
Research Progress in Epoxidation of Light Small-Molecule Olefins

 and
and
Abstract
1. Introduction
2. Overview of Major Light Olefin Epoxidation Processes
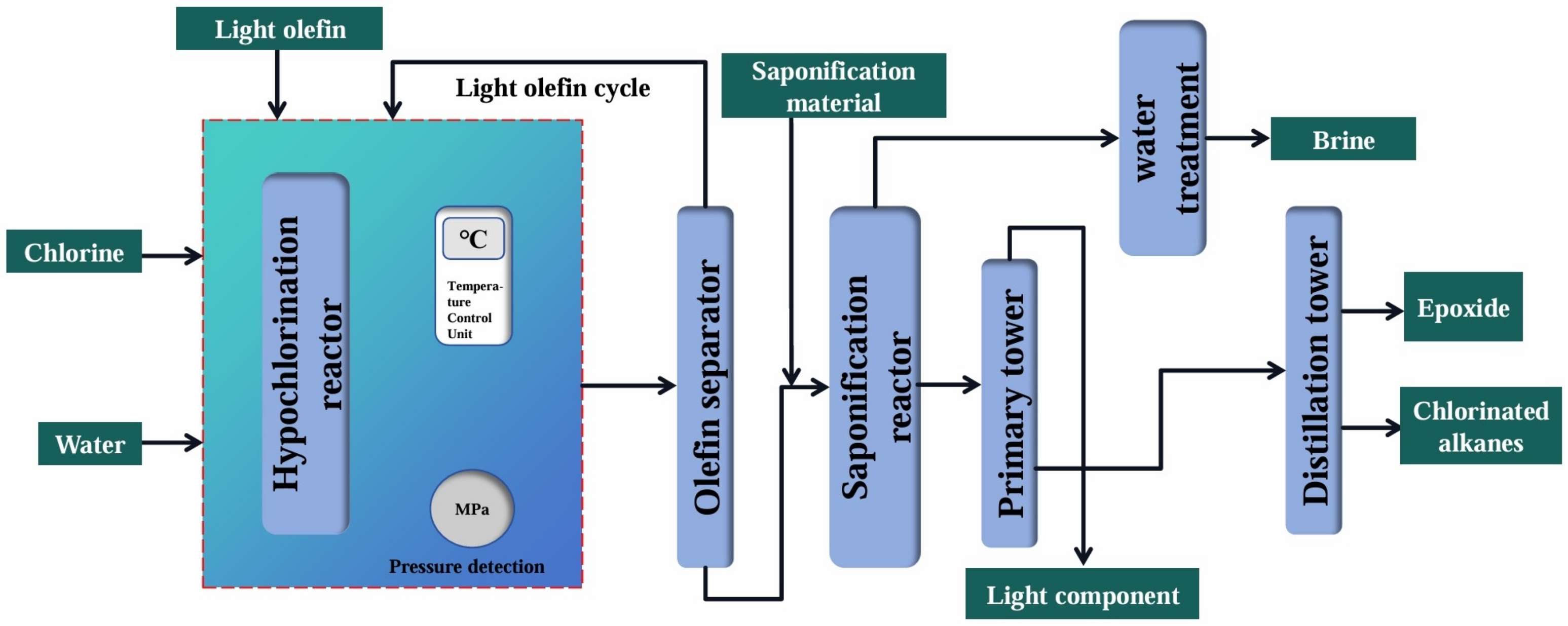
2.1. Chlorohydrin Method
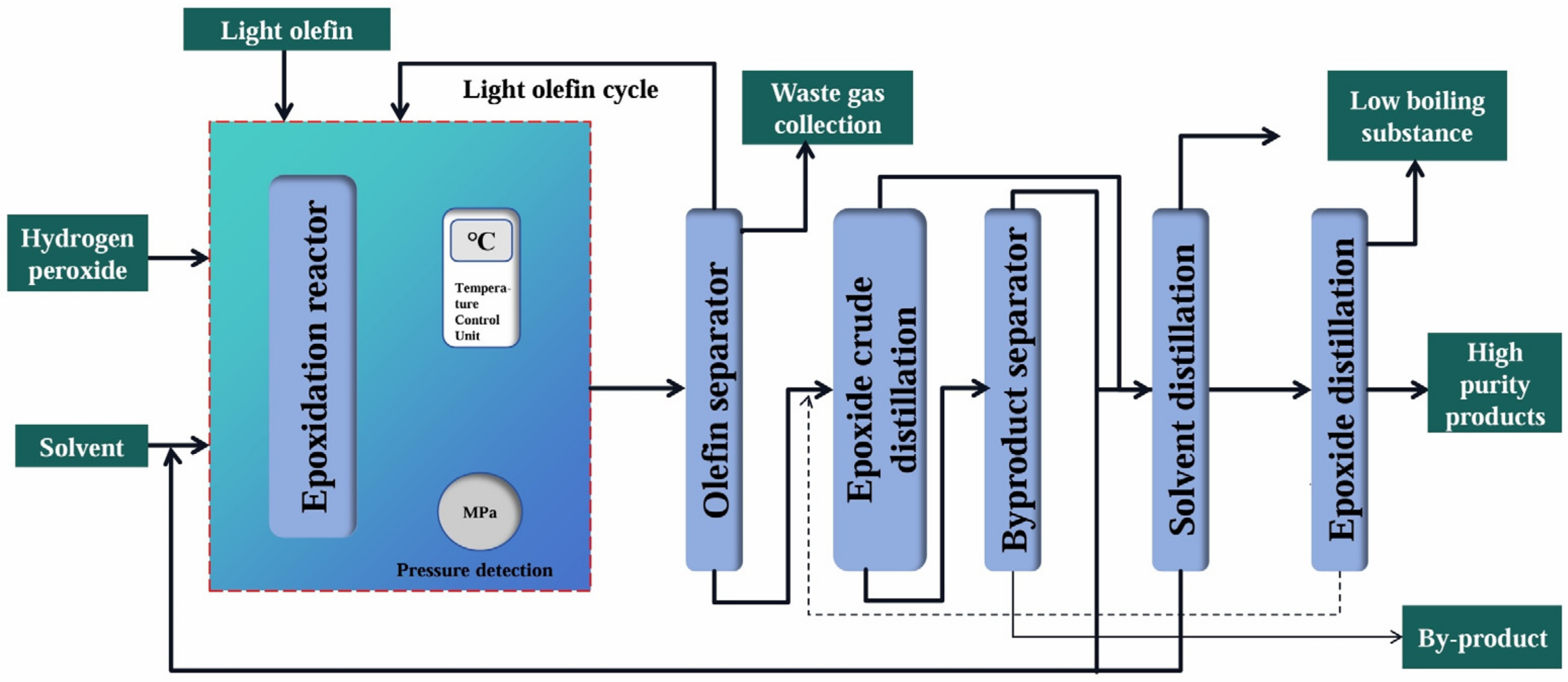
2.2. HPPO Method
2.3. Oxygen/Air Direct Oxidation Method
2.4. Co-Oxidation Method
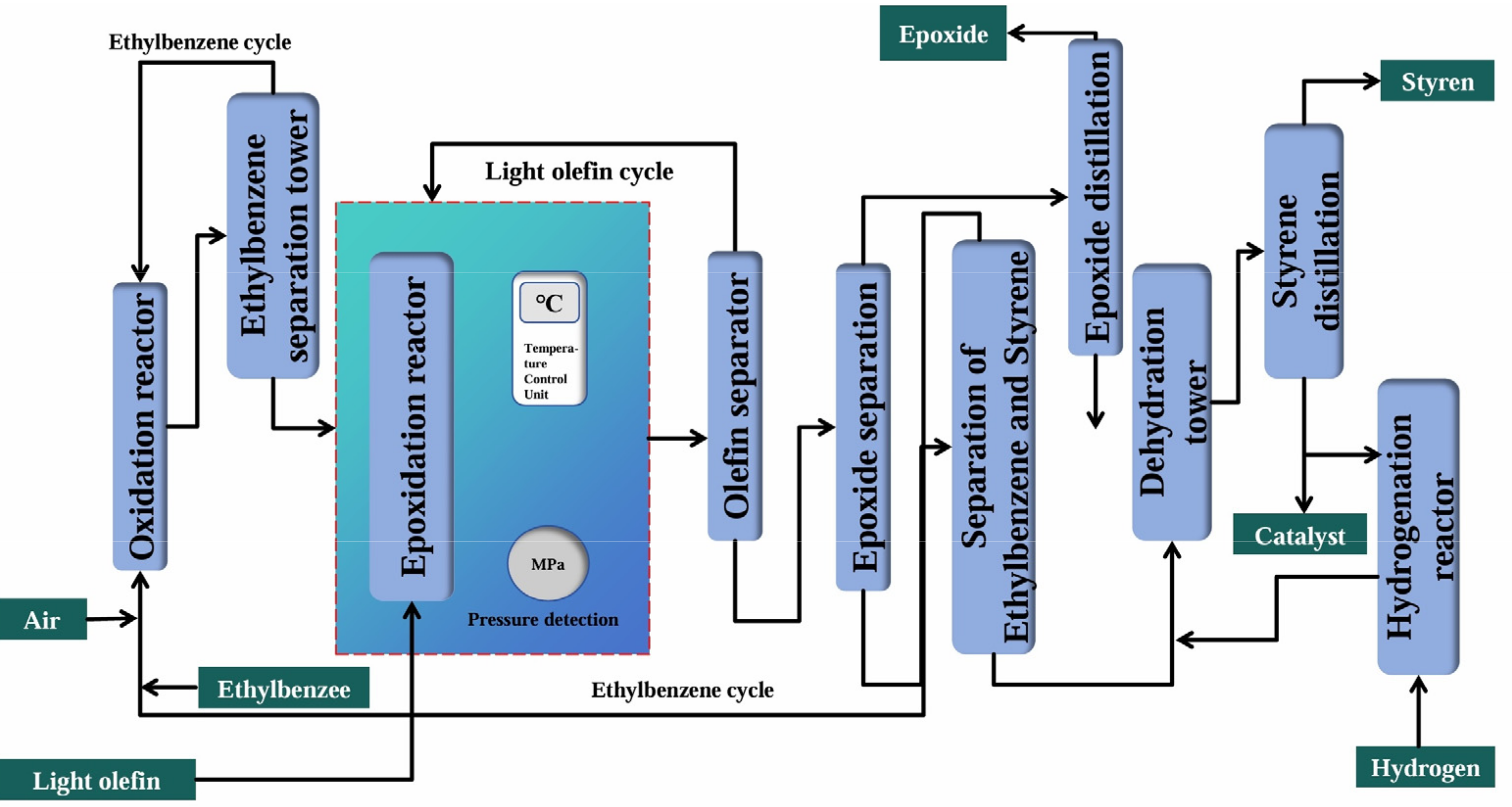
2.4.1. Ethylbenzene Co-Oxidation Method
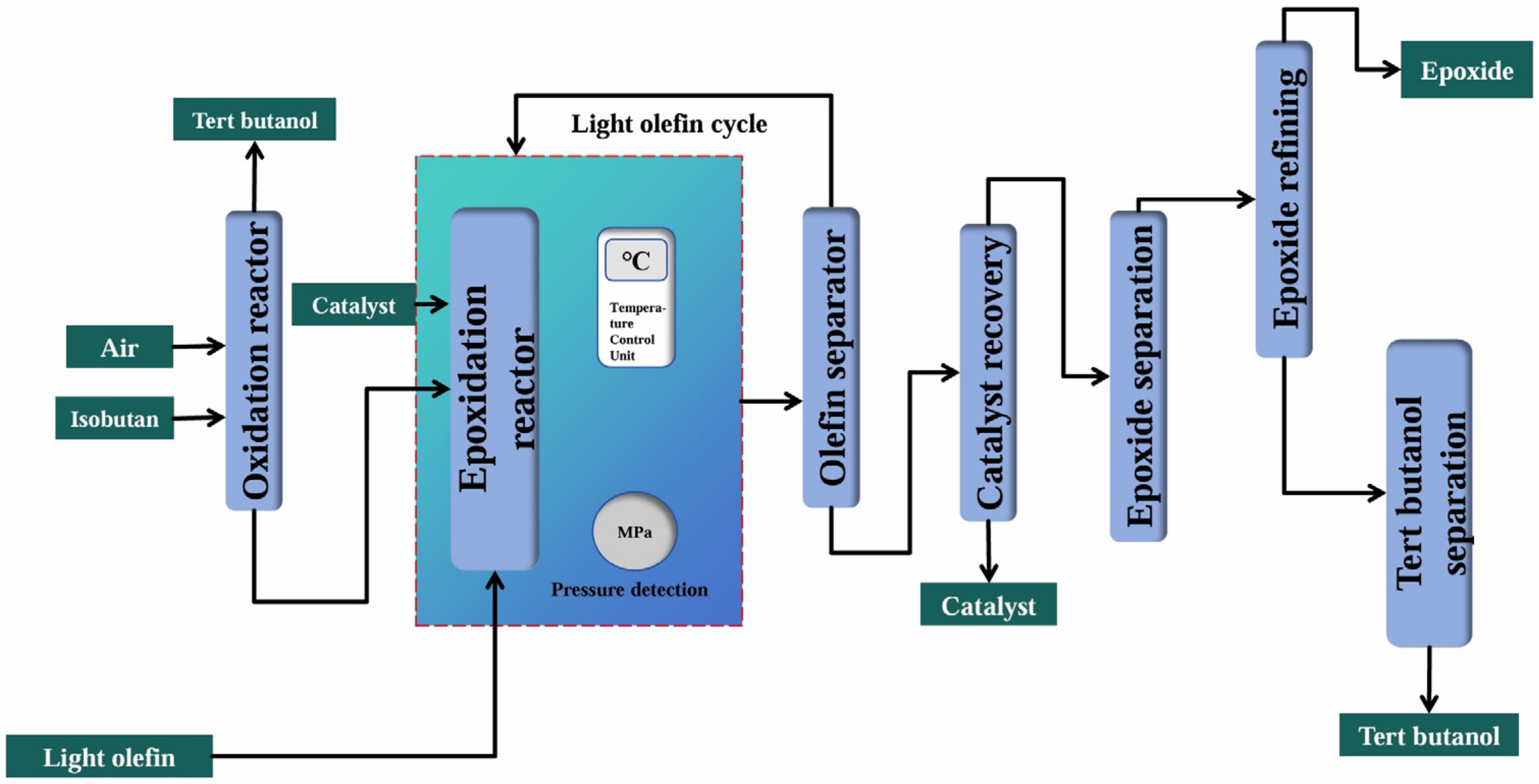
2.4.2. Isobutane Co-Oxidation Method
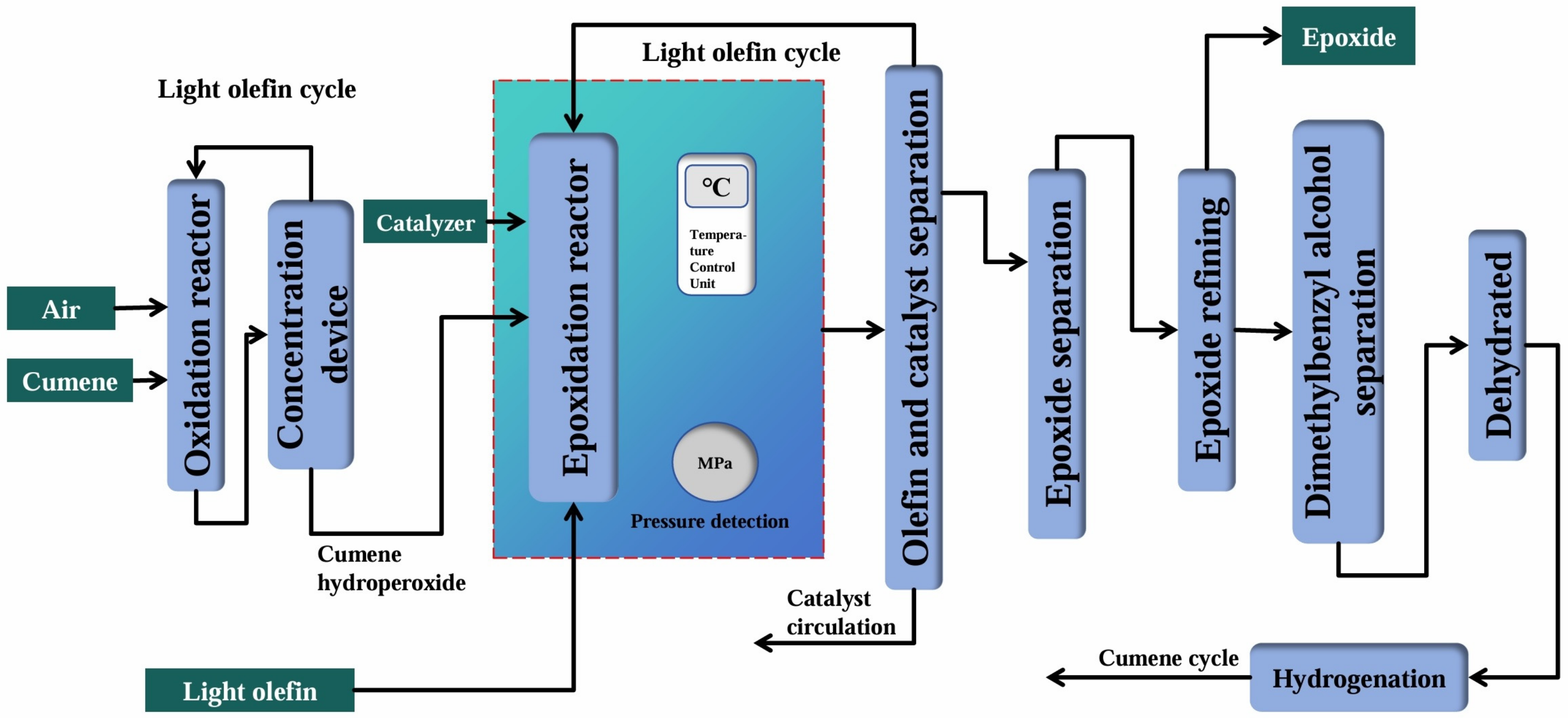
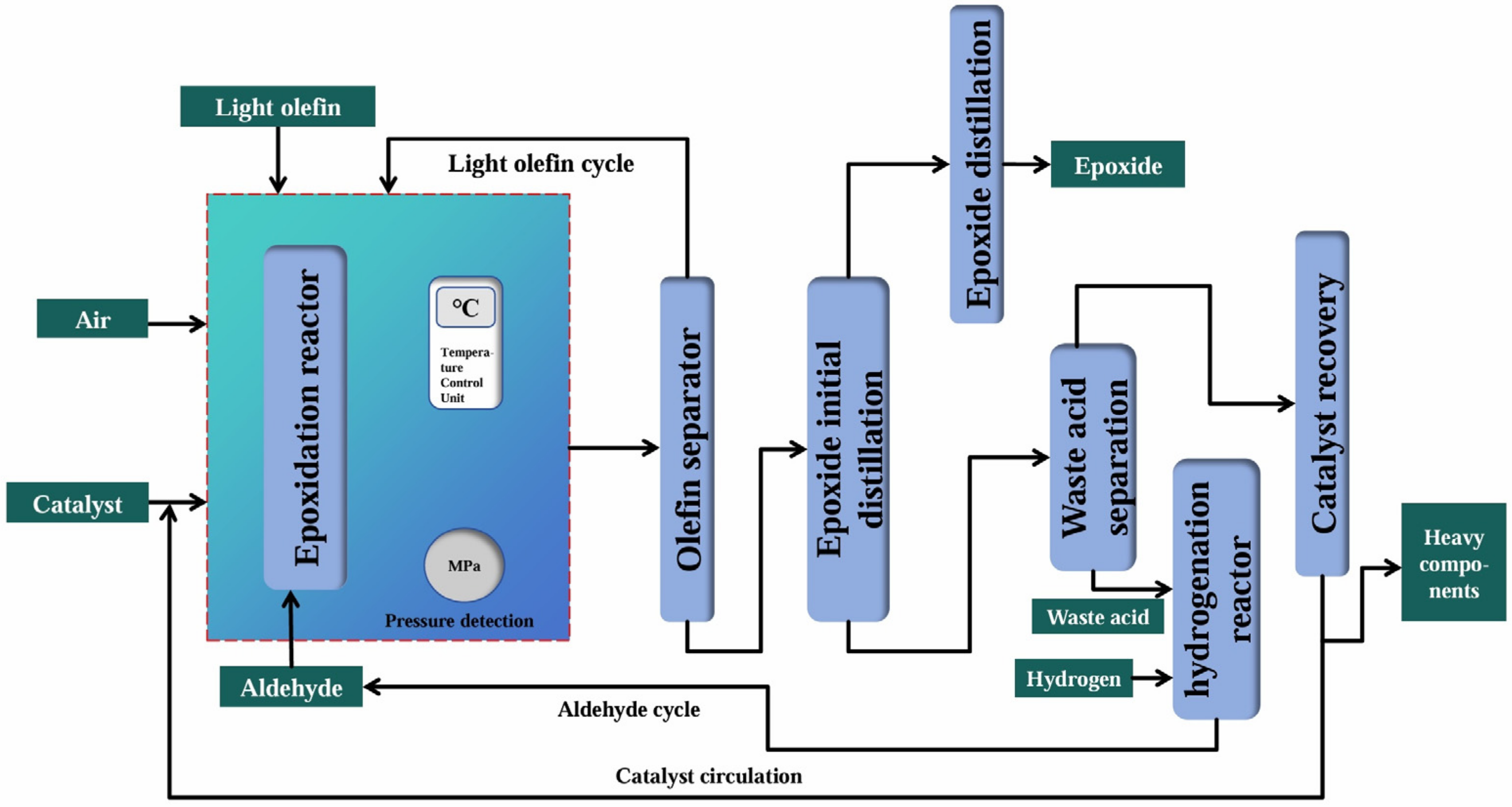
2.4.3. Cumene Co-Oxidation Method
2.5. Biomimetic Catalytic Oxidation Method
2.6. Application of Olefin Epoxidation Processes
2.6.1. Application of Light Olefin Epoxidation Processes
2.6.2. Application of Cyclic Olefin Epoxidation Processes
2.6.3. Application of Aromatic Olefin Epoxidation Processes
3. Overview of Catalysts for Light Olefin Epoxidation
3.1. Homogeneous Catalyst
3.1.1. Porphyrin Catalyst System
3.1.2. Schiff–Base-Based Metal Complexes
3.1.3. Other Metal Complex Catalysts
3.2. Heterogeneous Catalyst
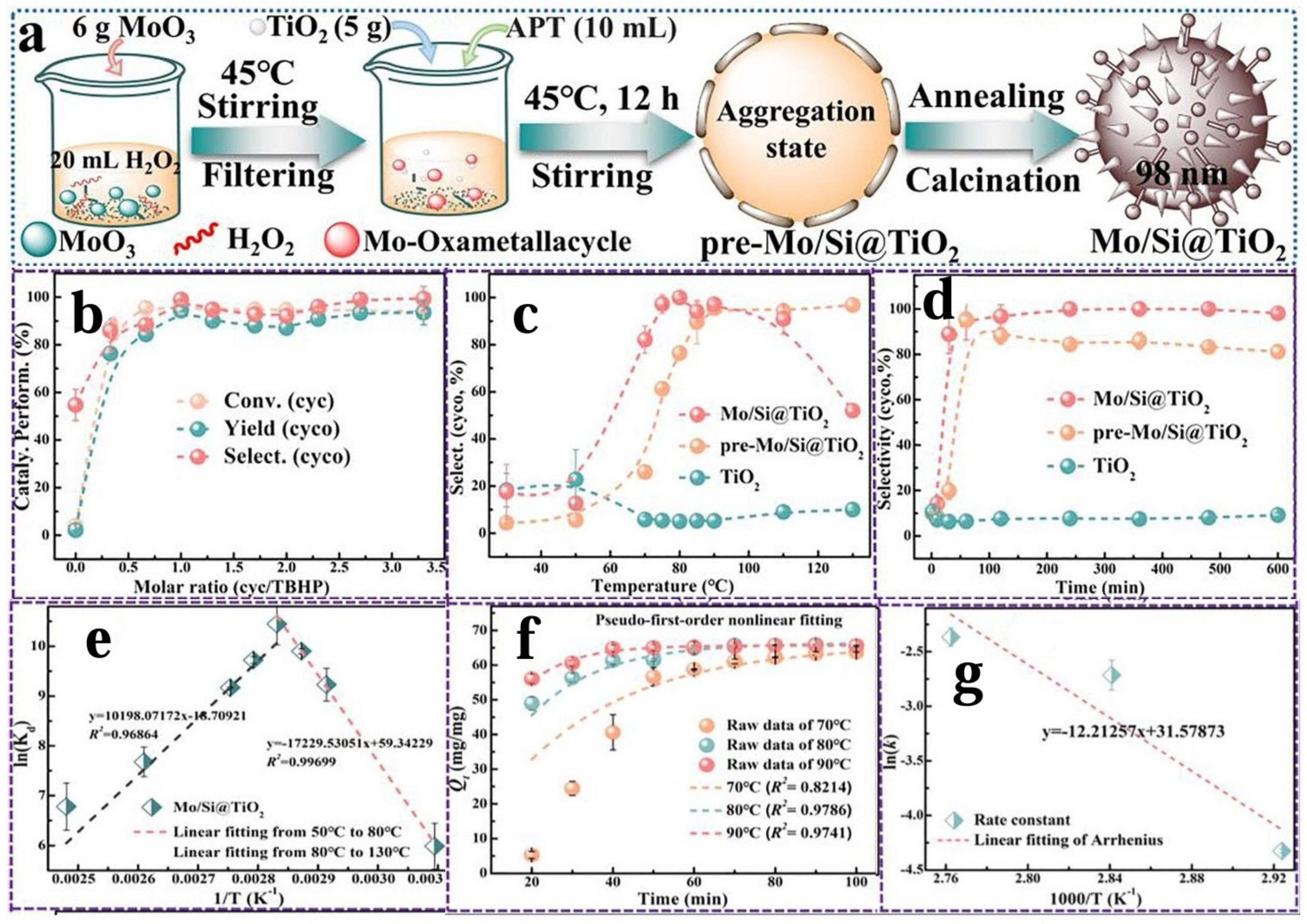
3.2.1. Supported Metal Nanocatalysts
Ag-Based Nanocatalysts
Au-Based Nanocatalysts
Cu-Based Nanocatalysts
Other Supported Metal Nanocatalysts
3.2.2. Other Heterogeneous Systems
4. Reaction Mechanism of Light Olefin Epoxidation
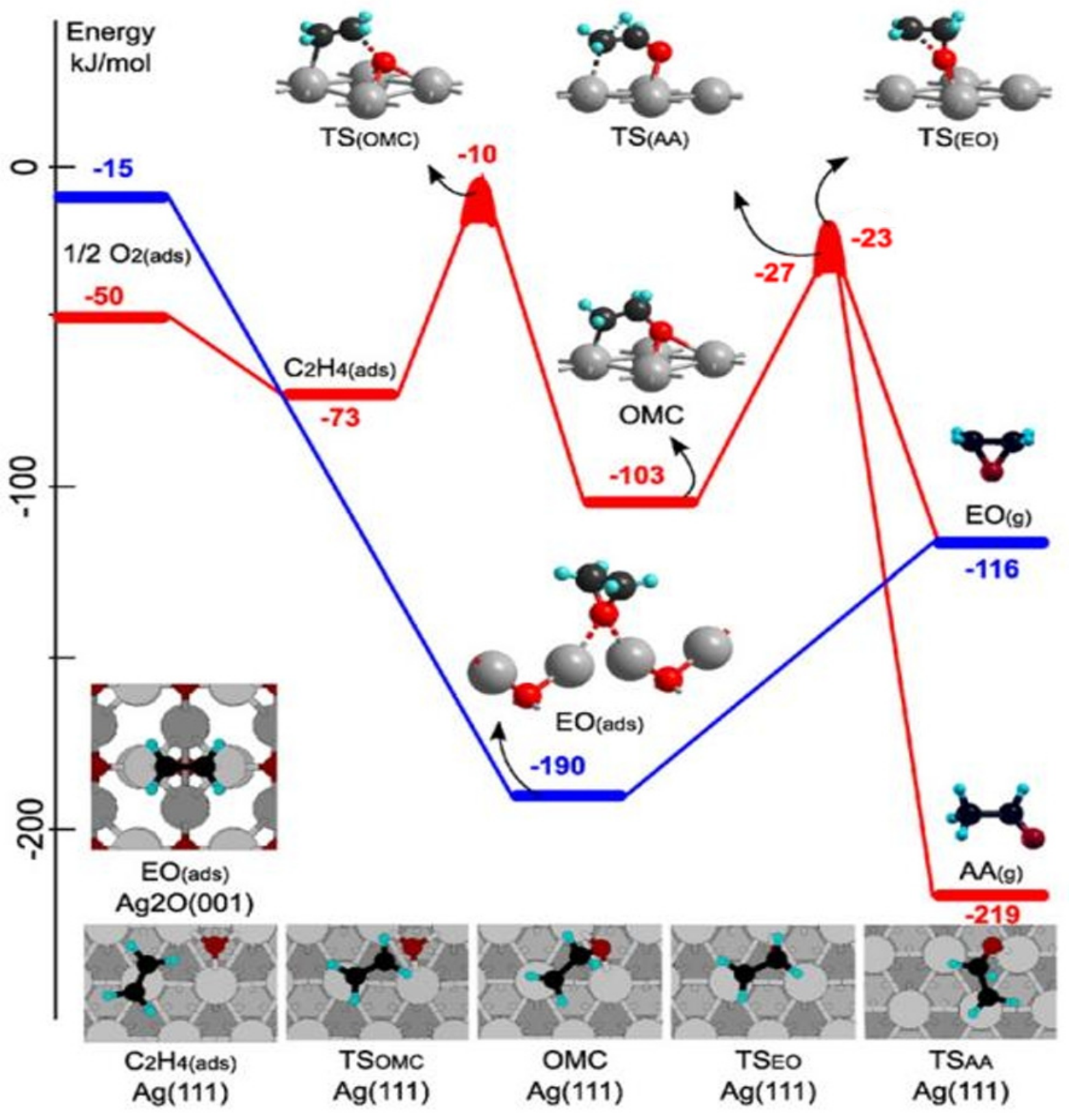
4.1. Ethylene Epoxidation Mechanism with Silver as the Catalyst (L-H Mechanism and E-R Mechanism)
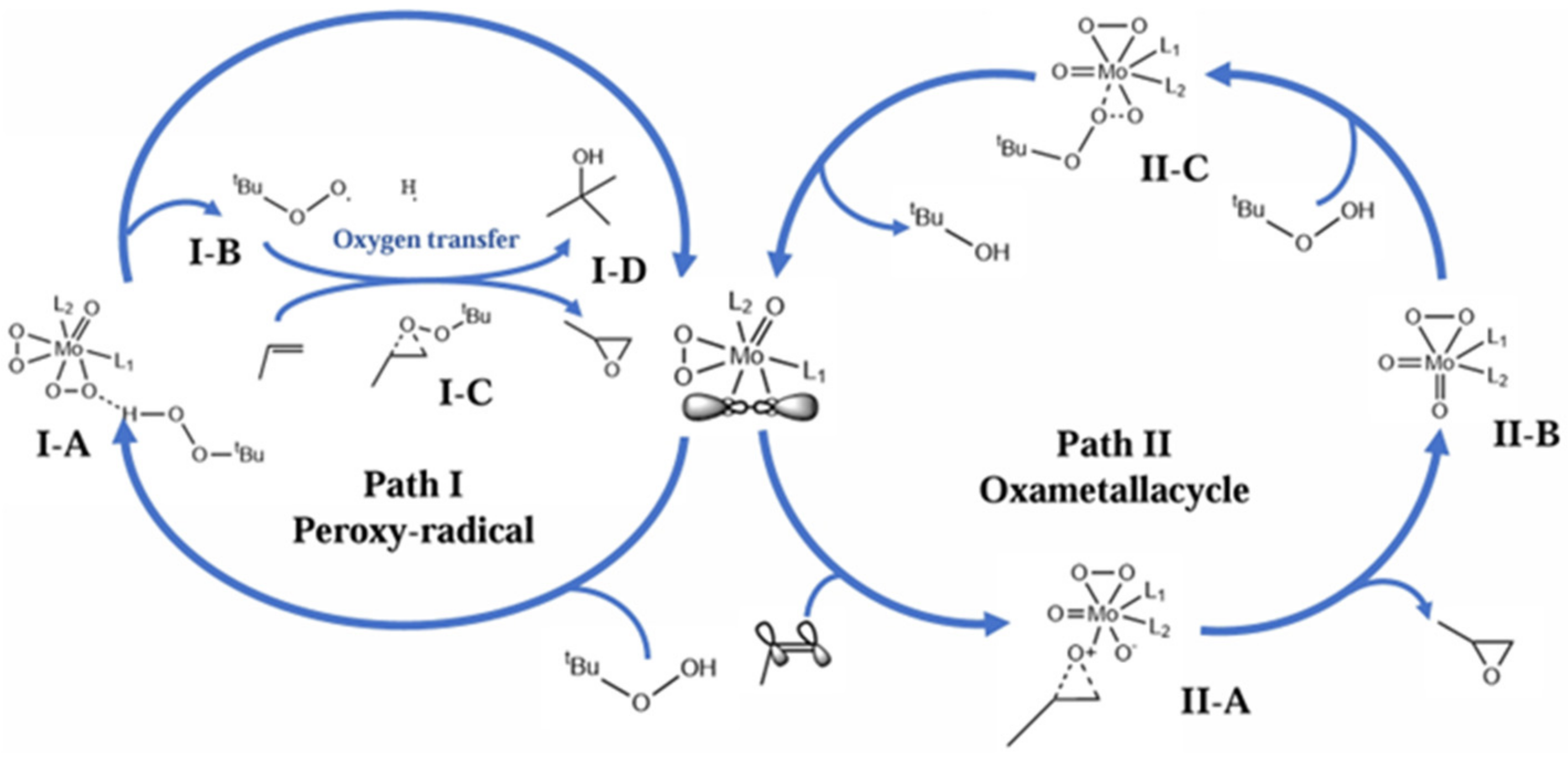
4.2. 2-Methylpropene Epoxidation Mechanism over Mo(O2)2@RT with TBHP as Oxidant (Free Radical Mechanism)
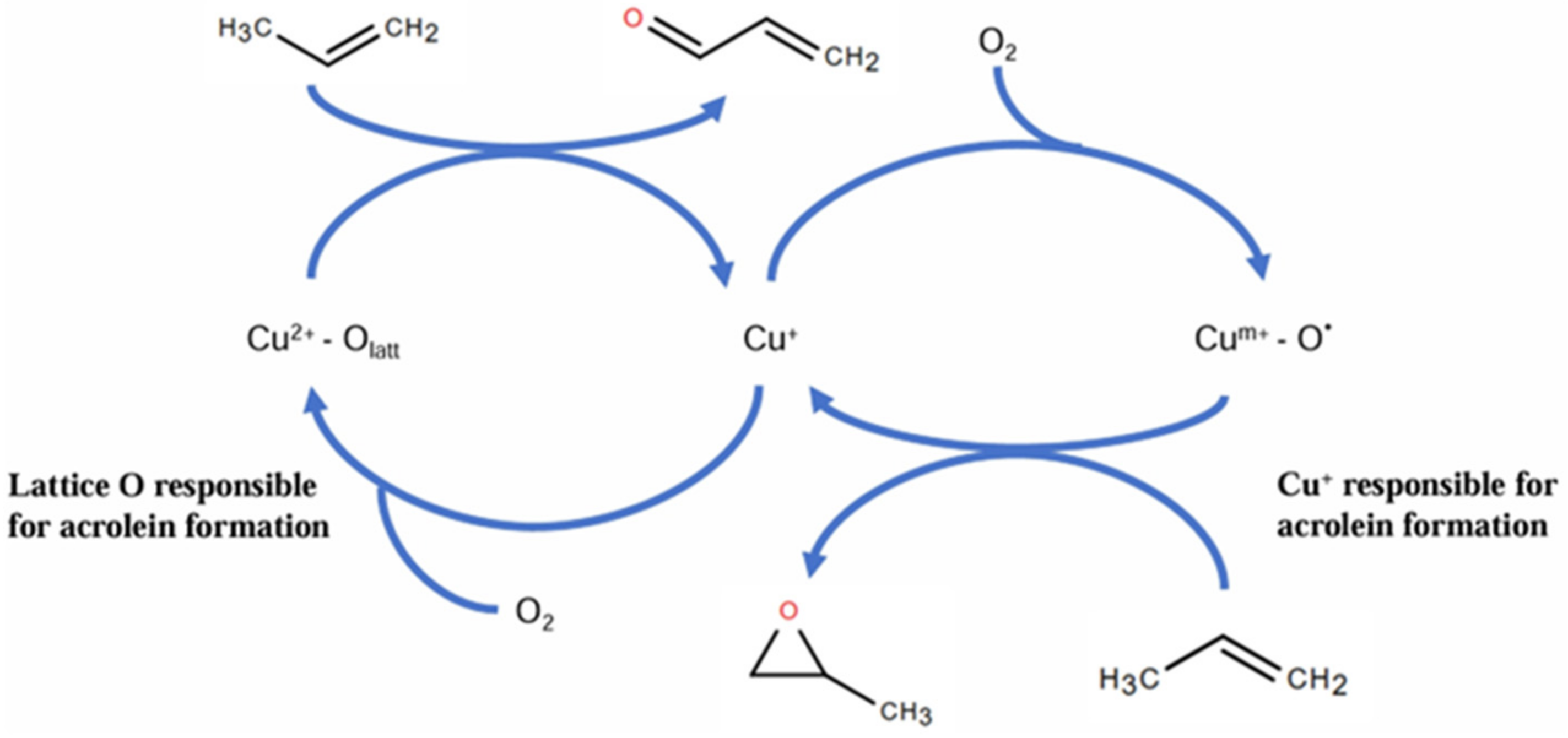
4.3. Propylene Epoxidation Mechanism over CuOx/SiO2 with O2 as Oxidant (High-Valence Metal Oxo Mechanism)
4.4. Propylene Epoxidation Mechanism over MoOO·DMF with TBHP as Oxidant (Free Radical and Peroxide Mechanisms)
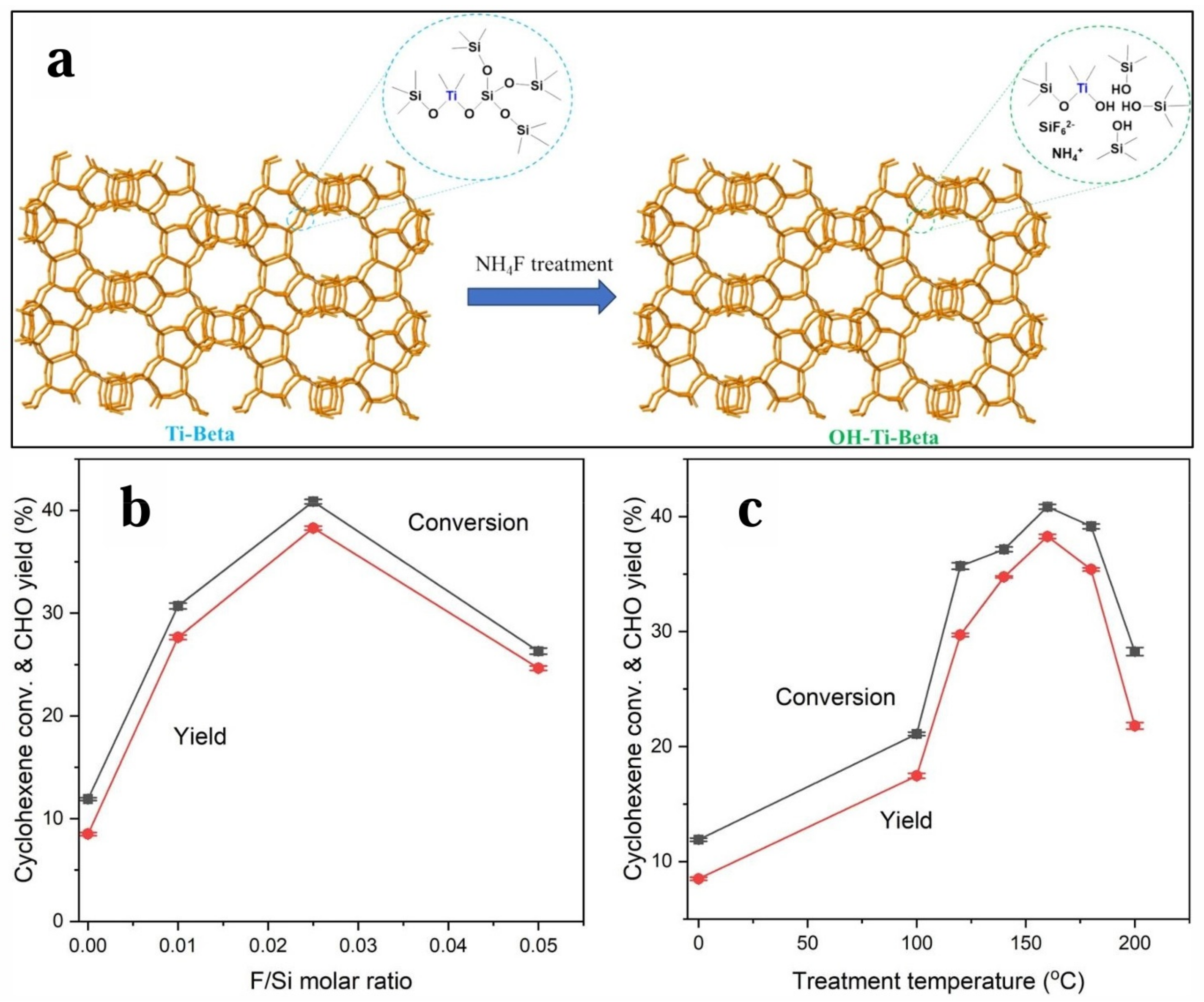
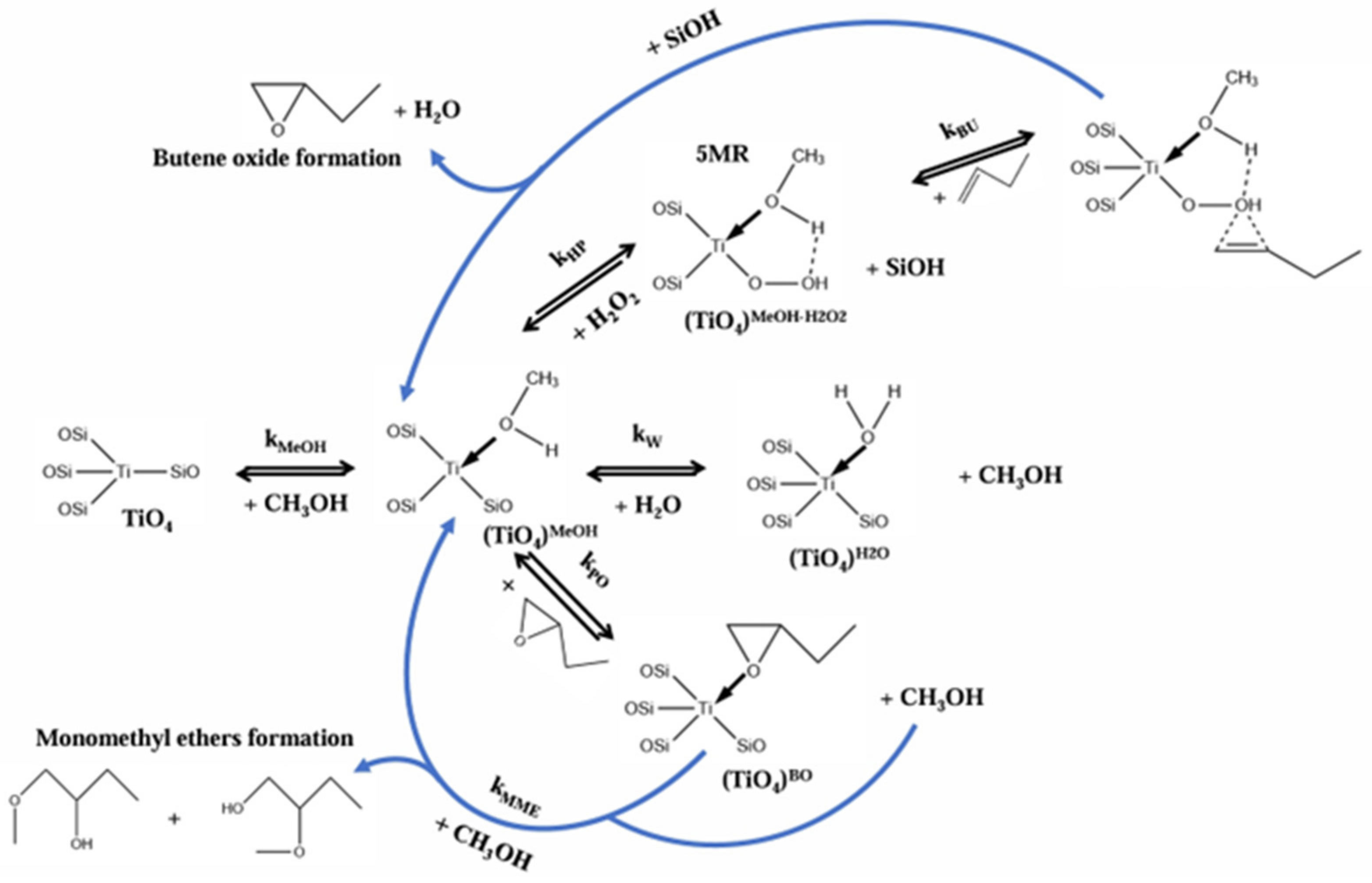
4.5. Butene Epoxidation Mechanism over TS-1 with H2O2 as Oxidant (L-H Mechanism)
5. Progress and Prospects of Green Chemistry in the Epoxidation of Olefins
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amini, M. Efficient and selective oxidation of olefins and alcohols using nanoparticles of WO3-supported manganese oxides (W1−xMnxO3). Korean J. Chem. Eng. 2016, 33, 126–131. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, L.; Zhang, J.; Cheng, L.; Zhai, Z.; Chen, J.; Hou, W. Template-free method to prepare porous Cu-containing nanotubes with a good catalytic performance for styrene epoxidation. Appl. Surf. Sci. 2014, 298, 116–124. [Google Scholar] [CrossRef]
- Amini, M.; Haghdoost, M.M.; Bagherzadeh, M. Monomeric and dimeric oxido–peroxido tungsten(VI) complexes in catalytic and stoichiometric epoxidation. Coord. Chem. Rev. 2014, 268, 83–100. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, G.; Yan, H.; Liu, Y.; Chen, X.; Feng, X.; Jin, X.; Yang, C. Liquid-Phase Epoxidation of Light Olefins over W and Nb Nanocatalysts. ACS Sustain. Chem. Eng. 2018, 6, 4423–4452. [Google Scholar] [CrossRef]
- Huš, M.; Grilc, M.; Teržan, J.; Gyergyek, S.; Likozar, B.; Hellman, A. Going Beyond Silver in Ethylene Epoxidation with First-Principles Catalyst Screening. Angew. Chem. Int. Ed. 2023, 62, e202305804. [Google Scholar] [CrossRef]
- Khatib, S.J.; Oyama, S.T. Direct Oxidation of Propylene to Propylene Oxide with Molecular Oxygen: A Review. Catal. Rev. 2015, 57, 306–344. [Google Scholar] [CrossRef]
- Li, T.; Zuo, Y.; Guo, Y.; Yang, H.; Liu, M.; Guo, X. Highly stable TS-1 extrudates for 1-butene epoxidation through improving the heat conductivity. Catal. Sci. Technol. 2020, 10, 6152–6160. [Google Scholar] [CrossRef]
- Pu, T.; Tian, H.; Ford, M.E.; Rangarajan, S.; Wachs, I.E. Overview of Selective Oxidation of Ethylene to Ethylene Oxide by Ag Catalysts. ACS Catal. 2019, 9, 10727–10750. [Google Scholar] [CrossRef]
- Pinaeva, L.G.; Noskov, A.S. Prospects for the Development of Ethylene Oxide Production Catalysts and Processes (Review). Pet. Chem. 2020, 60, 1191–1206. [Google Scholar] [CrossRef]
- Iyer, K.R.; Bhan, A. Particle size dependence of ethylene epoxidation rates on Ag/α-Al2O3 catalysts: Why particle size distributions matter. J. Catal. 2023, 420, 99–109. [Google Scholar] [CrossRef]
- Yu, B.; Ayvalı, T.; Raine, E.; Li, T.; Li, M.M.-J.; Zheng, J.; Wu, S.; Bagabas, A.A.; Tsang, S.C.E. Enhanced propylene oxide selectivity for gas phase direct propylene epoxidation by lattice expansion of silver atoms on nickel nanoparticles. Appl. Catal. B Environ. 2019, 243, 304–312. [Google Scholar] [CrossRef]
- Xiong, W.; Gu, X.-K.; Zhang, Z.; Chai, P.; Zang, Y.; Yu, Z.; Li, D.; Zhang, H.; Liu, Z.; Huang, W. Fine cubic Cu2O nanocrystals as highly selective catalyst for propylene epoxidation with molecular oxygen. Nat. Commun. 2021, 12, 5921. [Google Scholar]
- Arvay, J.W.; Hong, W.; Li, C.; Delgass, W.N.; Ribeiro, F.H.; Harris, J.W. Kinetics of Propylene Epoxidation over Extracrystalline Gold Active Sites on AU/TS-1 Catalysts. ACS Catal. 2022, 12, 10147–10160. [Google Scholar] [CrossRef]
- Zuo, Y.; Zhang, B.; Li, T.; Zhang, M.; Fan, J.; Liu, M.; Xu, J.; Yang, C.; Yang, H.; Guo, X. Highly stable monolithic titanium silicalite-1 catalyst for 1-butene epoxidation. AIChE J. 2023, 69, e17870. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, C.; Sun, B.; Zhu, H.; Xu, W. Preparation and modification of Au/TS-1 catalyst in the direct epoxidation of propylene with H2 and O2. Appl. Catal. A Gen. 2021, 624, 118329. [Google Scholar] [CrossRef]
- Horstmann, S.; Gardeler, H.; Fischer, K.; Köster, F.; Gmehling, J. Vapor Pressure, Vapor−Liquid Equilibrium, and Excess Enthalpy Data for Compounds and Binary Subsystems of the Chlorohydrin Process for Propylene Oxide Production. J. Chem. Eng. Data 2001, 46, 337–345. [Google Scholar] [CrossRef]
- Myszkowski, J.; Milchert, E.; Bartkowiak, M.; Pełech, R. Utilization of waste chloroorganic compounds. Pol. J. Chem. Technol. 2010, 12, 36–39. [Google Scholar] [CrossRef]
- Myszkowski, J.; Milchert, E.; Paździoch, W.; Pełech, R. Formation of environmentally friendly chloroorganic compounds technology by sewage and by-products utilization. Pol. J. Chem. Technol. 2007, 9, 118–121. [Google Scholar] [CrossRef]
- Liu, D.; Wang, R.; Yu, Y.; Chen, Z.; Fang, N.; Liu, Y.; He, M. Chemical deactivation of titanosilicate catalysts caused by propylene oxide in the HPPO process. Catal. Sci. Technol. 2023, 13, 1437–1447. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; de Frutos-Escrig, M.P.; Martín, H.; Campos-Martin, J.M.; Fierro, J.L.G. Selective hydrogenation of hydrogen peroxide in the epoxidation effluent of the HPPO process. Catal. Commun. 2012, 26, 83–87. [Google Scholar] [CrossRef]
- Liu, X.; Liu, J.; Xia, Y.; Yin, D.; Steven, R.K.; Mao, L. Catalytic performance of TS-1 in oxidative cleavage of 1-alkenes with H2O2. Catal. Commun. 2019, 126, 40–43. [Google Scholar] [CrossRef]
- Russo, V.; Tesser, R.; Santacesaria, E.; Di Serio, M. Chemical and Technical Aspects of Propene Oxide Production via Hydrogen Peroxide (HPPO Process). Ind. Eng. Chem. Res. 2013, 52, 1168–1178. [Google Scholar] [CrossRef]
- Zeb, A.; Park, C.; Son, M.; Baek, A.; Cho, Y.; Kim, D.; Rampogu, S.; Lee, G.; Kwak, Y.-S.; Park, S.J.; et al. Integration of virtual screening and computational simulation identifies photodynamic therapeutics against human Protoporphyrinogen Oxidase IX (HPPO). Arab. J. Chem. 2020, 13, 2245–2256. [Google Scholar] [CrossRef]
- Triandafillidi, I.; Kokotou, M.G.; Lotter, D.; Sparr, C.; Kokotos, C.G. Aldehyde-catalyzed epoxidation of unactivated alkenes with aqueous hydrogen peroxide. Chem. Sci. 2021, 12, 10191–10196. [Google Scholar] [CrossRef]
- Tang, K.; Hou, W.; Wang, X.; Xu, W.; Lu, X.; Ma, R.; Fu, Y.; Zhu, W. Enhanced catalytic performance of trimethylsilylated Ti-MWW zeolites for the liquid-phase epoxidation of propylene with H2O2. Microporous Mesoporous Mater. 2021, 328, 111492. [Google Scholar] [CrossRef]
- Sulimov, A.V.; Ovcharova, A.V.; Ovcharov, A.A.; Flid, V.R.; Leont’eva, S.V.; Bruk, L.G.; Pastukhova, Z.Y.; Flid, M.R. Study of alkene epoxidation in the presence of extruded silicalite titanium. Russ. Chem. Bull. 2016, 65, 2845–2849. [Google Scholar] [CrossRef]
- Cokoja, M.; Reich, R.M.; Wilhelm, M.E.; Kaposi, M.; Schäffer, J.; Morris, D.S.; Münchmeyer, C.J.; Anthofer, M.H.; Markovits, I.I.E.; Kühn, F.E.; et al. Olefin Epoxidation in Aqueous Phase Using Ionic-Liquid Catalysts. ChemSusChem 2016, 9, 1773–1776. [Google Scholar] [CrossRef]
- Engelmann, X.; Malik, D.D.; Corona, T.; Warm, K.; Farquhar, E.R.; Swart, M.; Nam, W.; Ray, K. Trapping of a Highly Reactive Oxoiron(IV) Complex in the Catalytic Epoxidation of Olefins by Hydrogen Peroxide. Angew. Chem. Int. Ed. 2019, 58, 4012–4016. [Google Scholar] [CrossRef]
