
Design, Synthesis, and Evaluation of Camptothecin-Based Antibody–Drug Conjugates with High Hydrophilicity and Structural Stability


Abstract
1. Introduction
2. Results
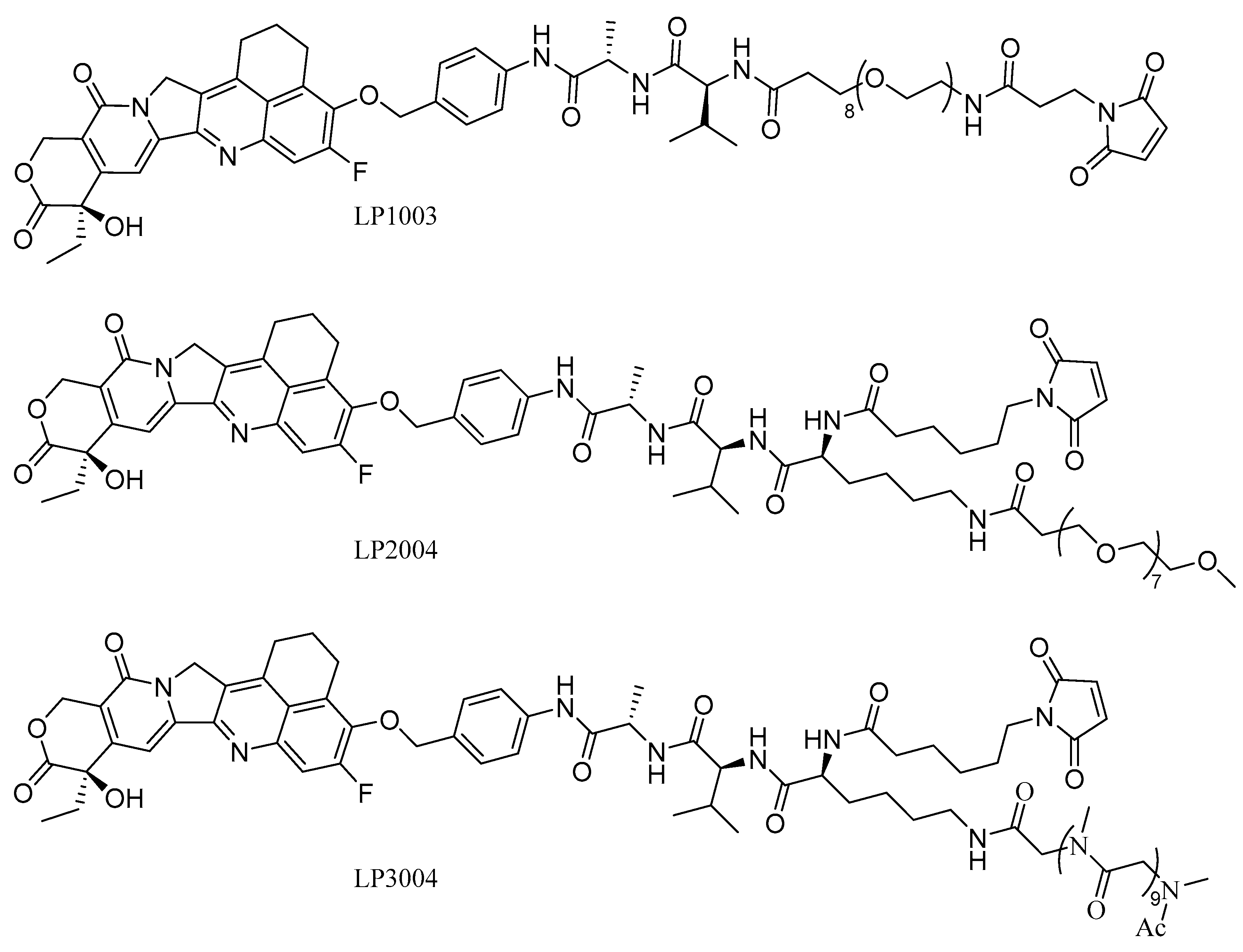
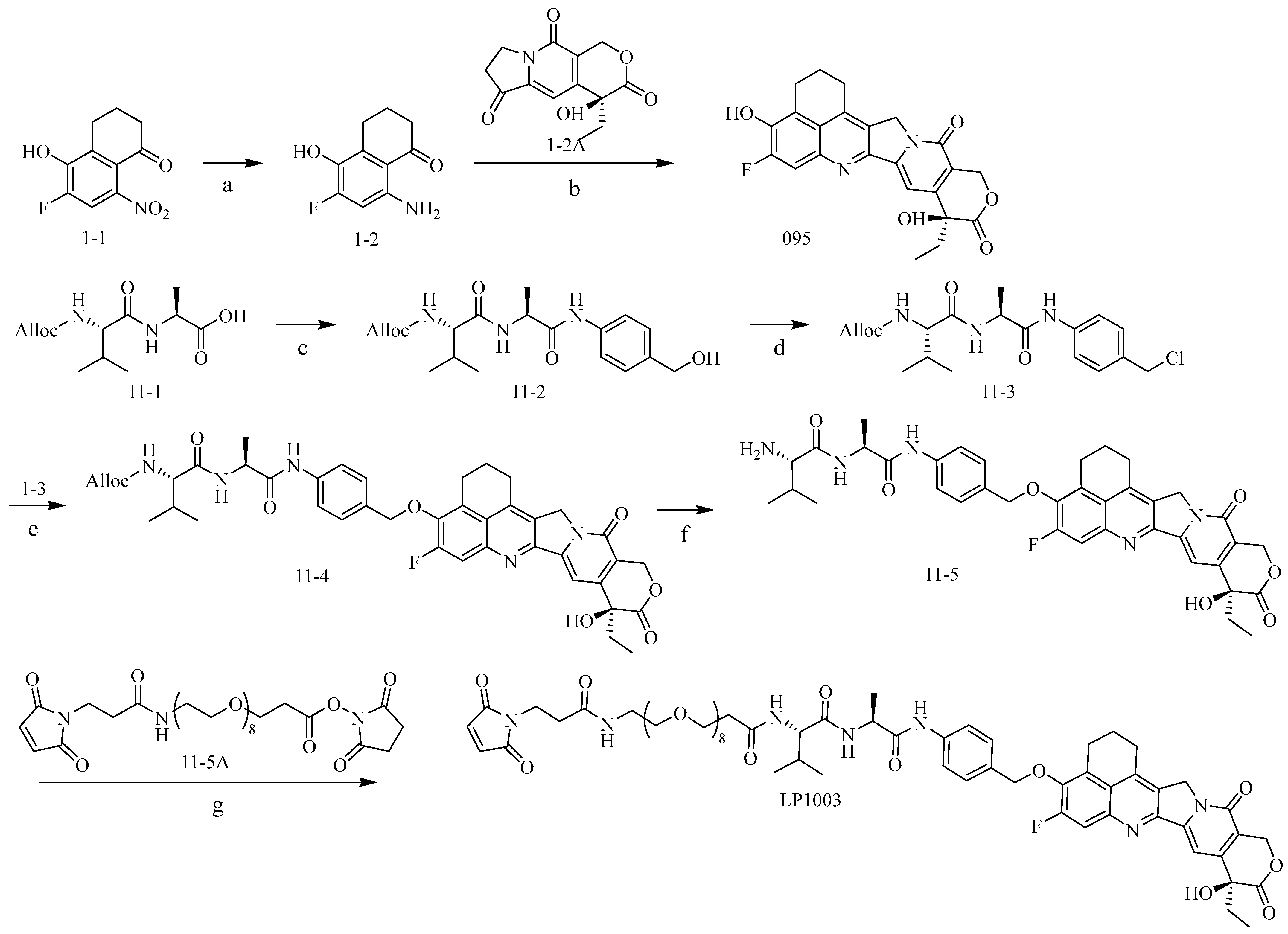
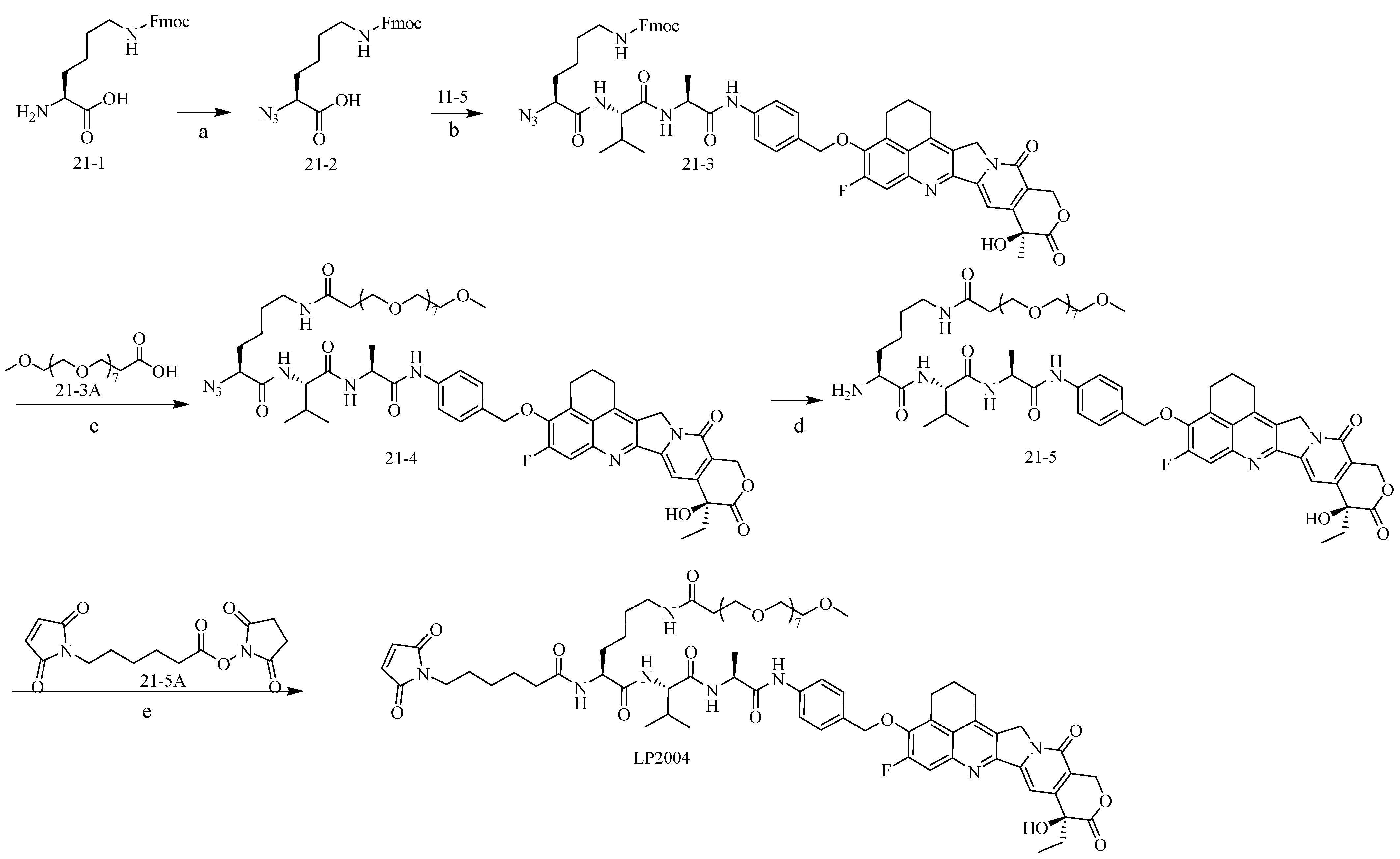
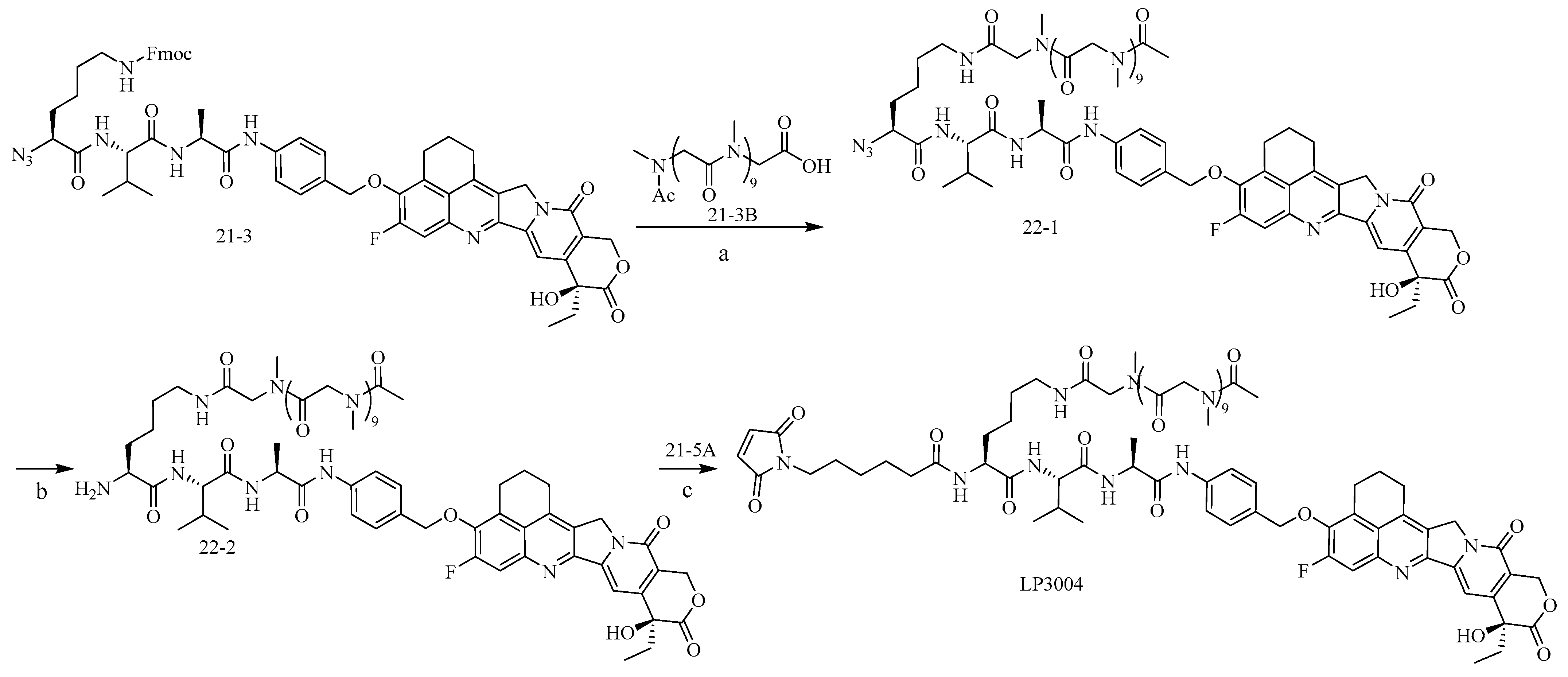
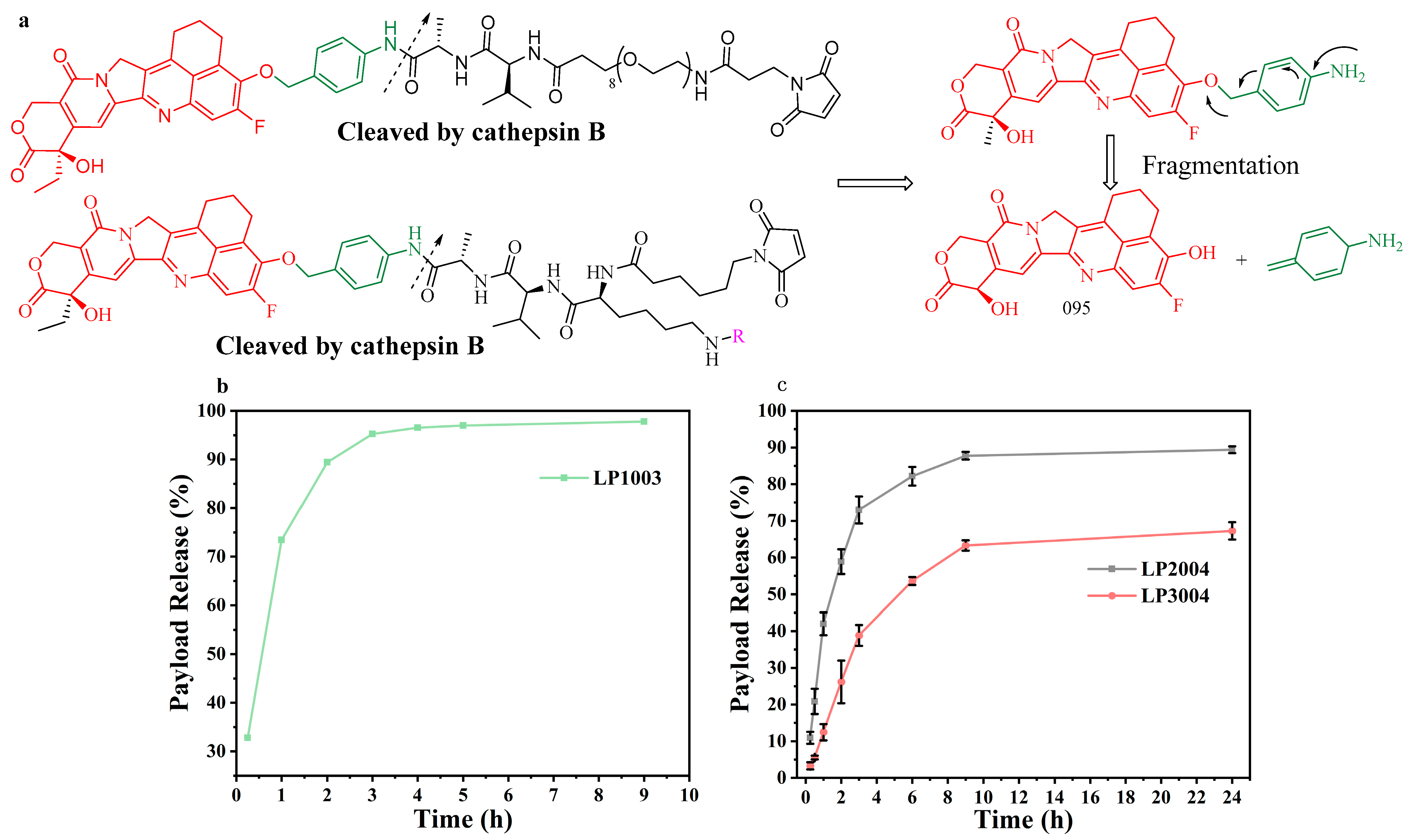
2.1. Construction and Synthesis of Linker–Payload (LP)
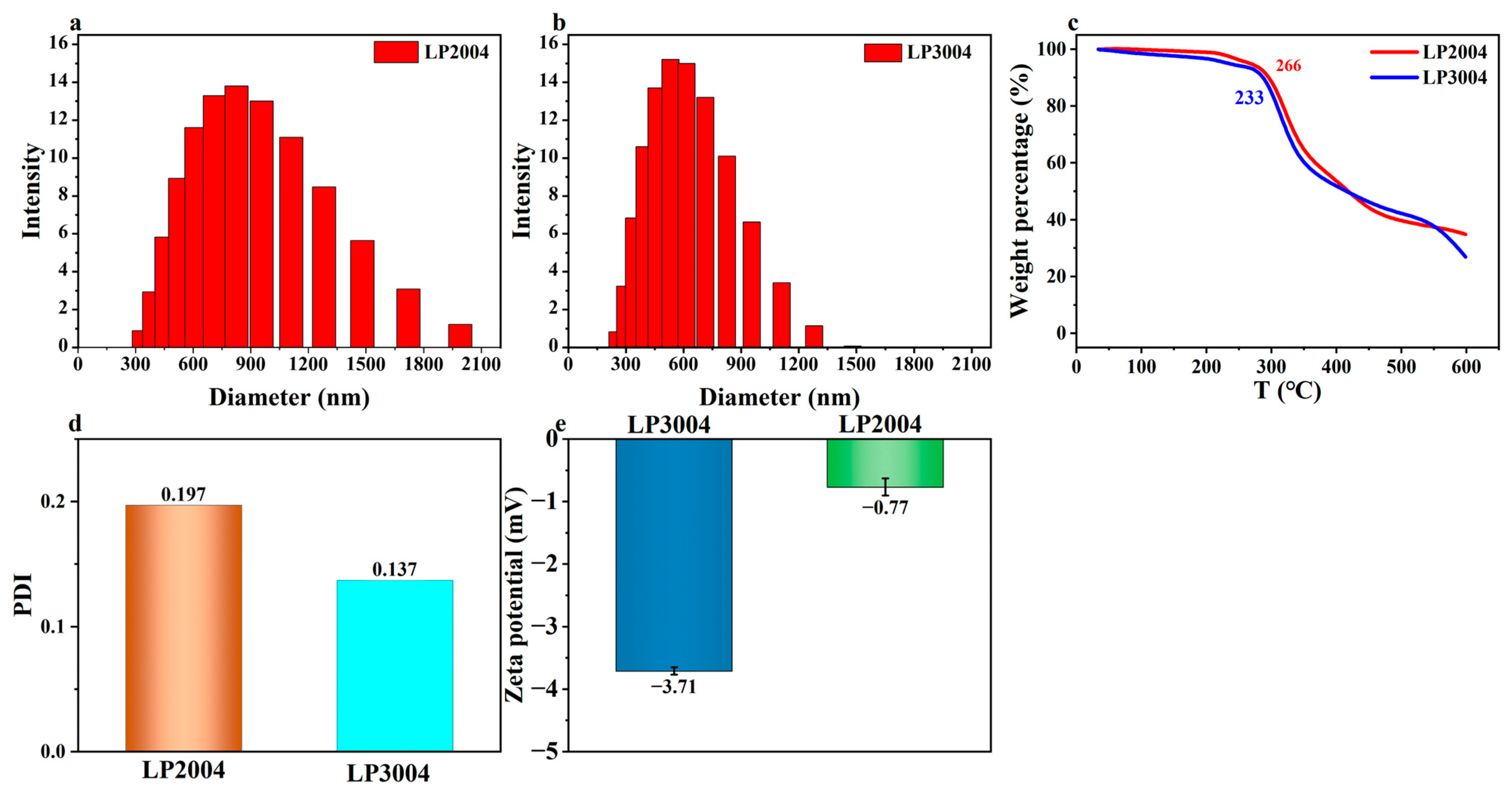
2.2. The Physical Characterization of LP2004 and LP3004
2.3. In Vitro Evaluation of Linker–Payload
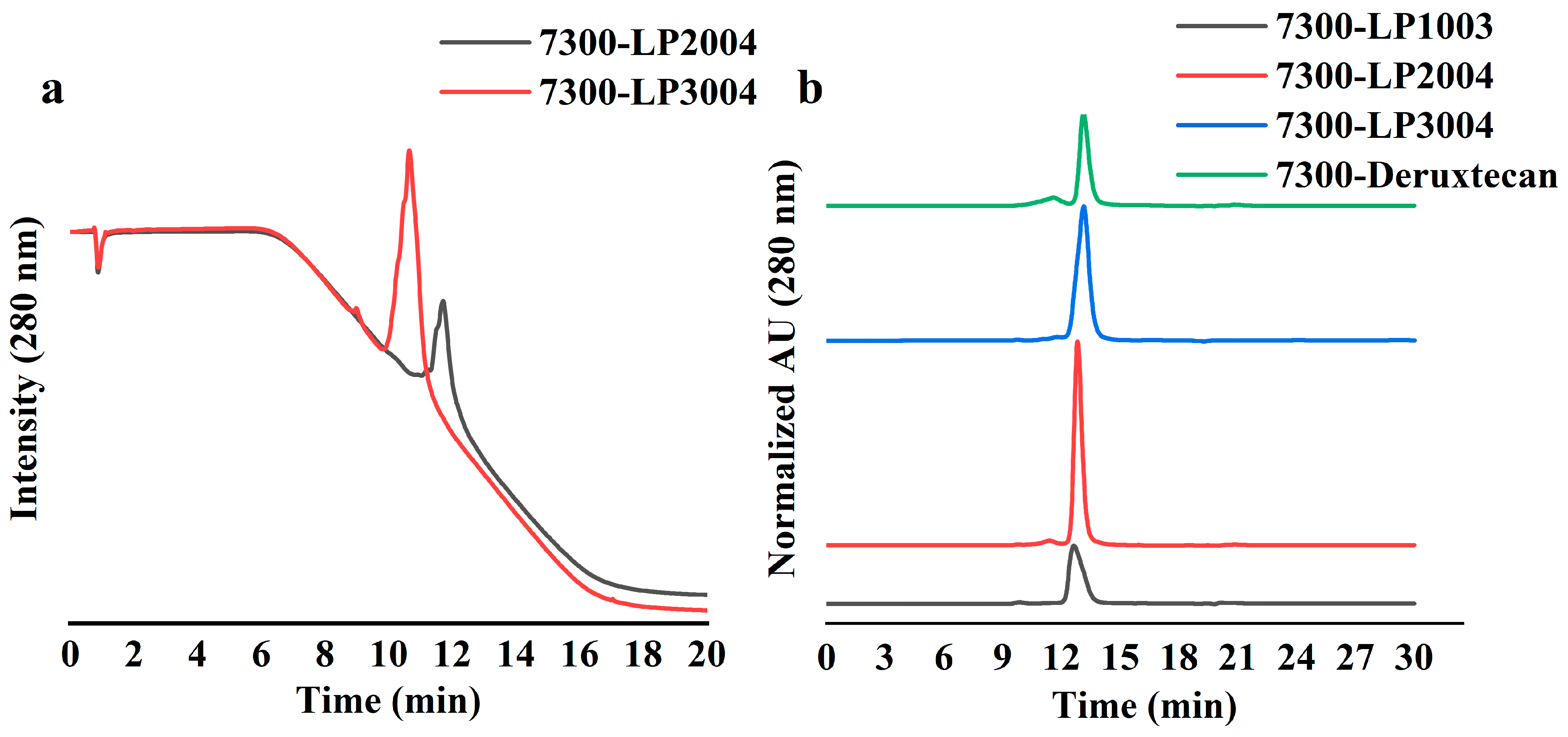
2.4. Bioconjugation and Physicochemical Characterization of ADCs
2.5. 7300-LP3004 Exhibited Strong Stability In Vitro
2.6. In Vitro Cytotoxicity of ADCs in the SHP-77 Cell Line
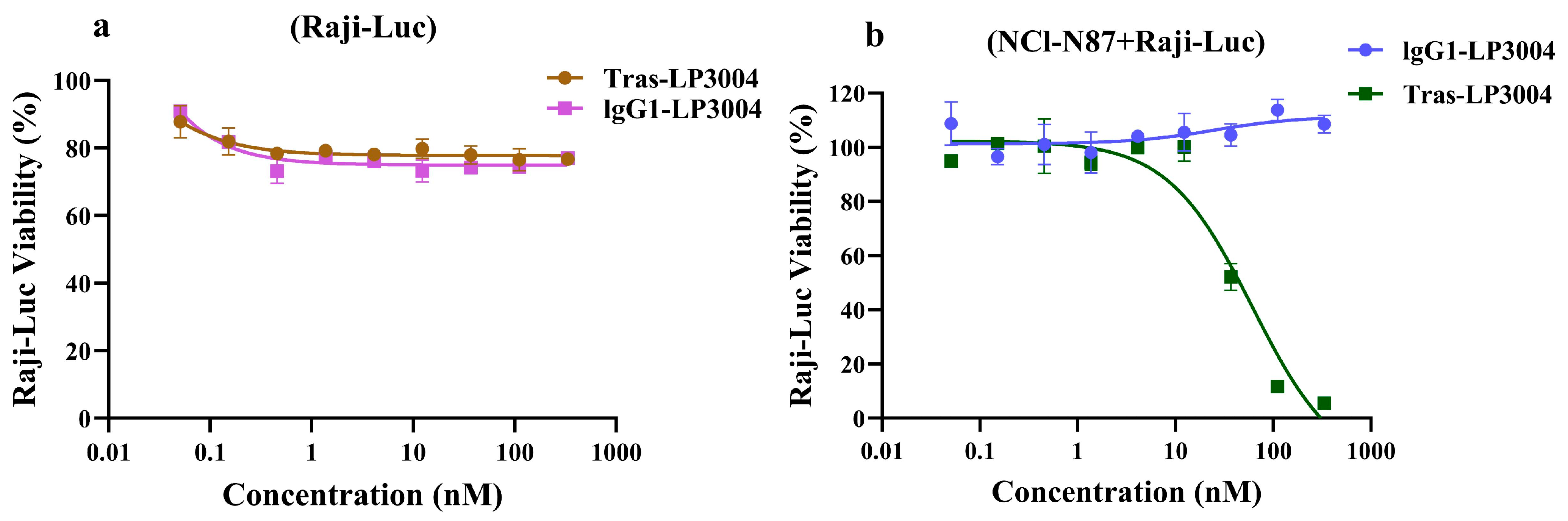
2.7. Tras-LP3004 Displayed Strong Bystander Activity In Vitro
2.8. Validation of ADCs In Vivo
3. Discussion
4. Materials and Methods
4.1. Preparation of ADCs
4.2. Characterization of Linker–Payload
4.3. Cathepsin B-Mediated Cleavage Assay of Linker–Payload
