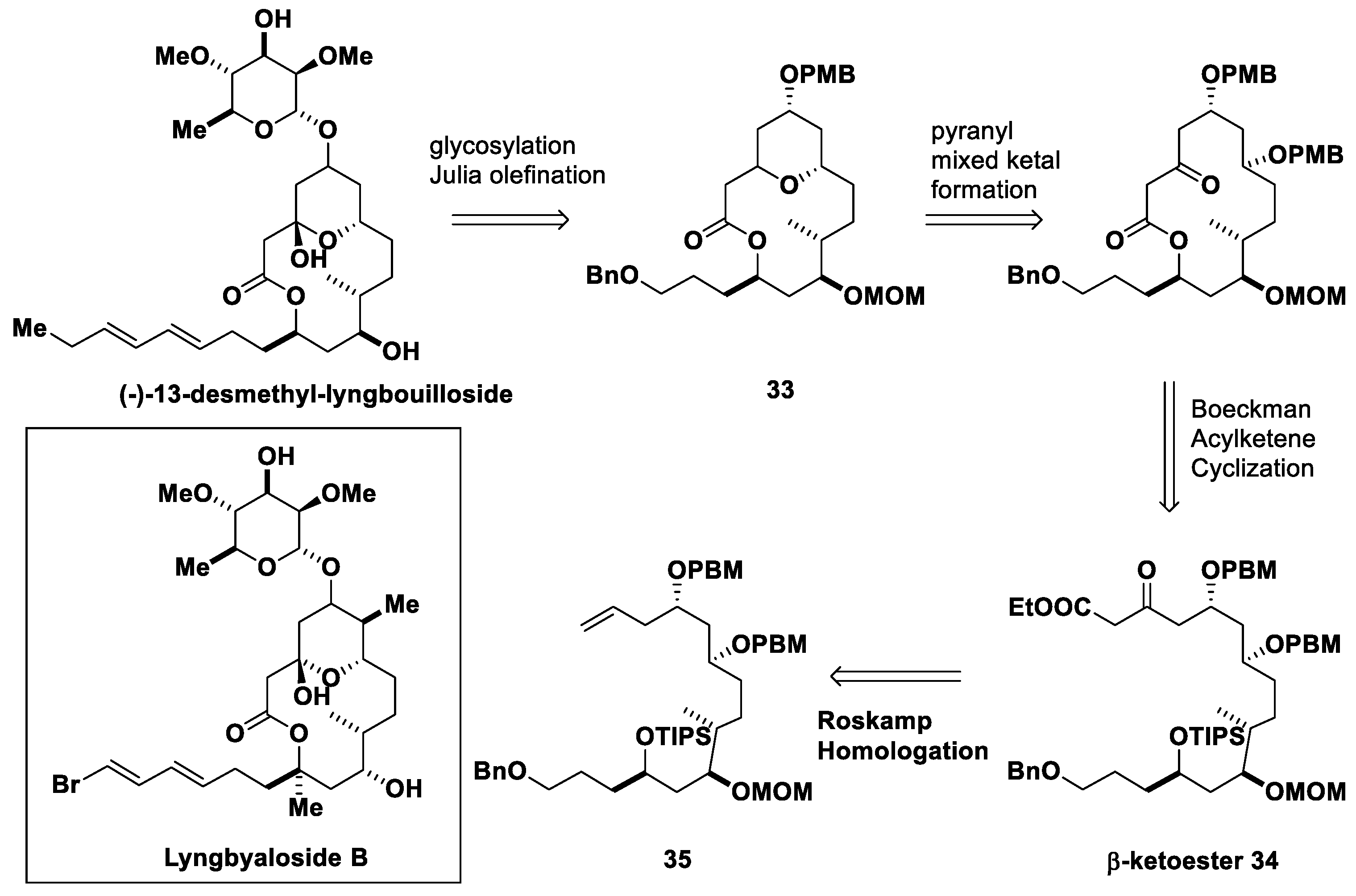
- Chegondi, R.; Hanson, P.R. Synthetic Studies to Lyngbouilloside: A Phosphate Tether-Mediated Synthesis of the Macrolactone Core. Tetrahedron Lett. 2015, 56, 3330–3333. [Google Scholar] [CrossRef]
- Ganguly, A.; Javed, S.; Bodugam, M.; Dissanayake, G.C.; Chegondi, R.; Hanson, P.R. Synthesis of the C1−C16 Polyol-Containing Macrolactone of 13-Desmethyl Lyngbouilloside, an Unnatural Analog of the Originally Assigned Structure of (−)-Lyngbouilloside. Isr. J. Chem. 2021, 61, 401–408. [Google Scholar] [CrossRef]
- Hashimoto, T.; Miyamoto, H.; Naganawa, Y.; Maruoka, K. Stereoselective Synthesis of α-Alkyl-β-keto Imides via Asymmetric Redox C−C Bond Formation between α-Alkyl-α-diazocarbonyl Compounds and Aldehydes. J. Am. Chem. Soc. 2009, 131, 11280–11281. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Kang, B.C.; Hwang, G.-S.; Ryu, D.H. Enantioselective Synthesis of α-Alkyl-β-ketoesters: Asymmetric Roskamp Reaction Catalyzed by an Oxazaborolidinium Ion. Angew. Chem. Int. Ed. 2012, 51, 8322–8325. [Google Scholar] [CrossRef]
- Corey, E.J. Enantioselective Catalysis Based on Cationic Oxazaborolidines. Angew. Chem. Int. Ed. 2009, 48, 2100–2117. [Google Scholar] [CrossRef]
- Gao, L.; Hwang, G.-S.; Ryu, D.H. Oxazaborolidinium Ion-Catalyzed Cyclopropanation of α-Substituted Acroleins: Enantioselective Synthesis of Cyclopropanes Bearing Two Chiral Quaternary Centers. J. Am. Chem. Soc. 2011, 133, 20708–20711. [Google Scholar] [CrossRef]
- Senapati, B.K.; Hwang, G.S.; Lee, S.; Ryu, D.H. Enantioselective synthesis of beta-iodo Morita-Baylis-Hillman esters by a catalytic asymmetric three-component coupling reaction. Angew. Chem. Int. Ed. Engl. 2009, 48, 4398–4401. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.H.; Corey, E.J. Enantioselective Cyanosilylation of Ketones Catalyzed by a Chiral Oxazaborolidinium Ion. J. Am. Chem. Soc. 2005, 127, 5384–5387. [Google Scholar] [CrossRef]