- Li, L.; Song, H.-J.; Meng, X.-G.; Yang, R.-Q.; Zhang, N. Efficient epoxidation reaction of terminal olefins with hydrogen peroxide catalyzed by an iron (II) complex. Tetrahedron Lett. 2018, 59, 2436–2439. [Google Scholar] [CrossRef]
- Majetich, G.; Hicks, R.; Sun, G.-r.; McGill, P. Carbodiimide-Promoted Olefin Epoxidation with Aqueous Hydrogen Peroxide. J. Org. Chem. 1998, 63, 2564–2573. [Google Scholar] [CrossRef]
- Li, P.; Gao, J.; Shi, J.; Wang, H.; Xing, X.; Ren, J.; Meng, Y.; Wang, L.; Lv, B. Insights into the effect of oxygen vacancies on the epoxidation of 1-hexene with hydrogen peroxide over WO3−x/SBA-15. Catal. Sci. Technol. 2022, 12, 6827–6837. [Google Scholar] [CrossRef]
- Rezaeifard, A.; Jafarpour, M.; Haddad, R.; Feizpour, F. {Mo72Cr30} nanocluster as a novel self-separating catalyst for hydrogen peroxide olefin epoxidation. Catal. Commun. 2017, 95, 88–91. [Google Scholar] [CrossRef]
- Borrell, M.; Costas, M. Greening Oxidation Catalysis: Iron Catalyzed Alkene syn-Dihydroxylation with Aqueous Hydrogen Peroxide in Green Solvents. ACS Sustain. Chem. Eng. 2018, 6, 8410–8416. [Google Scholar] [CrossRef]
- Ai, C.; Zhu, F.; Wang, Y.; Yan, Z.; Lin, S. SO2F2-Mediated Epoxidation of Olefins with Hydrogen Peroxide. J. Org. Chem. 2019, 84, 11928–11934. [Google Scholar] [CrossRef]
- Wang, T.; Jing, X.; Chen, C.; Yu, L. Organoselenium-Catalyzed Oxidative C-C Bond Cleavage: A Relatively Green Oxidation of Alkenes into Carbonyl Compounds with Hydrogen Peroxide. J. Org. Chem. 2017, 82, 9342–9349. [Google Scholar] [CrossRef]
- Chen, Z.; Luck, R.L. Oxidation of olefins using atmospheric oxygen atoms initiated by tert-butylhydroperoxide or hydrogen peroxide with silver nanoparticles deposited on MCM-41 as catalysts. Green Chem. 2016, 18, 3354–3359. [Google Scholar] [CrossRef]
- Mao, S.; Budweg, S.; Spannenberg, A.; Wen, X.; Yang, Y.; Li, Y.-W.; Junge, K.; Beller, M. Iron-Catalyzed Epoxidation of Linear α-Olefins with Hydrogen Peroxide. ChemCatChem 2022, 14, e202101668. [Google Scholar] [CrossRef]
- Hong, M.; Min, J.; Wang, S. Metal-Free Epoxidation of Internal and Terminal Alkenes with tert-Butyl Hydroperoxide/Isobutyraldehyde/Oxygen System. ChemistrySelect 2018, 3, 4818–4821. [Google Scholar] [CrossRef]
- Bose, I.; Zhao, Y. Site-Selective Catalytic Epoxidation of Alkenes with Tunable Atomic Precision by Molecularly Imprinted Artificial Epoxidases. ACS Catal. 2022, 12, 3444–3451. [Google Scholar] [CrossRef]
- Vanoye, L.; Wang, J.; Pablos, M.; de Bellefon, C.; Favre-Réguillon, A. Epoxidation using molecular oxygen in flow: Facts and questions on the mechanism of the Mukaiyama epoxidation. Catal. Sci. Technol. 2016, 6, 4724–4732. [Google Scholar] [CrossRef]
- Koya, S.; Nishioka, Y.; Mizoguchi, H.; Uchida, T.; Katsuki, T. Asymmetric Epoxidation of Conjugated Olefins with Dioxygen. Angew. Chem. Int. Ed. 2012, 51, 8243–8246. [Google Scholar] [CrossRef] [PubMed]
- Iwahama, T.; Hatta, G.; Sakaguchi, S.; Ishii, Y. Epoxidation of alkenes using alkyl hydroperoxides generated in situ by catalytic autoxidation of hydrocarbons with dioxygen. Chem. Commun. 2000, 2, 163–164. [Google Scholar] [CrossRef]
- Yamada, T.; Takai, T.; Rhode, O.; Mukaiyama, T. Direct Epoxidation of Olefins Catalyzed by Nickel(II) Complexes with Molecular Oxygen and Aldehydes. Bull. Chem. Soc. Jpn. 1991, 64, 2109–2117. [Google Scholar] [CrossRef]
- Abbasi, V.; Hosseini-Monfared, H.; Hosseini, S.M. A heterogenized chiral imino indanol complex of manganese as an efficient catalyst for aerobic epoxidation of olefins. New J. Chem. 2017, 41, 9866–9874. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, L.; Mi, Z. Influence of Pt-Pd/TS-1 Catalyst Preparation on Epoxidation of Olefins with Hydrogen Peroxide. Catal. Lett. 2005, 103, 161–164. [Google Scholar] [CrossRef]
- Luz, I.; León, A.; Boronat, M.; Llabrés i Xamena, F.X.; Corma, A. Selective aerobic oxidation of activated alkanes with MOFs and their use for epoxidation of olefins with oxygen in a tandem reaction. Catal. Sci. Technol. 2013, 3, 371–379. [Google Scholar] [CrossRef]
- Zheng, Y.; Shen, Q.; Li, Z.; Jing, X.; Duan, C. Two Copper-Containing Polyoxometalate-Based Metal-Organic Complexes as Heterogeneous Catalysts for the C-H Bond Oxidation of Benzylic Compounds and Olefin Epoxidation. Inorg. Chem. 2022, 61, 11156–11164. [Google Scholar] [CrossRef]
- Tebandeke, E.; Coman, C.; Guillois, K.; Canning, G.; Ataman, E.; Knudsen, J.; Wallenberg, L.R.; Ssekaalo, H.; Schnadt, J.; Wendt, O.F. Epoxidation of olefins with molecular oxygen as the oxidant using gold catalysts supported on polyoxometalates. Green Chem. 2014, 16, 1586–1593. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Z.; Gao, G.; Xiao, G.; Du, A.; Bottle, S.; Sarina, S.; Zhu, H. Stable Copper Nanoparticle Photocatalysts for Selective Epoxidation of Alkenes with Visible Light. ACS Catal. 2017, 7, 4975–4985. [Google Scholar] [CrossRef]
- Zwaschka, G.; Rondelli, M.; Krause, M.; Rötzer, M.D.; Hedhili, M.N.; Heiz, U.; Basset, J.M.; Schweinberger, F.F.; D’Elia, V. Supported sub-nanometer Ta oxide clusters as model catalysts for the selective epoxidation of cyclooctene. New J. Chem. 2018, 42, 3035–3041. [Google Scholar] [CrossRef]
- Schröder, K.; Join, B.; Amali, A.J.; Junge, K.; Ribas, X.; Costas, M.; Beller, M. A Biomimetic Iron Catalyst for the Epoxidation of Olefins with Molecular Oxygen at Room Temperature. Angew. Chem. Int. Ed. 2011, 50, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, E.; Laszlo, P.; Levart, M.; Montaufier, M.-T.; Singh, G.P. Epoxidation of olefins by molecular oxygen with clay-impregnated nickel catalysts. Tetrahedron Lett. 1993, 34, 1123–1126. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, W.-J.; Lu, X.; Yang, H.; Chen, Z.; Liao, R.-Z.; Yin, G. Nonredox Metal Ions Promoted Olefin Epoxidation by Iron(II) Complexes with H2O2: DFT Calculations Reveal Multiple Channels for Oxygen Transfer. Inorg. Chem. 2017, 56, 15138–15149. [Google Scholar] [CrossRef]
- Bregante, D.T.; Tan, J.Z.; Schultz, R.L.; Ayla, E.Z.; Potts, D.S.; Torres, C.; Flaherty, D.W. Catalytic Consequences of Oxidant, Alkene, and Pore Structures on Alkene Epoxidations within Titanium Silicates. ACS Catal. 2020, 10, 10169–10184. [Google Scholar] [CrossRef]
- Neves, P.; Nogueira, L.S.; Gomes, A.C.; Oliveira, T.S.M.; Lopes, A.D.; Valente, A.A.; Gonçalves, I.S.; Pillinger, M. Chemistry and Catalytic Performance of Pyridyl-Benzimidazole Oxidomolybdenum(VI) Compounds in (Bio)Olefin Epoxidation. Eur. J. Inorg. Chem. 2017, 2017, 2617–2627. [Google Scholar] [CrossRef]
- Zwettler, N.; Schachner, J.A.; Belaj, F.; Mösch-Zanetti, N.C. Hydrogen bond donor functionalized dioxido-molybdenum(VI) complexes as robust and highly efficient precatalysts for alkene epoxidation. Mol. Catal. 2017, 443, 209–219. [Google Scholar] [CrossRef]
- Rossi-Fernández, L.; Dorn, V.; Radivoy, G. A new and efficient methodology for olefin epoxidation catalyzed by supported cobalt nanoparticles. Beilstein J. Org. Chem. 2021, 17, 519–526. [Google Scholar] [CrossRef]
- Bafti, A.; Razum, M.; Topić, E.; Agustin, D.; Pisk, J.; Vrdoljak, V. Implication of oxidant activation on olefin epoxidation catalysed by Molybdenum catalysts with aroylhydrazonato ligands: Experimental and theoretical studies. Mol. Catal. 2021, 512, 111764. [Google Scholar] [CrossRef]
- Bhuiyan, M.M.; Mohammed, M.L.; Saha, B. Greener and Efficient Epoxidation of 1,5-Hexadiene with tert-Butyl Hydroperoxide (TBHP) as an Oxidising Reagent in the Presence of Polybenzimidazole Supported Mo(VI) Catalyst. Reactions 2022, 3, 537–552. [Google Scholar] [CrossRef]
- Nunes, M.S.; Gomes, A.C.; Neves, P.; Mendes, R.F.; Almeida Paz, F.A.; Lopes, A.D.; Pillinger, M.; Gonçalves, I.S.; Valente, A.A. Molybdenum(VI) complexes with ligands derived from 5-(2-pyridyl)-2H-tetrazole as catalysts for the epoxidation of olefins. Catal. Today 2023, 423, 114273. [Google Scholar] [CrossRef]
- Samani, M.; Ardakani, M.H.; Sabet, M. Efficient and selective oxidation of hydrocarbons with tert-butyl hydroperoxide catalyzed by oxidovanadium(IV) unsymmetrical Schiff base complex supported on γ-Fe2O3 magnetic nanoparticles. Res. Chem. Intermed. 2022, 48, 1481–1494. [Google Scholar] [CrossRef]
- Fadaei Sarabi, M.; Bezaatpour, A.; Mahmoudi, A. Anchoring of a terpyridine-based Mo(VI) complex on manganese ferrite as a recoverable catalyst for epoxidation of olefins under solvent-free conditions. J. Coord. Chem. 2021, 74, 1597–1612. [Google Scholar] [CrossRef]
- Mirdarvatan, V.; Bahramian, B.; Khalaji, A.D.; Vaclavu, T.; Gómez-García, C.J.; Benmansour, S.; Triki, S. A novel mixed azido/phenoxido bridged 1D CuII coordination polymer containing o-vanillin-based compartmental ligand: Synthesis, magnetism, phenoxazinone synthase and catalytic activity in epoxidation of alkenes. Polyhedron 2023, 244, 116581. [Google Scholar] [CrossRef]