4.4. Characterization of Antibody–Drug Conjugates
4.5. Stability Study
4.6. Cell Culture
4.7. In Vitro Cytotoxicity Assays
4.8. In Vitro Bystander Killing Assay
4.9. In Vivo Efficacy Experiments
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruan, D.-Y.; Wu, H.-X.; Meng, Q.; Xu, R.-H. Development of antibody-drug conjugates in cancer: Overview and prospects. Cancer Commun. 2024, 44, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Goldmacher, V.S. Cell killing by antibody–drug conjugates. Cancer Lett. 2007, 255, 232–240. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jia, L.; He, Z.; Wang, Y. Recent advances in SN-38 drug delivery system. Int. J. Pharm. 2023, 637, 122886. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park Yeon, H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Weng, W.; Meng, T.; Zhao, Q.; Shen, Y.; Fu, G.; Shi, J.; Zhang, Y.; Wang, Z.; Wang, M.; Pan, R.; et al. Antibody-Exatecan Conjugates with a Novel Self-immolative Moiety Overcome Resistance in Colon and Lung Cancer. Cancer Discov. 2023, 13, 950–973. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016, 9, 33–46. [Google Scholar] [CrossRef]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Liu, L.; Xie, F.; Xiao, D.; Xu, X.; Su, Z.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Synthesis and evaluation of highly releasable and structurally stable antibody-SN-38-conjugates. Drug Deliv. 2021, 28, 2603–2617. [Google Scholar] [CrossRef]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal Antibody Conjugates of Doxorubicin Prepared with Branched Peptide Linkers: Inhibition of Aggregation by Methoxytriethyleneglycol Chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [PubMed]
- Khutoryanskiy, V.V. Beyond PEGylation: Alternative surface-modification of nanoparticles with mucus-inert biomaterials. Adv. Drug Deliv. Rev. 2018, 124, 140–149. [Google Scholar] [PubMed]
- Hu, Y.; Hou, Y.; Wang, H.; Lu, H. Polysarcosine as an Alternative to PEG for Therapeutic Protein Conjugation. Bioconjugate Chem. 2018, 29, 2232–2238. [Google Scholar]
- Prakash, A.; Baer, M.D.; Mundy, C.J.; Pfaendtner, J. Peptoid Backbone Flexibilility Dictates Its Interaction with Water and Surfaces: A Molecular Dynamics Investigation. Biomacromolecules 2018, 19, 1006–1015. [Google Scholar]