- Senatore, R.; Ielo, L.; Monticelli, S.; Castoldi, L.; Pace, V. Weinreb Amides as Privileged Acylating Agents for Accessing α-Substituted Ketones. Synthesis 2019, 51, 2792–2808. [Google Scholar] [CrossRef]
- Miele, M.; Castiglione, D.; Holzer, W.; Castoldi, L.; Pace, V. Chemoselective homologative preparation of trisubstituted alkenyl halides from carbonyls and carbenoids. Chem. Commun. 2025, 61, 1180–1183. [Google Scholar] [CrossRef]
- Miele, M.; Citarella, A.; Micale, N.; Holzer, W.; Pace, V. Direct and Chemoselective Synthesis of Tertiary Difluoroketones via Weinreb Amide Homologation with a CHF2-Carbene Equivalent. Org. Lett. 2019, 21, 8261–8265. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, S.; Holzer, W.; Langer, T.; Roller, A.; Olofsson, B.; Pace, V. Sustainable Asymmetric Organolithium Chemistry: Enantio- and Chemoselective Acylations through Recycling of Solvent, Sparteine, and Weinreb “Amine”. ChemSusChem 2019, 12, 1147–1154. [Google Scholar] [CrossRef]
- Miele, M.; Pillari, V.; Pace, V.; Alcántara, A.R.; de Gonzalo, G. Application of biobased solvents in asymmetric catalysis. Molecules 2022, 27, 6701. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Baek, E.H.; Hwang, G.-S.; Ryu, D.H. Enantioselective Synthesis of syn-α-Aryl-β-hydroxy Weinreb Amides: Catalytic Asymmetric Roskamp Reaction of α-Aryl Diazo Weinreb Amides. Org. Lett. 2015, 17, 4746–4749. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Hedley, S.J.; Bohall, B.R. Asymmetric Intermolecular C−H Functionalization of Benzyl Silyl Ethers Mediated by Chiral Auxiliary-Based Aryldiazoacetates and Chiral Dirhodium Catalysts. J. Org. Chem. 2005, 70, 10737–10742. [Google Scholar] [CrossRef]