- Marreiros, J.; Diaz-Couce, M.; Ferreira, M.J.; Vaz, P.D.; Calhorda, M.J.; Nunes, C.D. Synthesis and catalytic activity of Mo(II) complexes of α-diimines intercalated in layered double hydroxides. Inorganica Chim. Acta 2019, 486, 274–282. [Google Scholar] [CrossRef]
- Aigner, M.; Grosso-Giordano, N.A.; Okrut, A.; Zones, S.; Katz, A. Epoxidation of 1-octene under harsh tail-end conditions in a flow reactor I: A comparative study of crystalline vs. amorphous catalysts. React. Chem. Eng. 2017, 2, 842–851. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; Capel-Sanchez, M.C.; Campos-Martin, J.M.; Fierro, J.L.G. Effect of precursor nature on the behavior of titanium-polysiloxane homogeneous catalysts in primary alkene epoxidation. J. Mol. Catal. A Chem. 2007, 269, 133–140. [Google Scholar] [CrossRef]
- Bento, A.; Sanches, A.; Vaz, P.D.; Nunes, C.D. Catalytic Application of Fe-doped MoO2 Tremella-Like Nanosheets. Top. Catal. 2016, 59, 1123–1131. [Google Scholar] [CrossRef]
- Bento, A.; Sanches, A.; Medina, E.; Nunes, C.D.; Vaz, P.D. MoO2 nanoparticles as highly efficient olefin epoxidation catalysts. Appl. Catal. A Gen. 2015, 504, 399–407. [Google Scholar] [CrossRef]
- Naja I, M.; Abbasi, A.; Masteri-Farahani, M. Preparation of MoO3/CuMoO4 nanoparticles as selective catalyst for olefin epoxidation. Sci. Iran. 2017, 24, 1203–1208. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Liu, L.; Chen, Z. The interaction of molybdenum and titanium in mesoporous materials for olefin epoxidation. React. Kinet. Mech. Catal. 2022, 135, 317–331. [Google Scholar] [CrossRef]
- Martins, A.M.; Romão, C.C.; Abrantes, M.; Azevedo, M.C.; Cui, J.; Dias, A.R.; Duarte, M.T.; Lemos, M.A.; Lourenço, T.; Poli, R. Mononuclear and Binuclear Cyclopentadienyl Oxo Molybdenum and Tungsten Complexes: Syntheses and Applications in Olefin Epoxidation Catalysis. Organometallics 2005, 24, 2582–2589. [Google Scholar] [CrossRef]
- Huang, J.; Yuan, L.; Cai, J.; Liu, Z. MoO2(acac)2 anchored on organic copolymer-inorganic hybrid zirconium phosphonate-phosphate functionalized by pyridines as highly efficient, reusable catalysts for alkene epoxidation. Microporous Mesoporous Mater. 2015, 214, 121–126. [Google Scholar] [CrossRef]
- Masteri-Farahani, M.; Modarres, M. Superiority of Activated Carbon versus MCM-41 for the Immobilization of Molybdenum Dithiocarbamate Complex as Heterogeneous Epoxidation Catalyst. ChemistrySelect 2017, 2, 1163–1169. [Google Scholar] [CrossRef]
- Nunes, M.S.; Gomes, D.M.; Gomes, A.C.; Neves, P.; Mendes, R.F.; Paz, F.A.A.; Lopes, A.D.; Valente, A.A.; Gonçalves, I.S.; Pillinger, M. A 5-(2-Pyridyl)tetrazolate Complex of Molybdenum(VI), Its Structure, and Transformation to a Molybdenum Oxide-Based Hybrid Heterogeneous Catalyst for the Epoxidation of Olefins. Catalysts 2021, 11, 1407. [Google Scholar] [CrossRef]
- Ambroziak, K.; Mbeleck, R.; He, Y.; Saha, B.; Sherrington, D.C. Investigation of Batch Alkene Epoxidations Catalyzed by Polymer-Supported Mo(VI) Complexes. Ind. Eng. Chem. Res. 2009, 48, 3293–3302. [Google Scholar] [CrossRef]
- Ambroziak, K.; Mbeleck, R.; Saha, B.; Sherrington, D. Greener and Sustainable Method for Alkene Epoxidations by Polymer-Supported Mo(VI) Catalysts. Int. J. Chem. React. Eng. 2010, 8, 1–13. [Google Scholar] [CrossRef]
- Liu, X.-H.; Yu, H.-Y.; Xue, C.; Zhou, X.-T.; Ji, H.-B. Cyclohexene Promoted Efficient Biomimetic Oxidation of Alcohols to Carbonyl Compounds Catalyzed by Manganese Porphyrin under Mild Conditions. Chin. J. Chem. 2020, 38, 458–464. [Google Scholar] [CrossRef]
- Zhou, X.-T.; Tang, Q.-H.; Ji, H.-B. Remarkable enhancement of aerobic epoxidation reactivity for olefins catalyzed by μ-oxo-bisiron(III) porphyrins under ambient conditions. Tetrahedron Lett. 2009, 50, 6601–6605. [Google Scholar] [CrossRef]
- Liu, X.-H.; Huang, J.-Y.; Tao, L.-M.; Yu, H.-Y.; Zhou, X.-T.; Xue, C.; Han, Q.; Zou, W.; Ji, H.-B. Oxygen Atom Transfer Mechanism for Vanadium-Oxo Porphyrin Complexes Mediated Aerobic Olefin Epoxidation. Chin. J. Chem. 2022, 40, 115–122. [Google Scholar] [CrossRef]
- Celano, G.; Šmejkalová, D.; Spaccini, R.; Piccolo, A. Reduced Toxicity of Olive Mill Waste Waters by Oxidative Coupling with Biomimetic Catalysis. Environ. Sci. Technol. 2008, 42, 4896–4901. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, D.; Li, Y.; Zhao, X.; Wang, B.; Qu, J. Biomimetic catalytic oxidative coupling of thiols using thiolate-bridged dinuclear metal complexes containing iron in water under mild conditions. Catal. Sci. Technol. 2019, 9, 6492–6502. [Google Scholar] [CrossRef]
- Esmelindro, M.C.; Oestreicher, E.G.; Márquez-Alvarez, H.; Dariva, C.; Egues, S.M.S.; Fernandes, C.; Bortoluzzi, A.J.; Drago, V.; Antunes, O.A.C. Catalytic oxidation of cyclohexane by a binuclear Fe(III) complex biomimetic to methane monooxygenase. J. Inorg. Biochem. 2005, 99, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Liu, D.; Sun, J.; Deng, W.; Sheng, W.; Liu, Q.; Guo, C. Effects of Oxygen Transfer Limitation and Kinetic Control on Biomimetic Catalytic Oxidation of Toluene. Chin. J. Chem. Eng. 2014, 22, 509–515. [Google Scholar] [CrossRef]
- Ibrahem, I.; Samec, J.S.M.; Bäckvall, J.E.; Córdova, A. Enantioselective addition of aldehydes to amines via combined catalytic biomimetic oxidation and organocatalytic C-C bond formation. Tetrahedron Lett. 2005, 46, 3965–3968. [Google Scholar] [CrossRef]
- Jana, N.C.; Patra, M.; Brandão, P.; Panja, A. Synthesis, structure and diverse coordination chemistry of cobalt(III) complexes derived from a Schiff base ligand and their biomimetic catalytic oxidation of o-aminophenols. Polyhedron 2019, 164, 23–34. [Google Scholar] [CrossRef]
- Murahashi, S.-I.; Zhang, D. Ruthenium catalyzed biomimetic oxidation in organic synthesis inspired by cytochrome P-450. Chem. Soc. Rev. 2008, 37, 1490–1501. [Google Scholar] [CrossRef]
- Fang, H.; Wang, M.; Yi, H.; Zhang, Y.; Li, X.; Yan, F.; Zhang, L. Electrostatic Assembly of Porphyrin-Functionalized Porous Membrane toward Biomimetic Photocatalytic Degradation Dyes. ACS Omega 2020, 5, 8707–8720. [Google Scholar] [CrossRef]
- Meng, X.; Yu, C.; Chen, G.; Zhao, P. Heterogeneous biomimetic aerobic synthesis of 3-iodoimidazo [1,2-a]pyridines via CuOx/OMS-2-catalyzed tandem cyclization/iodination and their late-stage functionalization. Catal. Sci. Technol. 2015, 5, 372–379. [Google Scholar] [CrossRef]
- Yan, Z.; Tian, J.; Wang, K.; Nigam, K.D.P.; Luo, G. Microreaction processes for synthesis and utilization of epoxides: A review. Chem. Eng. Sci. 2021, 229, 116071. [Google Scholar] [CrossRef]
- Grigoropoulou, G.; Clark, J.H.; Elings, J.A. Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant. Green Chem. 2003, 5, 1–7. [Google Scholar] [CrossRef]
- Mandelli, D.; van Vliet, M.C.A.; Sheldon, R.A.; Schuchardt, U. Alumina-catalyzed alkene epoxidation with hydrogen peroxide. Appl. Catal. A Gen. 2001, 219, 209–213. [Google Scholar] [CrossRef]
- Lane, B.S.; Burgess, K. Metal-Catalyzed Epoxidations of Alkenes with Hydrogen Peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef]
- Stoica, G.; Santiago, M.; Jacobs, P.A.; Pérez-Ramírez, J.; Pescarmona, P.P. Epoxidation catalysts derived from aluminium and gallium dawsonites. Appl. Catal. A Gen. 2009, 371, 43–53. [Google Scholar] [CrossRef]
- Tse, M.K.; Klawonn, M.; Bhor, S.; Döbler, C.; Anilkumar, G.; Hugl, H.; Mägerlein, W.; Beller, M. Convenient Method for Epoxidation of Alkenes Using Aqueous Hydrogen Peroxide. Org. Lett. 2005, 7, 987–990. [Google Scholar] [CrossRef]
- Alvear, M.; Fortunato, M.E.; Russo, V.; Eränen, K.; Di Serio, M.; Lehtonen, J.; Rautiainen, S.; Murzin, D.; Salmi, T. Continuous Liquid-Phase Epoxidation of Ethylene with Hydrogen Peroxide on a Titanium-Silicate Catalyst. Ind. Eng. Chem. Res. 2021, 60, 9429–9436. [Google Scholar] [CrossRef]
- Yan, W.; Ramanathan, A.; Ghanta, M.; Subramaniam, B. Towards highly selective ethylene epoxidation catalysts using hydrogen peroxide and tungsten or niobium-incorporated mesoporous silicate (KIT-6). Catal. Sci. Technol. 2014, 4, 4433–4439. [Google Scholar] [CrossRef]
- Lu, X.; Zhou, W.-J.; Guan, Y.; Liebens, A.; Wu, P. Enhancing ethylene epoxidation of a MWW-type titanosilicate/H2O2 catalytic system by fluorine implanting. Catal. Sci. Technol. 2017, 7, 2624–2631. [Google Scholar] [CrossRef]
- Wang, B.; Zhu, Y.; Han, H.; Qin, Q.; Zhang, Z.; Zhu, J. Preparation and Catalytic Performance in Propylene Epoxidation of Hydrophobic Hierarchical Porous TS-1 Zeolite. Catal. Lett. 2022, 152, 3076–3088. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Q.; Li, Y.; Feng, X.; Chai, Y.; Liu, C. Seed-assisted synthesis of hierarchical nanosized TS-1 in a low-cost system for propylene epoxidation with H2O2. Appl. Surf. Sci. 2019, 483, 652–660. [Google Scholar] [CrossRef]