- Lau, K.H.A.; Ren, C.; Sileika, T.S.; Park, S.H.; Szleifer, I.; Messersmith, P.B. Surface-Grafted Polysarcosine as a Peptoid Antifouling Polymer Brush. Langmuir 2012, 28, 16099–16107. [Google Scholar]
- Skoulas, D.; Stuettgen, V.; Gaul, R.; Cryan, S.-A.; Brayden, D.J.; Heise, A. Amphiphilic Star Polypept(o)ides as Nanomeric Vectors in Mucosal Drug Delivery. Biomacromolecules 2020, 21, 2455–2462. [Google Scholar]
- Stace, A. Preferential solvation of hydrogen ions in mixed water-amine ion clusters. J. Am. Chem. Soc. 1984, 106, 2306–2315. [Google Scholar] [CrossRef]
- Morrell, A.H.; Warren, N.J.; Thornton, P.D. The Production of Polysarcosine-Containing Nanoparticles by Ring-Opening Polymerization-Induced Self-Assembly. Macromol. Rapid Commun. 2024, 45, 2400103. [Google Scholar]
- Khuphe, M.; Mahon, C.S.; Thornton, P.D. Glucose-bearing biodegradable poly (amino acid) and poly (amino acid)-poly (ester) conjugates for controlled payload release. Biomater. Sci. 2016, 4, 1792–1801. [Google Scholar]