- Rossi, S.; Benaglia, M.; Cozzi, F.; Genoni, A.; Benincori, T. Organocatalytic Stereoselective Direct Aldol Reaction of Trifluoroethyl Thioesters. Adv. Synth. Catal. 2011, 353, 848–854. [Google Scholar] [CrossRef]
- Wang, H.; Li, G.; Engle, K.M.; Yu, J.-Q.; Davies, H.M.L. Sequential C–H Functionalization Reactions for the Enantioselective Synthesis of Highly Functionalized 2,3-Dihydrobenzofurans. J. Am. Chem. Soc. 2013, 135, 6774–6777. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kang, B.C.; Ryu, D.H. Catalytic Asymmetric Roskamp Reaction of Silyl Diazoalkane: Synthesis of Enantioenriched α-Silyl Ketone. Org. Lett. 2017, 19, 5936–5939. [Google Scholar] [CrossRef]
- Audubert, C.; Gamboa Marin, O.J.; Lebel, H. Batch and Continuous-Flow One-Pot Processes using Amine Diazotization to Produce Silylated Diazo Reagents. Angew. Chem. Int. Ed. Engl. 2017, 56, 6294–6297. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, X.-M.; Bordwell, F.G. .alpha.-Silyl Effects on the Acidities of Carbon Acids and the Homolytic Bond Dissociation Enthalpies of Their Acidic C-H Bonds. J. Am. Chem. Soc. 1995, 117, 602–606. [Google Scholar] [CrossRef]
- Showell, G.A.; Mills, J.S. Chemistry challenges in lead optimization: Silicon isosteres in drug discovery. Drug Discov. Today 2003, 8, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.I.; Rychnovsky, S.D. Total Synthesis of Filipin III. J. Am. Chem. Soc. 1997, 119, 12360–12361. [Google Scholar] [CrossRef]