- Maiti, S.K.; Ramanathan, A.; Subramaniam, B. 110th Anniversary: Near-Total Epoxidation Selectivity and Hydrogen Peroxide Utilization with Nb-EISA Catalysts for Propylene Epoxidation. Ind. Eng. Chem. Res. 2019, 58, 17727–17735. [Google Scholar] [CrossRef]
- Zuo, Y.; Liu, M.; Ma, M.; Song, C.; Guo, X. Improved Catalytic Performance for 1-Butene Epoxidation over Titanium Silicalite-1 Extrudates by Using SBA-15 or Carborundum as Additives. Ind. Eng. Chem. Res. 2017, 56, 7462–7467. [Google Scholar] [CrossRef]
- Egelske, B.T.; Xiong, W.; Zhou, H.; Monnier, J.R. Effects of the method of active site characterization for determining structure-sensitivity in Ag-catalyzed ethylene epoxidation. J. Catal. 2022, 410, 221–235. [Google Scholar] [CrossRef]
- Serafin, J.G.; Liu, A.C.; Seyedmonir, S.R. Surface science and the silver-catalyzed epoxidation of ethylene: An industrial perspective. J. Mol. Catal. A Chem. 1998, 131, 157–168. [Google Scholar] [CrossRef]
- Song, Z.; Yuan, J.; Cai, Z.; Lin, D.; Feng, X.; Sheng, N.; Liu, Y.; Chen, X.; Jin, X.; Chen, D.; et al. Engineering three-layer core-shell S-1/TS-1@dendritic-SiO2 supported Au catalysts towards improved performance for propene epoxidation with H2 and O2. Green Energy Environ. 2020, 5, 473–483. [Google Scholar] [CrossRef]
- Hu, S.; Li, J.; Wang, Q.; Yang, W. Design and optimization of an integrated process for the purification of propylene oxide and the separation of propylene glycol by-product. Chin. J. Chem. Eng. 2022, 45, 111–120. [Google Scholar] [CrossRef]
- Liu, J.; Ji, X.; Shi, J.; Wang, L.; Jian, P.; Yan, X.; Wang, D. Experimental and theoretical investigation of the tuning of electronic structure in SnO2 via Co doping for enhanced styrene epoxidation catalysis. Catal. Sci. Technol. 2022, 12, 1499–1511. [Google Scholar] [CrossRef]
- Tian, S.; Peng, C.; Dong, J.; Xu, Q.; Chen, Z.; Zhai, D.; Wang, Y.; Gu, L.; Hu, P.; Duan, H.; et al. High-Loading Single-Atomic-Site Silver Catalysts with an Ag1-C2N1 Structure Showing Superior Performance for Epoxidation of Styrene. ACS Catal. 2021, 11, 4946–4954. [Google Scholar] [CrossRef]
- Gupta, R.; Uslu, H.; Majumder, S. Production of Styrene from Dehydrogenation of Ethylbenzene. Chem. Eng. Technol. 2022, 45, 817–823. [Google Scholar] [CrossRef]
- Buijink, J.K.F.; van Vlaanderen, J.J.M.; Crocker, M.; Niele, F.G.M. Propylene epoxidation over titanium-on-silica catalyst-the heart of the SMPO process. Catal. Today 2004, 93–95, 199–204. [Google Scholar] [CrossRef]
- De Vos, D.E.; Sels, B.F.; Jacobs, P.A. Practical Heterogeneous Catalysts for Epoxide Production. Adv. Synth. Catal. 2003, 345, 457–473. [Google Scholar] [CrossRef]
- Calvente, R.M.; Campos-Martin, J.M.; Fierro, J.L.G. Effective homogeneous molybdenum catalyst for linear terminal alkenes epoxidation with organic hydroperoxide. Catal. Commun. 2002, 3, 247–251. [Google Scholar] [CrossRef]
- Barrio, L.; Campos-Martín, J.M.; de Frutos, M.P.; Fierro, J.L.G. Alkene Epoxidation with Ethylbenzene Hydroperoxides Using Molybdenum Heterogeneous Catalysts. Ind. Eng. Chem. Res. 2008, 47, 8016–8024. [Google Scholar] [CrossRef]
- Levchuk, I.; Bhatnagar, A.; Sillanpää, M. Overview of technologies for removal of methyl tert-butyl ether (MTBE) from water. Sci. Total Environ. 2014, 476–477, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Fadhli, M.; Khedher, I.; Fraile, J.M. Enantioselective epoxidation of styrene with TBHP catalyzed by bis(oxazoline)–vanadyl–laponite materials. Catal. Commun. 2018, 117, 90–93. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, F.; Gao, R.; Dai, W.-L. Manganese-doped CeO2 nanocubes as highly efficient catalysts for styrene epoxidation with TBHP. Appl. Surf. Sci. 2019, 471, 767–775. [Google Scholar] [CrossRef]
- Mohammed, M.L.; Saha, B. Recent Advances in Greener and Energy Efficient Alkene Epoxidation Processes. Energies 2022, 15, 2858. [Google Scholar] [CrossRef]
- Willms, T.; Kryk, H.; Hampel, U. Partial Isobutane Oxidation to tert-Butyl Hydroperoxide in a Micro Reactor-Comparison of DTBP and Aqueous TBHP as Initiator. Chem. Ing. Tech. 2018, 90, 731–735. [Google Scholar] [CrossRef]
- Wang, Z.; Balkus, K.J. Liquid phase propylene oxidation with tert-butyl hydroperoxide over titanium containing wrinkled mesoporous silica. Catal. Commun. 2017, 96, 15–18. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Jiang, H.; Yuan, X. Catalytic epoxidation of propylene over a Schiff-base molybdenum complex supported on a silanized mesostructured cellular foam. Res. Chem. Intermed. 2020, 46, 4705–4721. [Google Scholar] [CrossRef]
- Chen, M.; Dai, C.; Yu, G.; Liu, N.; Xu, R.; Wang, N.; Chen, B. Highly efficient absorption of methyl tert-butyl ether with ionic liquids. Sep. Purif. Technol. 2022, 282, 120108. [Google Scholar] [CrossRef]
- Ji, B.; Shao, F.; Hu, G.; Zheng, S.; Zhang, Q.; Xu, Z. Adsorption of methyl tert-butyl ether (MTBE) from aqueous solution by porous polymeric adsorbents. J. Hazard. Mater. 2009, 161, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, A.; Vismara, R.; Mercuri, G.; Moroni, M.; Palomino, M.; Domasevitch, K.V.; Di Nicola, C.; Pettinari, C.; Giambastiani, G.; Llabrés i Xamena, F.X.; et al. Cobalt(II) Bipyrazolate Metal-Organic Frameworks as Heterogeneous Catalysts in Cumene Aerobic Oxidation: A Tag-Dependent Selectivity. Inorg. Chem. 2020, 59, 8161–8172. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, A.; Briantais, P.; Prestipino, C.; Llabrés i Xamena, F.X. Selective Aerobic Oxidation of Cumene to Cumene Hydroperoxide over Mono- and Bimetallic Trimesate Metal-Organic Frameworks Prepared by a Facile “Green” Aqueous Synthesis. ACS Sustain. Chem. Eng. 2019, 7, 7708–7715. [Google Scholar] [CrossRef]
- Li, K.-T.; Lin, P.-H.; Lin, S.-W. Preparation of Ti/SiO2 catalysts by chemical vapor deposition method for olefin epoxidation with cumene hydroperoxide. Appl. Catal. A Gen. 2006, 301, 59–65. [Google Scholar] [CrossRef]
- Xia, Z.; Li, F.; Xu, L.; Feng, P. A stable and highly selective metalloporphyrin based framework for the catalytic oxidation of cyclohexene. Dalton Trans. 2020, 49, 11157–11162. [Google Scholar] [CrossRef]
- Bach, R.D.; Canepa, C.; Winter, J.E.; Blanchette, P.E. Mechanism of Acid-Catalyzed Epoxidation of Alkenes with Peroxy Acids. J. Org. Chem. 1997, 62, 5191–5197. [Google Scholar] [CrossRef]
- Murphy, A.; Pace, A.; Stack, T.D.P. Ligand and pH Influence on Manganese-Mediated Peracetic Acid Epoxidation of Terminal Olefins. Org. Lett. 2004, 6, 3119–3122. [Google Scholar] [CrossRef]
- Wang, Q.; Gu, Q.; You, S.-L. Enantioselective Carbonyl Catalysis Enabled by Chiral Aldehydes. Angew. Chem. Int. Ed. 2019, 58, 6818–6825. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, X.; Chen, S.; Luo, R.; Jiang, J.; Liang, Z.; Ji, H. Direct aerobic liquid phase epoxidation of propylene catalyzed by Mn(iii) porphyrin under mild conditions: Evidence for the existence of both peroxide and Mn(iv)-oxo species from in situ characterizations. RSC Adv. 2015, 5, 30014–30020. [Google Scholar] [CrossRef]
- Xu, D.; He, Y.; Liu, X.; Xiong, C.; Zhou, X.; Xue, C.; Ji, H. N-Hydroxyphthalimide-Catalyzed Epoxidation of Inactive Aliphatic Olefins with Air at Room Temperature. Asian J. Org. Chem. 2021, 10, 3349–3354. [Google Scholar] [CrossRef]
- Xiong, C.; He, Y.; Xu, D.; Liu, X.; Xue, C.; Zhou, X.; Ji, H. Enhanced oxygen transfer over bifunctional Mo-based oxametallacycle catalyst for epoxidation of propylene. J. Colloid Interface Sci. 2022, 611, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Liang, Y.; Zhou, X.; Xue, C.; Ji, H. Facile synthesis of a Mo-based TiO2 catalyst via a redox strategy for high value-added conversion of olefin. Fuel 2023, 332, 126172. [Google Scholar] [CrossRef]
- Xu, B.; Deng, M.; Lin, K.; Wang, Y.; Lu, X.; Ma, R.; Fu, Y.; Zhu, W. Ti-Beta zeotypes with open Ti(OSi)3OH sites for the efficient epoxidation of cyclohexene with H2O2. J. Catal. 2024, 439, 115748. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, S.; Liu, Y.; Lu, B.; Wang, Y.; Li, H.; Wang, Y. Tandem catalytic efficient olefin epoxidation with integrated production of nicotinamide derivatives. Chem Catal. 2023, 3, 100691. [Google Scholar] [CrossRef]
- Xiong, C.; Xue, C.; Yu, X.; He, Y.; Liang, Y.; Zhou, X.; Ji, H. Tuning the olefin-VOCs epoxidation performance of ceria by mechanochemical loading of coinage metal. J. Hazard. Mater. 2023, 441, 129888. [Google Scholar] [CrossRef]
- Aoto, H.; Matsui, K.; Sakai, Y.; Kuchizi, T.; Sekiya, H.; Osada, H.; Yoshida, T.; Matsunaga, S.; Nomiya, K. Zirconium(IV) and hafnium(IV)-containing polyoxometalates as oxidation precatalysts: Homogeneous catalytic epoxidation of cyclooctene by hydrogen peroxide. J. Mol. Catal. A Chem. 2014, 394, 224–231. [Google Scholar] [CrossRef]
- Neves, P.; Gomes, A.C.; Paz, F.A.A.; Valente, A.A.; Gonçalves, I.S.; Pillinger, M. Synthesis, structure and catalytic olefin epoxidation activity of a dinuclear oxo-bridged oxodiperoxomolybdenum(VI) complex containing coordinated 4,4′-bipyridinium. Mol. Catal. 2017, 432, 104–114. [Google Scholar] [CrossRef]