- Yao, X.; Sun, C.; Xiong, F.; Zhang, W.; Yao, W.; Xu, Y.; Fan, W.; Huo, F. Polysarcosine as PEG Alternative for Enhanced Camptothecin-Induced Cancer Immunogenic Cell Death. ACS Appl. Mater. Interfaces 2024, 16, 19472–19479. [Google Scholar] [PubMed]
- Zhao, Y.; Chen, F.; Pan, Y.; Li, Z.; Xue, X.; Okeke, C.I.; Wang, Y.; Li, C.; Peng, L.; Wang, P.C.; et al. Nanodrug Formed by Coassembly of Dual Anticancer Drugs to Inhibit Cancer Cell Drug Resistance. ACS Appl. Mater. Interfaces 2015, 7, 19295–19305. [Google Scholar] [PubMed]
- Mallick, A.; More, P.; Ghosh, S.; Chippalkatti, R.; Chopade, B.A.; Lahiri, M.; Basu, S. Dual Drug Conjugated Nanoparticle for Simultaneous Targeting of Mitochondria and Nucleus in Cancer Cells. ACS Appl. Mater. Interfaces 2015, 7, 7584–7598. [Google Scholar]
- Tiberghien, A.C.; Levy, J.-N.; Masterson, L.A.; Patel, N.V.; Adams, L.R.; Corbett, S.; Williams, D.G.; Hartley, J.A.; Howard, P.W. Design and Synthesis of Tesirine, a Clinical Antibody–Drug Conjugate Pyrrolobenzodiazepine Dimer Payload. ACS Med. Chem. Lett. 2016, 7, 983–987. [Google Scholar] [PubMed]