- Herb, C.; Maier, M.E. A Formal Total Synthesis of the Salicylihalamides. J. Org. Chem. 2003, 68, 8129–8135. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Adams, D.J. Total Synthesis of (+)-Galbulimima Alkaloid 13 and (+)-Himgaline. J. Am. Chem. Soc. 2007, 129, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T.; Márquez, B.L.; Gerwick, W.H. Lyngbouilloside, a Novel Glycosidic Macrolide from the Marine Cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2002, 65, 925–928. [Google Scholar] [CrossRef]
- ElMarrouni, A.; Lebeuf, R.; Gebauer, J.; Heras, M.; Arseniyadis, S.; Cossy, J. Total Synthesis of Nominal Lyngbouilloside Aglycon. Org. Lett. 2012, 14, 314–317. [Google Scholar] [CrossRef]
- Sabitha, G.; Rammohan reddy, T.; Yadav, J.S.; Sirisha, K. Stereoselective synthesis of the C1–C8 and C9–C16 fragments of revised structure of (−)-lyngbouilloside. RSC Adv. 2014, 4, 3149–3152. [Google Scholar] [CrossRef]
- Fuwa, H.; Okuaki, Y.; Yamagata, N.; Sasaki, M. Total Synthesis, Stereochemical Reassignment, and Biological Evaluation of (−)-Lyngbyaloside B. Angew. Chem. Int. Ed. 2015, 54, 868–873. [Google Scholar] [CrossRef] [PubMed]



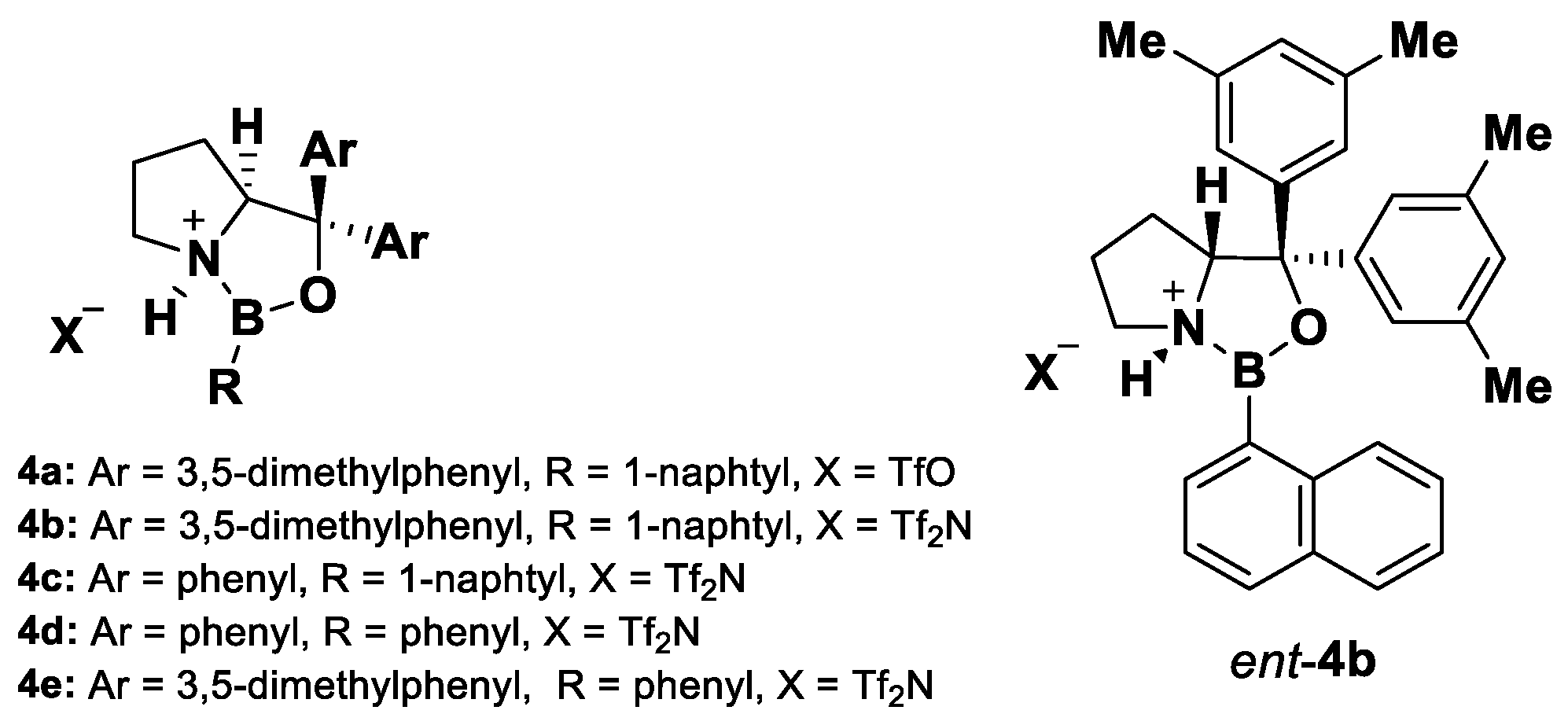










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
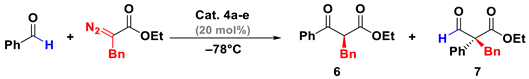
| Entry | Solvent | Cat. | Time [h] | 6/7 | %Yield | ee [%] |
|---|---|---|---|---|---|---|
| 1 | DCM | 4a | 1 | 90:10 | 82 | 87 |
| 2 | DCM | 4b | 1 | 89:11 | 80 | 90 |
| 3 | DCM | 4c | 1.5 | 82:18 | 77 | 82 |
| 4 | Propionitrile | 4c | 1 | 67:33 | 60 | 79 |
| 5 | Toluene | 4c | 1.5 | 91:9 | 87 | 88 |
| 6 | Toluene | 4d | 1 | 93:7 | 90 | 92 |
| 7 | Toluene | 4e | 2 | 94:6 | 92 | 95 |
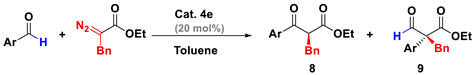
| Entry | Ar | T(°C) | Time [h] | 8/9 | %Yield | ee [%] |
|---|---|---|---|---|---|---|
| 1 | Ph | −78 | 2 | 94:6 | 92 | 95 |
| 2 | 4-MeC6H4 | −78 | 2 | 90:10 | 87 | 95 |