- Priyarega, S.; Haribabu, J.; Karvembu, R. Development of thiosemicarbazone-based transition metal complexes as homogeneous catalysts for various organic transformations. Inorganica Chim. Acta 2022, 532, 120742. [Google Scholar] [CrossRef]
- Nogueira, L.S.; Neves, P.; Gomes, A.C.; Valente, A.A.; Pillinger, M.; Gonçalves, I.S. Performance of a tetracarbonylmolybdenum(0) pyrazolylpyridine (pre)catalyst in olefin epoxidation and epoxide alcoholysis. J. Organomet. Chem. 2017, 846, 185–192. [Google Scholar] [CrossRef]
- Gomes, A.C.; Bruno, S.M.; Tomé, C.; Valente, A.A.; Pillinger, M.; Abrantes, M.; Gonçalves, I.S. Synthesis and characterization of CpMo(CO)3(CH2–pC6H4–CO2CH3) and its inclusion compounds with methylated cyclodextrins. Applications in olefin epoxidation catalysis. J. Organomet. Chem. 2013, 730, 116–122. [Google Scholar] [CrossRef]
- Sur, A.; Jernigan, N.B.; Powers, D.C. Kinetic Probes of the Origin of Activity in MOF-Based C-H Oxidation Catalysis. ACS Catal. 2022, 12, 3858–3867. [Google Scholar] [CrossRef]
- Karmadonova, I.E.; Zudin, V.N.; Kuznetsova, N.I.; Kuzhetsova, L.I.; Bal’zhinimaev, B.S. Preparation of Ethylbenzene and Isopropylbenzene Hydroperoxides in the N-Hydroxyphthalimide-Fe(III) Homogeneous Catalytic System and Use of Solutions in the Epoxidation of Olefins. Catal. Ind. 2020, 12, 216–225. [Google Scholar] [CrossRef]
- Chatterjee, D.; Mitra, A. Olefin epoxidation catalysed by Schiff-base complexes of Mn and Ni in heterogenised-homogeneous systems. J. Mol. Catal. A Chem. 1999, 144, 363–367. [Google Scholar] [CrossRef]
- Talzi, E.P.; Brylyakov, K.P.; Lyakin, O.Y.; Zima, A.M.; Soshnikov, I.E. NMR and EPR spectroscopy applied to homogeneous catalysis. Kinet. Catal. 2015, 56, 484–492. [Google Scholar] [CrossRef]
- Song, Z.; Liu, X.; Zhang, H.; Fang, D.; Ma, X. Investigation of physicochemical properties for novel perrhenate ionic liquid and its catalytic application towards epoxidation of olefins. J. Chem. Sci. 2021, 133, 23. [Google Scholar] [CrossRef]
- Escande, V.; Petit, E.; Garoux, L.; Boulanger, C.; Grison, C. Switchable Alkene Epoxidation/Oxidative Cleavage with H2O2/NaHCO3: Efficient Heterogeneous Catalysis Derived from Biosourced Eco-Mn. ACS Sustain. Chem. Eng. 2015, 3, 2704–2715. [Google Scholar] [CrossRef]
- Ren, J.; Wang, L.; Li, P.; Xing, X.; Wang, H.; Lv, B. Ag supported on alumina for the epoxidation of 1-hexene with molecular oxygen: The effect of Ag+/Ag0. New J. Chem. 2022, 46, 4792–4799. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, N.; Yu, J. Advances in Catalytic Applications of Zeolite-Supported Metal Catalysts. Adv. Mater. 2021, 33, 2104442. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Dai, Z.; Xiong, Y. Cobalt Porphyrin-Cross-Linked Poly(Ionic Liquid)s as Efficient Heterogeneous Catalysts for Carbon Dioxide Conversion under Mild Conditions. Ind. Eng. Chem. Res. 2021, 60, 18218–18229. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, H.; Guo, S.; Zeng, Y.; Ding, M. MOFs-induced high-amphiphilicity in hierarchical 3D reduced graphene oxide-based hydrogel. Appl. Surf. Sci. 2021, 540, 148303. [Google Scholar] [CrossRef]
- Mekrattanachai, P.; Liu, J.; Li, Z.; Cao, C.; Song, W. Extremely low loading of Ru species on hydroxyapatite as an effective heterogeneous catalyst for olefin epoxidation. Chem. Commun. 2018, 54, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Hazra, D.K.; Koner, S.; Helliwell, M.; Mukherjee, M.; Bhattacharjee, A. Hydrothermal synthesis of dimeric lanthanide compounds: X-ray structure, magnetic study and heterogeneous catalytic epoxidation of olefins. Polyhedron 2010, 29, 3183–3191. [Google Scholar] [CrossRef]
- Nur, H.; Ikeda, S.; Ohtani, B. Phase-Boundary Catalysis of Alkene Epoxidation with Aqueous Hydrogen Peroxide Using Amphiphilic Zeolite Particles Loaded with Titanium Oxide. J. Catal. 2001, 204, 402–408. [Google Scholar] [CrossRef]
- Duff, D.G.; Ohrenberg, A.; Voelkening, S.; Boll, M. A Screening Workflow for Synthesis and Testing of 10,000 Heterogeneous Catalysts per Day-Lessons Learned. Macromol. Rapid Commun. 2004, 25, 169–177. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, P.; Shen, Y.; Zhang, W.; Bian, G. Covalent anchoring of Mo(VI) Schiff base complex into SBA-15 as a novel heterogeneous catalyst for enhanced alkene epoxidation. J. Porous Mater. 2016, 23, 431–440. [Google Scholar] [CrossRef]
- Fernandes, C.I.; Silva, N.U.; Vaz, P.D.; Nunes, T.G.; Nunes, C.D. Bio-inspired Mo(II) complexes as active catalysts in homogeneous and heterogeneous olefin epoxidation. Appl. Catal. A Gen. 2010, 384, 84–93. [Google Scholar] [CrossRef]
- Torres, D.; Lopez, N.; Illas, F.; Lambert, R.M. Why Copper Is Intrinsically More Selective than Silver in Alkene Epoxidation: Ethylene Oxidation on Cu(111) versus Ag(111). J. Am. Chem. Soc. 2005, 127, 10774–10775. [Google Scholar] [CrossRef]
- Sha, S.; Yang, H.; Li, J.; Zhuang, C.; Gao, S.; Liu, S. Co(II) coordinated metal-organic framework: An efficient catalyst for heterogeneous aerobic olefins epoxidation. Catal. Commun. 2014, 43, 146–150. [Google Scholar] [CrossRef]
- Saha, D.; Gayen, S.; Koner, S. Cu(II)/Cu(II)-Mg(II) containing pyridine-2,5-dicarboxylate frameworks: Synthesis, structural diversity, inter-conversion and heterogeneous catalytic epoxidation. Polyhedron 2018, 146, 93–98. [Google Scholar] [CrossRef]
- Baumes, L.A.; Serra, J.M.; Serna, P.; Corma, A. Support Vector Machines for Predictive Modeling in Heterogeneous Catalysis: A Comprehensive Introduction and Overfitting Investigation Based on Two Real Applications. J. Comb. Chem. 2006, 8, 583–596. [Google Scholar] [CrossRef]
- Liu, T.-T.; Lin, Z.-J.; Shi, P.-C.; Ma, T.; Huang, Y.-B.; Cao, R. A Metallosalen-based Porous Organic Polymer for Olefin Epoxidation. ChemCatChem 2015, 7, 2340–2345. [Google Scholar] [CrossRef]
- Yang, F.; Wang, B.; Zhou, S.; Long, S.; Liu, X.; Kong, Y. Micropore-enriched CuO-based silica catalyst directly prepared by anionic template-induced method and its boosting catalytic activity in olefins epoxidation. Microporous Mesoporous Mater. 2017, 246, 215–224. [Google Scholar] [CrossRef]
- He, S.; Liu, X.; Zhao, H.; Zhu, Y.; Zhang, F. Zirconium phenylphosphonate-anchored methyltrioxorhenium as novel heterogeneous catalyst for epoxidation of cyclohexene. J. Colloid Interface Sci. 2015, 437, 58–64. [Google Scholar] [CrossRef]
- Baumes, L.A.; Blansché, A.; Serna, P.; Tchougang, A.; Lachiche, N.; Collet, P.; Corma, A. Using Genetic Programming for an Advanced Performance Assessment of Industrially Relevant Heterogeneous Catalysts. Mater. Manuf. Process. 2009, 24, 282–292. [Google Scholar] [CrossRef]
- Umsa, J.; Mingqiao, Z.; Xinzhi, C.; Zhangfa, T. Polyoxometalate-nano Gold Hybrid Mesostructured Catalyst for Green Cyclohexene Epoxidation. Curr. Org. Chem. 2017, 21, 2585–2596. [Google Scholar]
- He, X.; He, Q.; Deng, Y.; Peng, M.; Chen, H.; Zhang, Y.; Yao, S.; Zhang, M.; Xiao, D.; Ma, D.; et al. A versatile route to fabricate single atom catalysts with high chemoselectivity and regioselectivity in hydrogenation. Nat. Commun. 2019, 10, 3663. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, H.-Y.; Zhou, X.-T.; Chen, Y.-J.; Xue, C.; Ji, H.-B. Biomimetic Aerobic Epoxidation of Alkenes Catalyzed by Cobalt Porphyrin under Ambient Conditions in the Presence of Sunflower Seeds Oil as a Co-Substrate. ACS Omega 2020, 5, 4890–4899. [Google Scholar] [CrossRef]
- He, Q.; Zhang, Y.; Xiao, H.; He, X.; Zhou, X.; Ji, H. Facile Synthesis of Metalloporphyrins-Ba2+ Composites as Recyclable and Efficient Catalysts for Olefins Epoxidation Reactions. Chem. Res. Chin. Univ. 2019, 35, 251–255. [Google Scholar] [CrossRef]
- Ge, Y.; Su, X.; Li, G.; Yang, Y.-F.; She, Y. Oxidative functionalization of naphthalene derivatives catalyzed by Mn(III)-porphyrins. Tetrahedron Lett. 2023, 123, 154535. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, C.; She, Y.; Yang, Y.-F.; Houk, K.N. Molecular Dynamics of Iron Porphyrin-Catalyzed C-H Hydroxylation of Ethylbenzene. J. Am. Chem. Soc. 2023, 145, 14446–14455. [Google Scholar] [CrossRef]
- Yuan, R.; Wei, Y.; Xue, Z.; Wang, A.; Zhang, J.; Xu, H.; Zhao, L. Effects of support material and electrolyte on a triphenylamine substituted cobalt porphyrin catalytic oxygen reduction reaction. Colloids Surf. A Physicochem. Eng. Asp. 2023, 665, 131214. [Google Scholar] [CrossRef]
- Deeba, R.; Collard, A.; Rollin, C.; Molton, F.; Chardon-Noblat, S.; Costentin, C. Controlled Potential Electrolysis: Transition from Fast to Slow Regimes in Homogeneous Molecular Catalysis. Application to the Electroreduction of CO2 Catalyzed by Iron Porphyrin. ChemElectroChem 2023, 10, e202300350. [Google Scholar] [CrossRef]