- Best, R.L.; LaPointe, N.E.; Azarenko, O.; Miller, H.; Genualdi, C.; Chih, S.; Shen, B.-Q.; Jordan, M.A.; Wilson, L.; Feinstein, S.C.; et al. Microtubule and tubulin binding and regulation of microtubule dynamics by the antibody drug conjugate (ADC) payload, monomethyl auristatin E (MMAE): Mechanistic insights into MMAE ADC peripheral neuropathy. Toxicol. Appl. Pharmacol. 2021, 421, 115534. [Google Scholar]
- Reid, E.E.; Archer, K.E.; Shizuka, M.; McShea, M.A.; Maloney, E.K.; Ab, O.; Lanieri, L.; Wilhelm, A.; Ponte, J.F.; Yoder, N.C.; et al. Design, synthesis and evaluation of novel, potent DNA alkylating agents and their antibody-drug conjugates (ADCs). Bioorganic Med. Chem. Lett. 2019, 29, 2455–2458. [Google Scholar]
- Okajima, D.; Yasuda, S.; Yokouchi, Y.; Fujitani, T.; Sakurai, K.; Yamaguchi, J.; Kitamura, M.; Terauchi, T.; Shibutani, T.; Aida, T. Preclinical efficacy studies of DS-1062a, a novel TROP2-targeting antibody-drug conjugate with a novel DNA topoisomerase I inhibitor DXd. Am. Soc. Clin. Oncol. 2018, 36, e24206. [Google Scholar]