| 3 | 4-MeOC6H4 | −55 | 1 | 92:8 | 85 | 91 |
| 4 | 4-BrC6H4 | −78 | 1 | 94:6 | 91 | 94 |
| 5 | 4-CF3C6H4 | −95 | 1 | 89:11 | 82 | 96 |
| 6 | 3-OMe | −78 | 2 | 90:10 | 83 | 95 |
| 7 | 2-OMe | −55 | 5 | 76:24 | 69 | 80 |

| Entry | Cat. | X | Y | 11/12 | %Yield | ee [%] |
|---|---|---|---|---|---|---|
| 1 | 13a | OEt | Tf2N | 1:3 | 22 | - |
| 2 | 13a | t-BuO | Tf2N | 1:3 | 20 | - |
| 3 | 13a | NMe(OMe) | Tf2N | >20:1 | 47 | 80 |
| 4 | 13a | NMe(OMe) | TfO | >20:1 | 51 | 82 |
| 5 | 13b | NMe(OMe) | TfO | >20:1 | 92 | 89 |
| 6 | 13c | NMe(OMe) | TfO | >20:1 | 87 | 91 |
| 7 | 13d | NMe(OMe) | TfO | >20:1 | 90 | 91 |

| Entry | Cat. | R1 | 15/16 | %Yield | ee [%] |
|---|---|---|---|---|---|
| 1 | 17a | (CH3)2PhSi | 2:1 | 41 | −34 |
| 2 | 17b | (CH3)2PhSi | 6:1 | 40 | −50 |
| 3 | 17c | (CH3)2PhSi | 4:1 | 62 | 68 |
| 4 | 17d | (CH3)2PhSi | 5:1 | 68 | 81 |
| 5 | 17e | (CH3)2PhSi | 4:1 | 63 | 92 |
| 6 | 17f | (CH3)2PhSi | 9:1 | 73 | 92 |
| 7 | 17g | CH3Ph2Si | >20:1 | 79 | 98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miele, M.; Smajić, A.; Pace, V. The Versatility of the Roskamp Homologation in Synthesis. Molecules 2025, 30, 1192. https://doi.org/10.3390/molecules30061192
Miele M, Smajić A, Pace V. The Versatility of the Roskamp Homologation in Synthesis. Molecules. 2025; 30(6):1192. https://doi.org/10.3390/molecules30061192
Chicago/Turabian StyleMiele, Margherita, Aljoša Smajić, and Vittorio Pace. 2025. "The Versatility of the Roskamp Homologation in Synthesis" Molecules 30, no. 6: 1192. https://doi.org/10.3390/molecules30061192
APA StyleMiele, M., Smajić, A., & Pace, V. (2025). The Versatility of the Roskamp Homologation in Synthesis. Molecules, 30(6), 1192. https://doi.org/10.3390/molecules30061192