- Wu, F.; Jiang, F.; Yang, J.; Dai, W.; Lan, D.; Shen, J.; Fang, Z. Investigation of Molecular Mechanism of Cobalt Porphyrin Catalyzed CO2 Electrochemical Reduction in Ionic Liquid by In-Situ SERS. Molecules 2023, 28, 2747. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Chen, X.; Su, X.; She, Y.-B.; Yang, Y.-F. Computational insights into the dual reactivity of 1,2,3,4-tetrazole: A metalloporphyrin-catalyzed click reaction and denitrogenative annulation. Org. Chem. Front. 2023, 10, 5055–5063. [Google Scholar] [CrossRef]
- Fan, J.; Wang, Y.; Hu, X.; Liu, Y.; Che, C.-M. Iron porphyrin-catalysed C(sp3)–H amination with alkyl azides for the synthesis of complex nitrogen-containing compounds. Org. Chem. Front. 2023, 10, 1368–1374. [Google Scholar] [CrossRef]
- Murata, S.; Kurosawa, M.; Fujisawa, T. Efficient synthesis of carbon-14 labeled metabolites of the strobilurin fungicide mandestrobin using biomimetic iron-porphyrin catalyzed oxidation. J. Label. Compd. Radiopharm. 2023, 66, 290–297. [Google Scholar] [CrossRef]
- Cao, Y.-C.; Shi, L.-L.; Li, M.; You, B.; Liao, R.-Z. Deciphering the Selectivity of the Electrochemical CO2 Reduction to CO by a Cobalt Porphyrin Catalyst in Neutral Aqueous Solution: Insights from DFT Calculations. ChemistryOpen 2023, 12, e202200254. [Google Scholar] [CrossRef]
- Zhou, X.-T.; Wang, L.-L.; Li, Y.; Ji, H.-B. Liquid-phase epoxidation of propylene with molecular oxygen by chloride manganese meso-tetraphenylporphyrins. Chin. J. Chem. Eng. 2022, 48, 61–65. [Google Scholar] [CrossRef]
- Zhou, X.-T.; Yu, H.-Y.; Li, Y.; Xue, C.; Ji, H.-B. Cerium(IV) Sulfate as a Cocatalyst for Promoting the Direct Epoxidation of Propylene by Ruthenium Porphyrin with Molecular Oxygen. Ind. Eng. Chem. Res. 2020, 59, 19982–19988. [Google Scholar] [CrossRef]
- Mahmoudi, H.; Bagherzadeh, M.; Ataie, S.; Kia, R.; Moravej, S.H.; Zare, M.; Raithby, P.R.; Ferlin, F.; Vaccaro, L. Synthesis and X-ray Crystal Structure of a Molybdenum(VI) Schiff Base Complex: Design of a New Catalytic System for Sustainable Olefin Epoxidation. Inorganica Chim. Acta 2020, 511, 119775. [Google Scholar] [CrossRef]
- Adam, M.S.S.; Ahmed, M.S.M.; El-Hady, O.M.; Shaaban, S. Bis-dioxomolybdenum (VI) oxalyldihydrazone complexes: Synthesis, characterization, DFT studies, catalytic epoxidation potential, molecular modeling and biological evaluations. Appl. Organomet. Chem. 2020, 34, e5573. [Google Scholar] [CrossRef]
- Mandal, M.; Nagaraju, V.; Karunakar, G.V.; Sarma, B.; Borah, B.J.; Bania, K.K. Electronic, Conjugation, and Confinement Effects on Structure, Redox, and Catalytic Behavior of Oxido-Vanadium(IV) and -(V) Chiral Schiff Base Complexes. J. Phys. Chem. C 2015, 119, 28854–28870. [Google Scholar] [CrossRef]
- Lazar, A.; Thiel, W.R.; Singh, A.P. Synthesis and characterization of 3-[N,N′-bis-3-(salicylidenamino)ethyltriamine] Mo(vi)O2@SBA-15: A highly stable and reusable catalyst for epoxidation and sulfoxidation reactions. RSC Adv. 2014, 4, 14063–14073. [Google Scholar] [CrossRef]
- Mirzaee, M.; Bahramian, B.; Shahraki, M.; Moghadam, H.; Mirzaee, A. Molybdenum Containing Catalysts Grafted on Functionalized Hydrous Zirconia Nano-particles for Epoxidation of Alkenes. Catal. Lett. 2018, 148, 3003–3017. [Google Scholar] [CrossRef]
- Ramanathan, A.; Wu, J.-F.; Maheswari, R.; Hu, Y.; Subramaniam, B. Synthesis of molybdenum-incorporated mesoporous silicates by evaporation-induced self-assembly: Insights into surface oxide species and corresponding olefin metathesis activity. Microporous Mesoporous Mater. 2017, 245, 118–125. [Google Scholar] [CrossRef]
- Bagherzadeh, M.; Zare, M.; Amani, V.; Ellern, A.; Keith Woo, L. Dioxo and oxo-peroxo molybdenum(VI) complexes bearing salicylidene 2-picoloyl hydrazone: Structures and catalytic performances. Polyhedron 2013, 53, 223–229. [Google Scholar] [CrossRef]
- Neves, P.; Lysenko, A.B.; Gomes, A.C.; Pillinger, M.; Gonçalves, I.S.; Valente, A.A. Behavior of Triazolylmolybdenum(VI) Oxide Hybrids as Oxidation Catalysts with Hydrogen Peroxide. Catal. Lett. 2017, 147, 1133–1143. [Google Scholar] [CrossRef]
- Schachner, J.A.; Mösch-Zanetti, N.C.; Peuronen, A.; Lehtonen, A. Dioxidomolybdenum(VI) and tungsten(VI) complexes with tetradentate amino bisphenolates as catalysts for epoxidation. Polyhedron 2017, 134, 73–78. [Google Scholar] [CrossRef]
- Lysenko, A.B.; Senchyk, G.A.; Domasevitch, K.V.; Kobalz, M.; Krautscheid, H.; Cichos, J.; Karbowiak, M.; Neves, P.; Valente, A.A.; Gonçalves, I.S. Triazolyl, Imidazolyl, and Carboxylic Acid Moieties in the Design of Molybdenum Trioxide Hybrids: Photophysical and Catalytic Behavior. Inorg. Chem. 2017, 56, 4380–4394. [Google Scholar] [CrossRef]
- Deubel, D.V.; Frenking, G.; Gisdakis, P.; Herrmann, W.A.; Rösch, N.; Sundermeyer, J. Olefin Epoxidation with Inorganic Peroxides. Solutions to Four Long-Standing Controversies on the Mechanism of Oxygen Transfer. Acc. Chem. Res. 2004, 37, 645–652. [Google Scholar] [CrossRef]
- Liu, X.; Gu, Q.; Zhang, Y.; Xu, X.; Wang, H.; Sun, Z.; Cao, L.; Sun, Q.; Xu, L.; Wang, L.; et al. Atomically Thick Oxide Overcoating Stimulates Low-Temperature Reactive Metal–Support Interactions for Enhanced Catalysis. J. Am. Chem. Soc. 2023, 145, 6702–6709. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanloo, M.; Heydari, A.; Yahiro, H. Ag-nanoparticle embedded p(AA) hydrogel as an efficient green heterogeneous Nano-catalyst for oxidation and reduction of organic compounds. Appl. Organomet. Chem. 2018, 32, e3917. [Google Scholar] [CrossRef]
- Chakraborty, T.; Chakraborty, A.; Maity, S.; Das, D.; Chattopadhyay, T. Conglomerated system of Ag nanoparticles decorated Al2O3 supported cobalt and copper complexes with enhanced catalytic activity for oxidation reactions. Mol. Catal. 2019, 462, 104–113. [Google Scholar] [CrossRef]
- Hoseinzade, K.; Mousavi-Mashhadi, S.A.; Shiri, A. An efficient and green one-pot synthesis of tetrahydrobenzo[a]xanthenes, 1,8-dioxo-octahydroxanthenes and dibenzo[a,j]xanthenes by Fe3O4@Agar-Ag as nanocatalyst. Mol. Divers. 2022, 26, 2745–2759. [Google Scholar] [CrossRef]
- Khan, M.; Janjua, N.K.; Khan, S.; Qazi, I.; Ali, S.; Saad Algarni, T. Electro-Oxidation of Ammonia at Novel Ag2O−PrO2/γ-Al2O3 Catalysts. Coatings 2021, 11, 257. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, H.; Feng, Z.; Zhang, Y.; Cui, M.; Cao, D.; Cheng, D. Design of High-Performance Co-Based Alloy Nanocatalysts for the Oxygen Reduction Reaction. Chem.-A Eur. J. 2020, 26, 4128–4135. [Google Scholar] [CrossRef]
- Mullangi, D.; Chakraborty, D.; Pradeep, A.; Koshti, V.; Vinod, C.P.; Panja, S.; Nair, S.; Vaidhyanathan, R. Heterogenous Catalysts: Highly Stable COF-Supported Co/Co(OH)2 Nanoparticles Heterogeneous Catalyst for Reduction of Nitrile/Nitro Compounds under Mild Conditions (Small 37/2018). Small 2018, 14, 1870169. [Google Scholar] [CrossRef]
- Gholinejad, M.; Zareh, F.; Najera, C. Iron oxide modified with pyridyl-triazole ligand for stabilization of gold nanoparticles: An efficient heterogeneous catalyst for A3 coupling reaction in water. Appl. Organomet. Chem. 2018, 32, e4454. [Google Scholar] [CrossRef]
- Liu, Y.; Li, W.; Zhao, G.; Qin, G.; Li, Y.; Liu, Y. Self-driven microstructural evolution of Au@Pd core–shell nanoparticles for greatly enhanced catalytic performance during methanol electrooxidation. Nanoscale 2021, 13, 3528–3542. [Google Scholar] [CrossRef]
- Yazdani, H.; Pardis, S.; Loni, M.; Bazgir, A. Gold nanoparticle as a Lewis acid catalyst in 1,3-dipolar cycloaddition reaction. Catal. Commun. 2020, 134, 105844. [Google Scholar] [CrossRef]
- Zhao, J.; Yuan, H.; Yang, G.; Liu, Y.; Qin, X.; Chen, Z.; Weng-Chon, C.; Zhou, L.; Fang, S. AuPt bimetallic nanoalloys supported on SBA-15: A superior catalyst for quinoline selective hydrogenation in water. Nano Res. 2022, 15, 1796–1802. [Google Scholar] [CrossRef]
- Chakraborty, D.; Nandi, S.; Mullangi, D.; Haldar, S.; Vinod, C.P.; Vaidhyanathan, R. Cu/Cu2O Nanoparticles Supported on a Phenol–Pyridyl COF as a Heterogeneous Catalyst for the Synthesis of Unsymmetrical Diynes via Glaser-Hay Coupling. ACS Appl. Mater. Interfaces 2019, 11, 15670–15679. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Jin, Y.; Ge, M. Simple and green method for preparing copper nanoparticles supported on carbonized cotton as a heterogeneous Fenton-like catalyst. Colloids Surf. A Physicochem. Eng. Asp. 2022, 647, 128978. [Google Scholar] [CrossRef]
- Dehghani, F.; Sardarian, A.R.; Esmaeilpour, M. Salen complex of Cu(II) supported on superparamagnetic Fe3O4@SiO2 nanoparticles: An efficient and recyclable catalyst for synthesis of 1- and 5-substituted 1H-tetrazoles. J. Organomet. Chem. 2013, 743, 87–96. [Google Scholar] [CrossRef]
- Xu, Q.; Guo, C.; Li, B.; Zhang, Z.; Qiu, Y.; Tian, S.; Zheng, L.; Gu, L.; Yan, W.; Wang, D.; et al. Al3+ Dopants Induced Mg2+ Vacancies Stabilizing Single-Atom Cu Catalyst for Efficient Free-Radical Hydrophosphinylation of Alkenes. J. Am. Chem. Soc. 2022, 144, 4321–4326. [Google Scholar] [CrossRef]