- Yamato, M.; Hasegawa, J.; Maejima, T.; Hattori, C.; Kumagai, K.; Watanabe, A.; Nishiya, Y.; Shibutani, T.; Aida, T.; Hayakawa, I. DS-7300a, a DNA topoisomerase I inhibitor, DXd-based antibody–drug conjugate targeting B7-H3, exerts potent antitumor activities in preclinical models. Mol. Cancer Ther. 2022, 21, 635–646. [Google Scholar] [CrossRef]
- Timmins, P. Industry update: The latest developments in the field of therapeutic delivery, October 2023. Ther. Deliv. 2024, 15, 77–94. [Google Scholar]
- Buecheler, J.W.; Winzer, M.; Tonillo, J.; Weber, C.; Gieseler, H. Impact of payload hydrophobicity on the stability of antibody–drug conjugates. Mol. Pharm. 2018, 15, 2656–2664. [Google Scholar]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Smith, M.; Kolinsky, K.; Adames, V.; Mehta, N.; Fritzky, L.; Rashed, M.; Wheeldon, E.; Linn, M.; Higgins, B. Antitumor activity of HER1/EGFR tyrosine kinase inhibitor erlotinib, alone and in combination with CPT-11 (irinotecan) in human colorectal cancer xenograft models. Cancer Chemother. Pharmacol. 2007, 59, 651–659. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC | IC50 |
|---|---|
| 7300-LP2004 | 32.17 nM |
| 7300-LP3004 | 39.74 nM |
| 7300-LP1003 | 186.6 nM |
| 7300-Deruxtecan | 124.5 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, T.; Jin, J.; Liu, D.; Jin, C. Design, Synthesis, and Evaluation of Camptothecin-Based Antibody–Drug Conjugates with High Hydrophilicity and Structural Stability. Molecules 2025, 30, 1398. https://doi.org/10.3390/molecules30071398
Xiong T, Jin J, Liu D, Jin C. Design, Synthesis, and Evaluation of Camptothecin-Based Antibody–Drug Conjugates with High Hydrophilicity and Structural Stability. Molecules. 2025; 30(7):1398. https://doi.org/10.3390/molecules30071398
Chicago/Turabian StyleXiong, Tingyu, Jiyu Jin, Dongliang Liu, and Chen Jin. 2025. "Design, Synthesis, and Evaluation of Camptothecin-Based Antibody–Drug Conjugates with High Hydrophilicity and Structural Stability" Molecules 30, no. 7: 1398. https://doi.org/10.3390/molecules30071398
APA StyleXiong, T., Jin, J., Liu, D., & Jin, C. (2025). Design, Synthesis, and Evaluation of Camptothecin-Based Antibody–Drug Conjugates with High Hydrophilicity and Structural Stability. Molecules, 30(7), 1398. https://doi.org/10.3390/molecules30071398