- Geesi, M.H.; Ouerghi, O.; Elsanousi, A.; Kaiba, A.; Riadi, Y. Ultrasound-Assisted Preparation of Cu-Doped TiO2 Nanoparticles as a Nanocatalyst for Sonochemical Synthesis of Pyridopyrimidines. Polycycl. Aromat. Compd. 2021, 42, 80–90. [Google Scholar] [CrossRef]
- Liu, K.; Qin, R.; Zheng, N. Insights into the Interfacial Effects in Heterogeneous Metal Nanocatalysts toward Selective Hydrogenation. J. Am. Chem. Soc. 2021, 143, 4483–4499. [Google Scholar] [CrossRef]
- Imran, M.; Yousaf, A.B.; Zhou, X.; Jiang, Y.-F.; Yuan, C.-Z.; Zeb, A.; Jiang, N.; Xu, A.-W. Pd/TiO Nanocatalyst with Strong Metal-Support Interaction for Highly Efficient Durable Heterogeneous Hydrogenation. J. Phys. Chem. C 2017, 121, 1162–1170. [Google Scholar] [CrossRef]
- Das, M.R.; Hussain, N.; Duarah, R.; Sharma, N.; Sarmah, P.; Thakur, A.; Bhattacharjee, P.; Bora, U.; Boukherroub, R. Metal nanoparticles decorated two-dimensional nanosheets as heterogeneous catalysts for coupling reactions. Catal. Rev. 2024, 66, 923–995. [Google Scholar] [CrossRef]
- Bisio, C.; Carniato, F.; Palumbo, C.; Safronyuk, S.L.; Starodub, M.F.; Katsev, A.M.; Marchese, L.; Guidotti, M. Nanosized inorganic metal oxides as heterogeneous catalysts for the degradation of chemical warfare agents. Catal. Today 2016, 277, 192–199. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Navalon, S.; Alvaro, M.; Garcia, H. Metal Nanoparticles as Heterogeneous Fenton Catalysts. ChemSusChem 2012, 5, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Gawande, M.B.; Goswami, A.; Felpin, F.-X.; Asefa, T.; Huang, X.; Silva, R.; Zou, X.; Zboril, R.; Varma, R.S. Cu and Cu-Based Nanoparticles: Synthesis and Applications in Catalysis. Chem. Rev. 2016, 116, 3722–3811. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, T.; Wu, P.; Tatsumi, T. Selective oxidation of propylene to propylene oxide over Ti-MCM-41 supporting metal nitrate. Catal. Today 2001, 71, 169–176. [Google Scholar] [CrossRef]
- Sharma, A.S.; Sharma, V.S.; Kaur, H.; Varma, R.S. Supported heterogeneous nanocatalysts in sustainable, selective and eco-friendly epoxidation of olefins. Green Chem. 2020, 22, 5902–5936. [Google Scholar] [CrossRef]
- Tyablikov, I.; Romanovsky, B. A heterogeneous organocatalyst for olefin epoxidation. Catal. Today 2016, 278, 40–44. [Google Scholar] [CrossRef]
- van Hoof, A.J.F.; Filot, I.A.W.; Friedrich, H.; Hensen, E.J.M. Reversible Restructuring of Silver Particles during Ethylene Epoxidation. ACS Catal. 2018, 8, 11794–11800. [Google Scholar] [CrossRef]
- Lei, Y.; Mehmood, F.; Lee, S.; Greeley, J.; Lee, B.; Seifert, S.; Winans, R.E.; Elam, J.W.; Meyer, R.J.; Redfern, P.C.; et al. Increased Silver Activity for Direct Propylene Epoxidation via Subnanometer Size Effects. Science 2010, 328, 224–228. [Google Scholar] [CrossRef]
- Kim, K.J.; Rath, M.K.; Kwak, H.H.; Kim, H.J.; Han, J.W.; Hong, S.-T.; Lee, K.T. A Highly Active and Redox-Stable SrGdNi0.2Mn0.8O4±δ Anode with in Situ Exsolution of Nanocatalysts. ACS Catal. 2019, 9, 1172–1182. [Google Scholar] [CrossRef]
- Yang, H.; Li, G.; Jiang, G.; Zhang, Z.; Hao, Z. Heterogeneous selective oxidation over supported metal catalysts: From nanoparticles to single atoms. Appl. Catal. B Environ. 2023, 325, 122384. [Google Scholar] [CrossRef]
- Yang, H.; Ma, C.; Li, Y.; Wang, J.; Zhang, X.; Wang, G.; Qiao, N.; Sun, Y.; Cheng, J.; Hao, Z. Synthesis, characterization and evaluations of the Ag/ZSM-5 for ethylene oxidation at room temperature: Investigating the effect of water and deactivation. Chem. Eng. J. 2018, 347, 808–818. [Google Scholar] [CrossRef]
- Yang, H.; Ma, C.; Wang, G.; Sun, Y.; Cheng, J.; Zhang, Z.; Zhang, X.; Hao, Z. Fluorine-enhanced Pt/ZSM-5 catalysts for low-temperature oxidation of ethylene. Catal. Sci. Technol. 2018, 8, 1988–1996. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Y.; Zhang, X.; Deng, H.; He, H.; Li, Y. Promotion Effect of H2 on Ethanol Oxidation and NOx Reduction with Ethanol over Ag/Al2O3 Catalyst. Environ. Sci. Technol. 2015, 49, 481–488. [Google Scholar] [CrossRef]
- Yang, H.; Ma, C.; Zhang, X.; Li, Y.; Cheng, J.; Hao, Z. Understanding the Active Sites of Ag/Zeolites and Deactivation Mechanism of Ethylene Catalytic Oxidation at Room Temperature. ACS Catal. 2018, 8, 1248–1258. [Google Scholar] [CrossRef]
- Christopher, P.; Linic, S. Engineering Selectivity in Heterogeneous Catalysis: Ag Nanowires as Selective Ethylene Epoxidation Catalysts. J. Am. Chem. Soc. 2008, 130, 11264–11265. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Hueso, J.L.; Suarez, H.; Mallada, R.; Ibarra, A.; Irusta, S.; Santamaria, J. A Nanoarchitecture Based on Silver and Copper Oxide with an Exceptional Response in the Chlorine-Promoted Epoxidation of Ethylene. Angew. Chem. Int. Ed. 2016, 55, 11158–11161. [Google Scholar] [CrossRef]
- Jing, X.; Wang, H.; Chen, H.; Huang, J.; Li, Q.; Sun, D. Biosynthesized Ag/α-Al2O3 catalyst for ethylene epoxidation: The influence of silver precursors. RSC Adv. 2014, 4, 27597–27603. [Google Scholar] [CrossRef]
- Ghosh, S.; Acharyya, S.S.; Tiwari, R.; Sarkar, B.; Singha, R.K.; Pendem, C.; Sasaki, T.; Bal, R. Selective Oxidation of Propylene to Propylene Oxide over Silver-Supported Tungsten Oxide Nanostructure with Molecular Oxygen. ACS Catal. 2014, 4, 2169–2174. [Google Scholar] [CrossRef]
- Lu, G.; Zuo, X. Epoxidation of propylene by air over modified silver catalyst. Catal. Lett. 1999, 58, 67–70. [Google Scholar] [CrossRef]
- Lee, E.J.; Lee, J.; Seo, Y.-J.; Lee, J.W.; Ro, Y.; Yi, J.; Song, I.K. Direct epoxidation of propylene to propylene oxide with molecular oxygen over Ag-Mo-W/ZrO2 catalysts. Catal. Commun. 2017, 89, 156–160. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature far Below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Hutchings, G.J. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Haruta, M. Spiers Memorial Lecture Role of perimeter interfaces in catalysis by gold nanoparticles. Faraday Discuss. 2011, 152, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Uphade, B.S.; Tsubota, S.; Miyamoto, A. Selective oxidation of propylene over gold deposited on titanium-based oxides. Res. Chem. Intermed. 1998, 24, 329–336. [Google Scholar] [CrossRef]
- Uphade, B.S.; Akita, T.; Nakamura, T.; Haruta, M. Vapor-Phase Epoxidation of Propene Using H2 and O2 over Au/Ti–MCM-48. J. Catal. 2002, 209, 331–340. [Google Scholar] [CrossRef]
- Kapoor, M.P.; Sinha, A.K.; Seelan, S.; Inagaki, S.; Tsubota, S.; Yoshida, H.; Haruta, M. Hydrophobicity induced vapor-phase oxidation of propene over gold supported on titanium incorporated hybrid mesoporous silsesquioxane. Chem. Commun. 2002, 23, 2902–2903. [Google Scholar] [CrossRef]
- Chen, S.; Li, D.; Cao, T.; Huang, W. Size-Dependent Structures and Catalytic Performances of Au/TiO2-{001} Catalysts for Propene Epoxidation. J. Phys. Chem. C 2020, 124, 15264–15274. [Google Scholar] [CrossRef]
- Uphade, B.S.; Okumura, M.; Tsubota, S.; Haruta, M. Effect of physical mixing of CsCl with Au/Ti-MCM-41 on the gas-phase epoxidation of propene using H2 and O2: Drastic depression of H2 consumption. Appl. Catal. A Gen. 2000, 190, 43–50. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, Y.; Du, W.; Xu, J.; Wang, Q.; Song, N.; Qian, G.; Duan, X.; Zhou, X. Engineering gold impregnated uncalcined TS-1 to boost catalytic formation of propylene oxide. Appl. Catal. B: Environ. 2022, 319, 121837. [Google Scholar] [CrossRef]
- Pulido, A.; Boronat, M.; Corma, A. Propene Epoxidation with H2/H2O/O2 Mixtures Over Gold Atoms Supported on Defective Graphene: A Theoretical Study. J. Phys. Chem. C 2012, 116, 19355–19362. [Google Scholar] [CrossRef]
- Lu, J.; Luo, M.; Lei, H.; Bao, X.; Li, C. Epoxidation of Propylene on NaCl-Modified VCe1-x Cux Oxide Catalysts with Direct Molecular Oxygen as the Oxidant. J. Catal. 2002, 211, 552–555. [Google Scholar] [CrossRef]
- Monnier, J.R.; Hartley, G.W. Comparison of Cu and Ag Catalysts for Epoxidation of Higher Olefins. J. Catal. 2001, 203, 253–256. [Google Scholar] [CrossRef]
- Hua, Q.; Cao, T.; Gu, X.-K.; Lu, J.; Jiang, Z.; Pan, X.; Luo, L.; Li, W.-X.; Huang, W. Crystal-Plane-Controlled Selectivity of Cu2O Catalysts in Propylene Oxidation with Molecular Oxygen. Angew. Chem. Int. Ed. 2014, 53, 4856–4861. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Zhai, Q.; He, J.; Zhang, Q.; Deng, W.; Wang, Y. Significant Synergistic Effect between Supported Ruthenium and Copper Oxides for Propylene Epoxidation by Oxygen. ChemPlusChem 2012, 77, 27–30. [Google Scholar] [CrossRef]
- Seubsai, A.; Uppala, C.; Tiencharoenwong, P.; Chukeaw, T.; Chareonpanich, M.; Zohour, B.; Noon, D.; Senkan, S. High Stability of Ruthenium-Copper-Based Catalysts for Epoxidation of Propylene. Catal. Lett. 2018, 148, 586–600. [Google Scholar] [CrossRef]
- Lei, J.; Dai, J.; Tan, K.B.; Huang, J.; Zhan, G.; Li, Q. Insight into the Effect of Copper Substitution on the Catalytic Performance of LaCoO3-Based Catalysts for Direct Epoxidation of Propylene with Molecular Oxygen. ACS Sustain. Chem. Eng. 2021, 9, 794–808. [Google Scholar] [CrossRef]
- He, J.; Zhai, Q.; Zhang, Q.; Deng, W.; Wang, Y. Active site and reaction mechanism for the epoxidation of propylene by oxygen over CuOx/SiO2 catalysts with and without Cs+ modification. J. Catal. 2013, 299, 53–66. [Google Scholar] [CrossRef]
- Song, Z.; Mimura, N.; Bravo-Suárez, J.J.; Akita, T.; Tsubota, S.; Oyama, S.T. Gas-phase epoxidation of propylene through radicals generated by silica-supported molybdenum oxide. Appl. Catal. A Gen. 2007, 316, 142–151. [Google Scholar] [CrossRef]
- Murata, K.; Liu, Y.; Mimura, N.; Inaba, M. Direct vapor phase oxidation of propylene by molecular oxygen over MCM-41 or MCM-22 based catalysts. Catal. Commun. 2003, 4, 385–391. [Google Scholar] [CrossRef]
- Yan, W.; Ramanathan, A.; Patel, P.D.; Maiti, S.K.; Laird, B.B.; Thompson, W.H.; Subramaniam, B. Mechanistic insights for enhancing activity and stability of Nb-incorporated silicates for selective ethylene epoxidation. J. Catal. 2016, 336, 75–84. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Jin, H.; Zhou, K.; Zhang, R.; Cui, H.; Yu, Y.; Cui, P.; Song, W.; Cao, C. Regulating the electronic structure through charge redistribution in dense single-atom catalysts for enhanced alkene epoxidation. Nat. Commun. 2023, 14, 2494. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xu, D.; Wang, L.; Cui, S.; Liu, Q.; Han, X.; Wang, Z.; Ning, H.; Wu, M. A core-shell confinement strategy towards single-atom Fe-N/S-C bifunctional catalyst for selective nitroarene reduction and olefin epoxidation. J. Alloys Compd. 2025, 1012, 178488. [Google Scholar] [CrossRef]
- Niatouri, A.D.; Yadollahi, B. A novel POM/LDH/GO nanocomposite as highly efficient heterogeneous catalyst in green epoxidation of alkenes with hydrogen peroxide. Catal. Commun. 2023, 185, 106808. [Google Scholar] [CrossRef]
- Yuan, J.; Meng, H.; Li, Y.; Liu, Y.; Wang, Y.; Liu, J.; Zhou, Z. An ionic liquid in Core-shell structure: Halogen-free, metal-free bifunctional catalyst for olefin epoxidation and CO2 cycloaddition. J. CO2 Util. 2024, 86, 102906. [Google Scholar] [CrossRef]
- Ozbek, M.O.; Onal, I.; van Santen, R.A. Effect of Surface and Oxygen Coverage on Ethylene Epoxidation. Top. Catal. 2012, 55, 710–717. [Google Scholar] [CrossRef]
- Linic, S.; Barteau, M.A. Formation of a Stable Surface Oxametallacycle that Produces Ethylene Oxide. J. Am. Chem. Soc. 2002, 124, 310–317. [Google Scholar] [CrossRef]
- Linic, S.; Barteau, M.A. Control of Ethylene Epoxidation Selectivity by Surface Oxametallacycles. J. Am. Chem. Soc. 2003, 125, 4034–4035. [Google Scholar] [CrossRef]
- Linic, S.; Piao, H.; Adib, K.; Barteau, M.A. Ethylene Epoxidation on Ag: Identification of the Crucial Surface Intermediate by Experimental and Theoretical Investigation of its Electronic Structure. Angew. Chem. Int. Ed. 2004, 43, 2918–2921. [Google Scholar] [CrossRef]
- Christopher, P.; Linic, S. Shape and Size-Specific Chemistry of Ag Nanostructures in Catalytic Ethylene Epoxidation. ChemCatChem 2010, 2, 78–83. [Google Scholar] [CrossRef]
- Özbek, M.O.; van Santen, R.A. The Mechanism of Ethylene Epoxidation Catalysis. Catal. Lett. 2013, 143, 131–141. [Google Scholar] [CrossRef]
- Ozbek, M.O.; Onal, I.; Van Santen, R.A. Ethylene epoxidation catalyzed by chlorine-promoted silver oxide. J. Phys. Condens. Matter 2011, 23, 404202. [Google Scholar] [CrossRef] [PubMed]
- Ozbek, M.O.; Onal, I.; van Santen, R.A. Why silver is the unique catalyst for ethylene epoxidation. J. Catal. 2011, 284, 230–235. [Google Scholar] [CrossRef]
- Clerici, M.G.; Ingallina, P. Epoxidation of Lower Olefins with Hydrogen Peroxide and Titanium Silicalite. J. Catal. 1993, 140, 71–83. [Google Scholar] [CrossRef]
- Xiong, C.; Liu, H.; Zhou, J.; Xu, D.; Liang, Y.; Zhou, X.; Xue, C.; Ji, H. High selective epoxidation of 2-methylpropene over a Mo-based oxametallacycle reinforced nano composite. Nano Res. 2023, 16, 209–218. [Google Scholar] [CrossRef]
- Li, J.; Zhou, X.; Ao, J.; Gao, J.; Wang, A.; Shen, Z.; Gu, Y.; Zhou, J.; Chen, Y. P450DA Monooxygenase-Catalyzed Chemoselective and Enantiodivergent Epoxidation of Unactivated Alkenes. ACS Catal. 2024, 14, 16175–16183. [Google Scholar] [CrossRef]
- Wu, H.; Xu, Y.; Ao, J.; Guo, P.; Xu, Y.; Huang, Z.; Zhang, L. Electrochemical epoxidation of alkene with high faradaic efficiencies using water as an oxygen source. Green Chem. 2024, 26, 2922–2927. [Google Scholar] [CrossRef]
- Kim, S.S.; Hong, S.; Surendran, A.K.; Roy, A.; Malik, D.D.; Chun, D.; Kim, S.; Kim, Y.; Lee, Y.M.; Lee, Y.H.; et al. Electrochemically Driven Selective Olefin Epoxidation by Cobalt-TAML Catalyst. J. Am. Chem. Soc. 2025, 147, 5269–5278. [Google Scholar] [CrossRef]
- Gadolini, S.; Kerber, R.N.; Seljamäe-Green, R.T.; Tong, W.; Farràs, P.; Corbos, E.C. Covalently Anchored Molecular Catalyst onto a Graphitic Carbon Nitride Surface for Photocatalytic Epoxidation of Olefins. ACS Catal. 2024, 14, 14639–14651. [Google Scholar] [CrossRef]



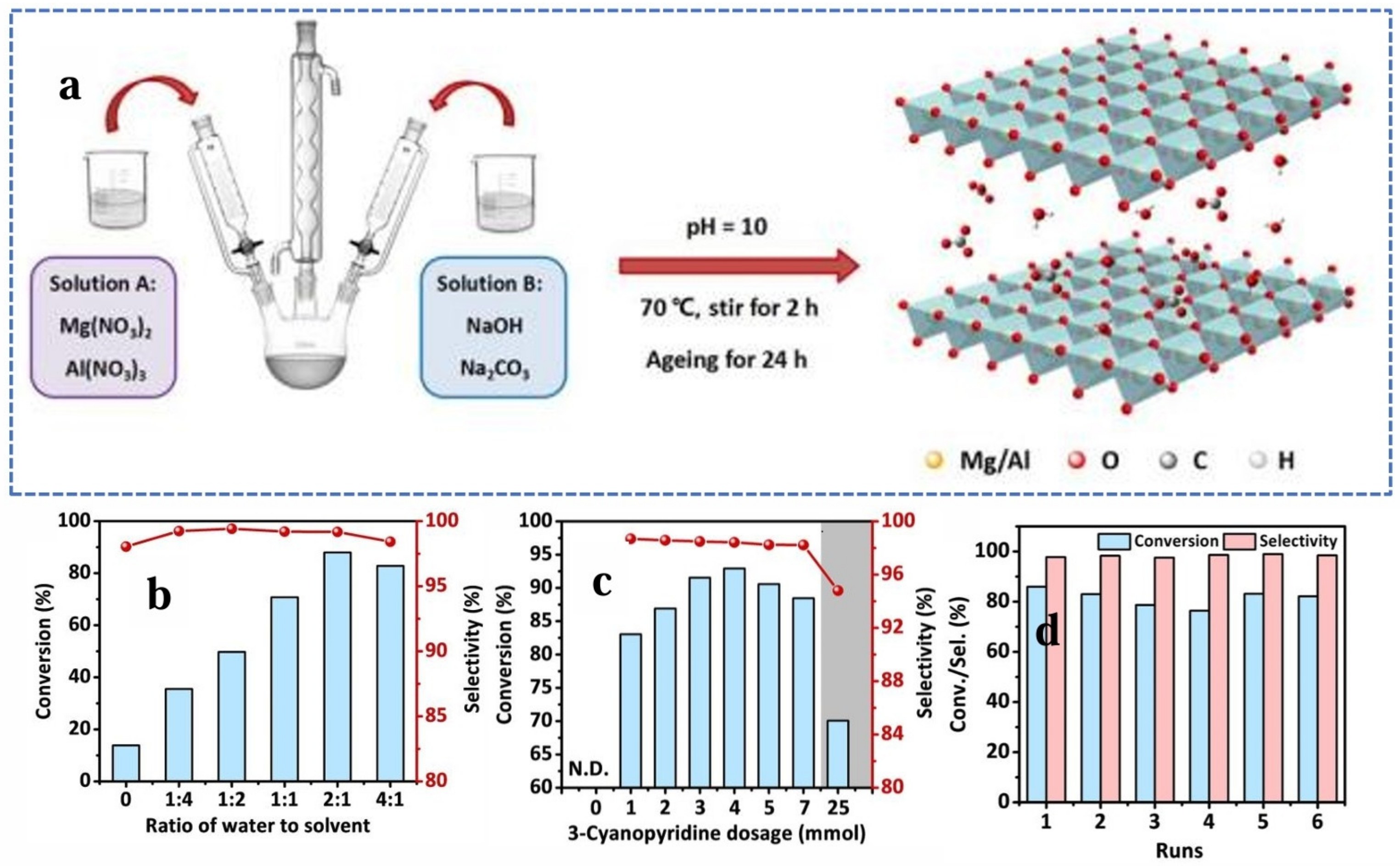

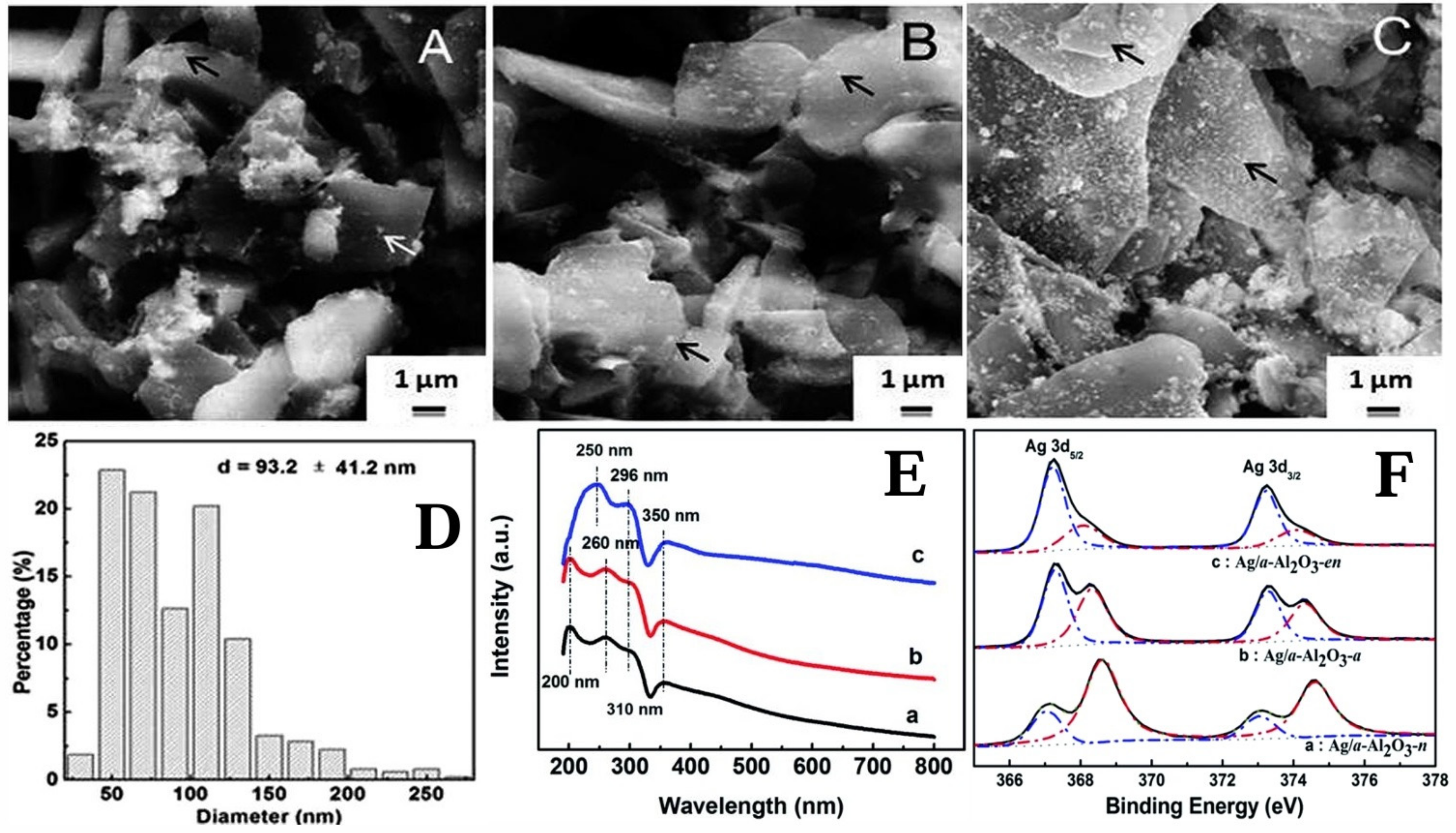
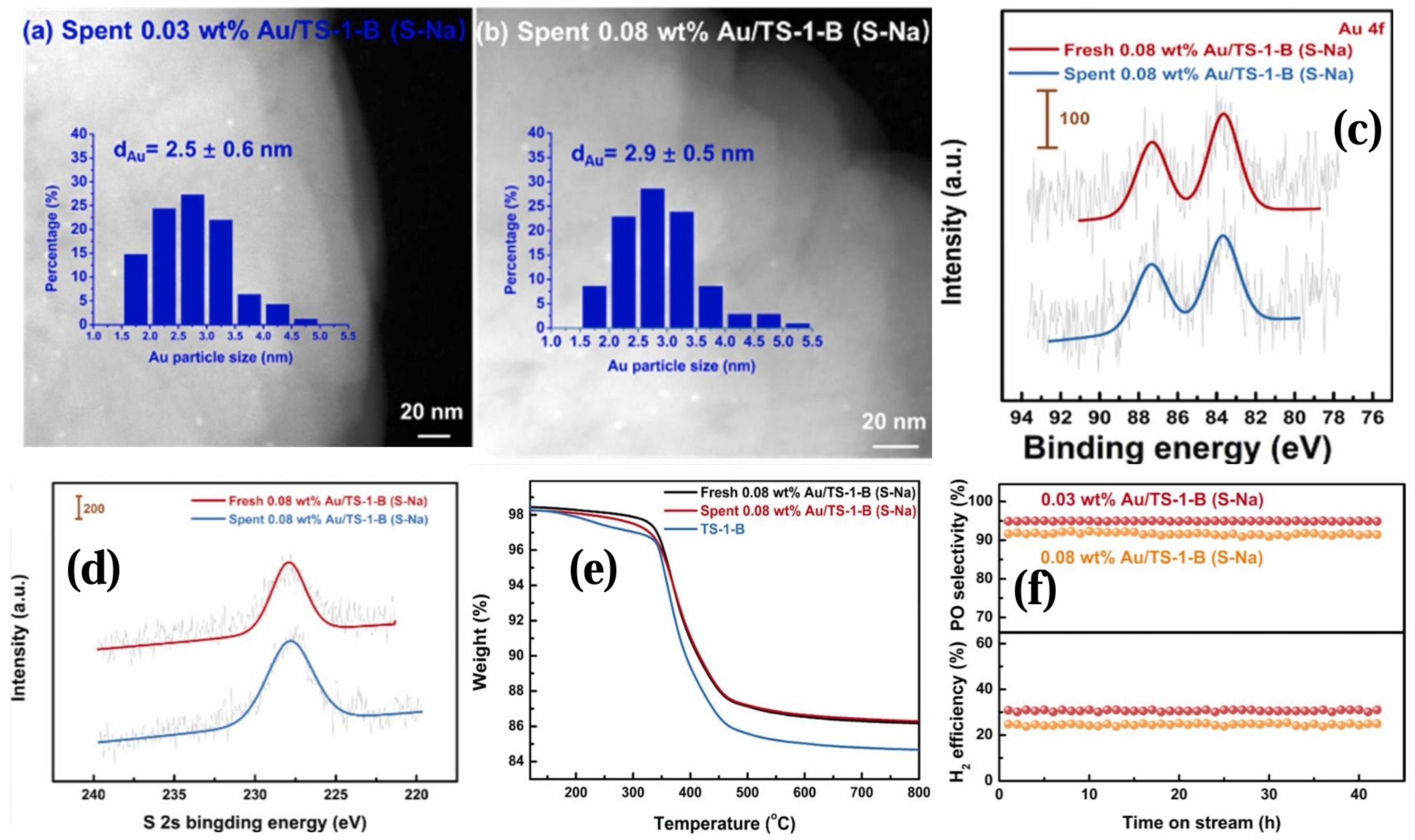
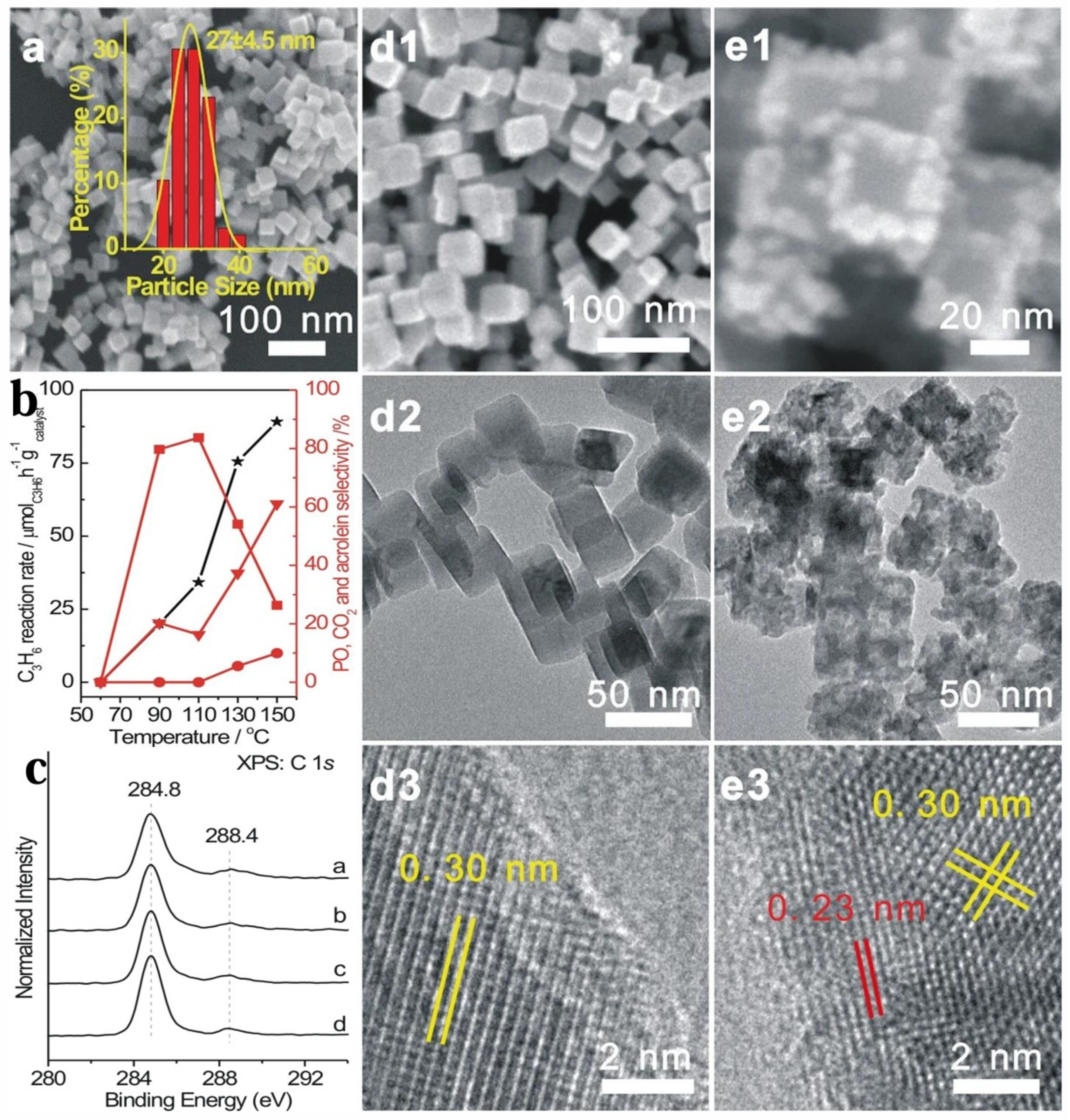


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epoxidation Processes | Items | Details |
|---|---|---|
| Chlorohydrin Method | Reaction equation | |
| Advantages | The process was mature, the flow was simple, the selectivity was high, and the requirement for raw material purity was low. | |
| Disadvantages | The process consumed a large amount of water, generated a great deal of wastewater and waste residue, polluted the environment, corroded the equipment, and had high energy consumption. | |
| HPPO Method | Reaction equation | |
| Advantages | The process flow was simple, the product yield was high, few by-products were generated, and basically no pollutants such as waste residue were generated | |
| Disadvantages | The product was single, the risk-resistance ability was weak, there were transportation safety problems and storage difficulties, the process flow was long, and the investment cost was high. | |
| Oxygen/Air Direct Oxidation Method | Reaction equation | |
| Advantages | The process had a high atomic utilization rate, a short process flow, no by-products, a wide range of raw material sources and relatively low costs. | |
| Disadvantages | The process had relatively harsh reaction conditions, under—developed catalysts and technical bottlenecks in industrial promotion. | |
| Ethylbenzene Co-oxidation Method | Reaction equation | |
| Advantages | The process had little environmental pollution, and the co-products like styrene could share the cost. | |
| Disadvantages | The technological process was long, the pressure was high, and the reaction conditions were harsh. | |
| Isobutane Co-oxidation Method | Reaction equation | |
| Advantages | The process had little environmental pollution, and the co-products like tert-butanol could share the cost. | |
| Disadvantages | The technological process was long, the pressure was high, and the reaction conditions were harsh. | |
| Cumene Co-oxidation Method | Reaction equation | |
| Advantages | There was less wastewater, less corrosion of the equipment, and there were no by-products (cumene could be recycled). | |
| Disadvantages | The technological process was complex, and the investment cost was high. | |
| Biomimetic Catalytic Oxidation Method | Reaction equation | |
| Advantages | The reaction conditions were mild, the selectivity was high, and it was relatively environmentally friendly. | |
| Disadvantages | The cost was relatively high, and the catalytic efficiency in large-scale industrial production might be insufficient to meet the requirements, with the presence of a scale-up effect. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, G.; Yang, T.; Liu, J.; Xu, X.; Wang, Y.; Zhang, Y.; Gao, M.; Xiong, C.; Ji, H. Research Progress in Epoxidation of Light Small-Molecule Olefins. Molecules 2025, 30, 1340. https://doi.org/10.3390/molecules30061340
Zhao G, Yang T, Liu J, Xu X, Wang Y, Zhang Y, Gao M, Xiong C, Ji H. Research Progress in Epoxidation of Light Small-Molecule Olefins. Molecules. 2025; 30(6):1340. https://doi.org/10.3390/molecules30061340
Chicago/Turabian StyleZhao, Guanghui, Tianfu Yang, Jincheng Liu, Xianming Xu, Yulong Wang, Yongjun Zhang, Meng Gao, Chao Xiong, and Hongbing Ji. 2025. "Research Progress in Epoxidation of Light Small-Molecule Olefins" Molecules 30, no. 6: 1340. https://doi.org/10.3390/molecules30061340
APA StyleZhao, G., Yang, T., Liu, J., Xu, X., Wang, Y., Zhang, Y., Gao, M., Xiong, C., & Ji, H. (2025). Research Progress in Epoxidation of Light Small-Molecule Olefins. Molecules, 30(6), 1340. https://doi.org/10.3390/molecules30061340