
Towards Metabolic Organic Radical Contrast Agents (mORCAs) for Magnetic Resonance Imaging

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Synthesis
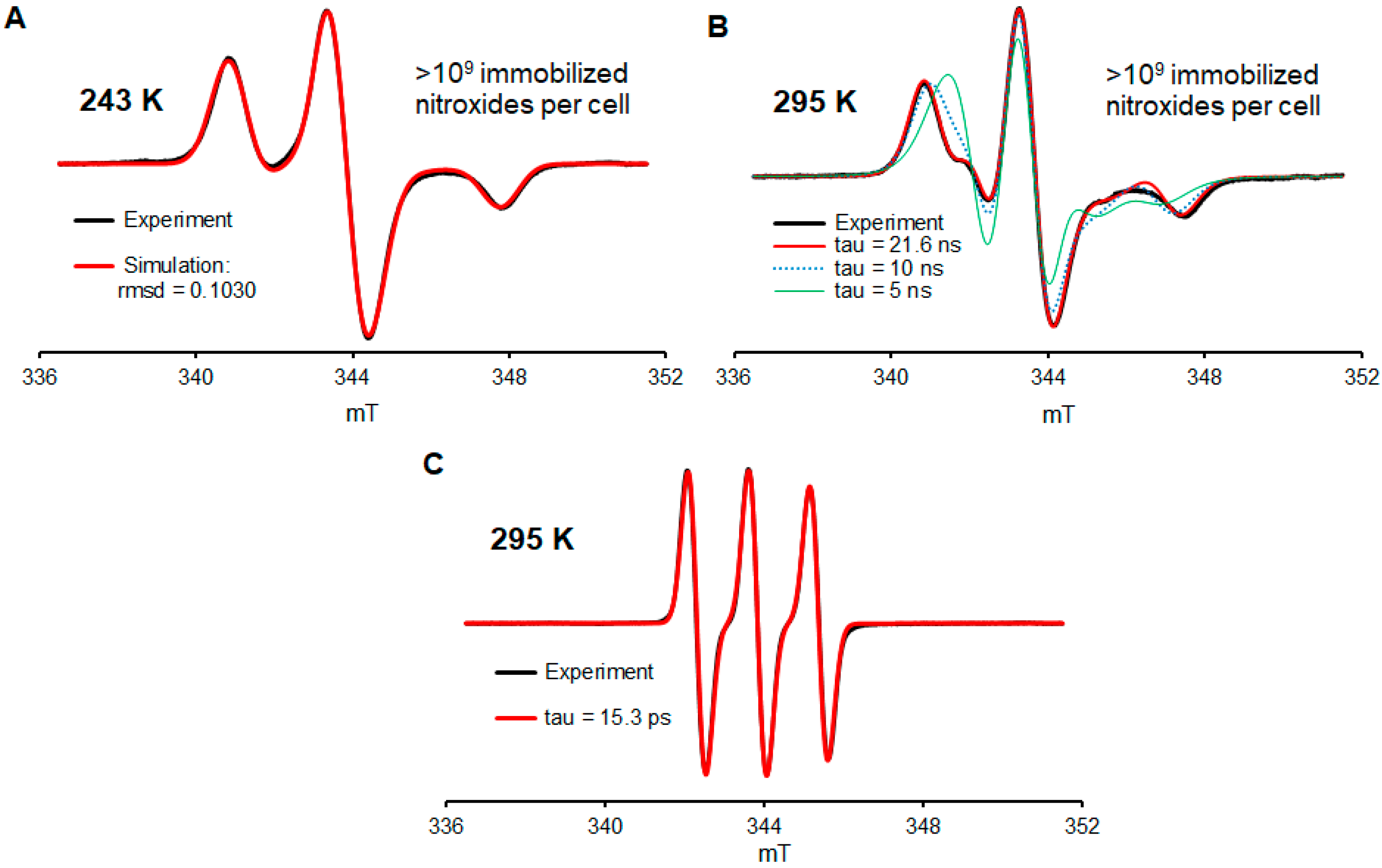
2.2. EPR Spectroscopy of 2a in Jurkat Cells
- Will most of the nitroxide radicals remain intact after a few days of incubation at 37 °C?
- How many nitroxide radicals per cell may be incorporated into the cells?
- Will the nitroxide radical be well immobilized inside the cell?
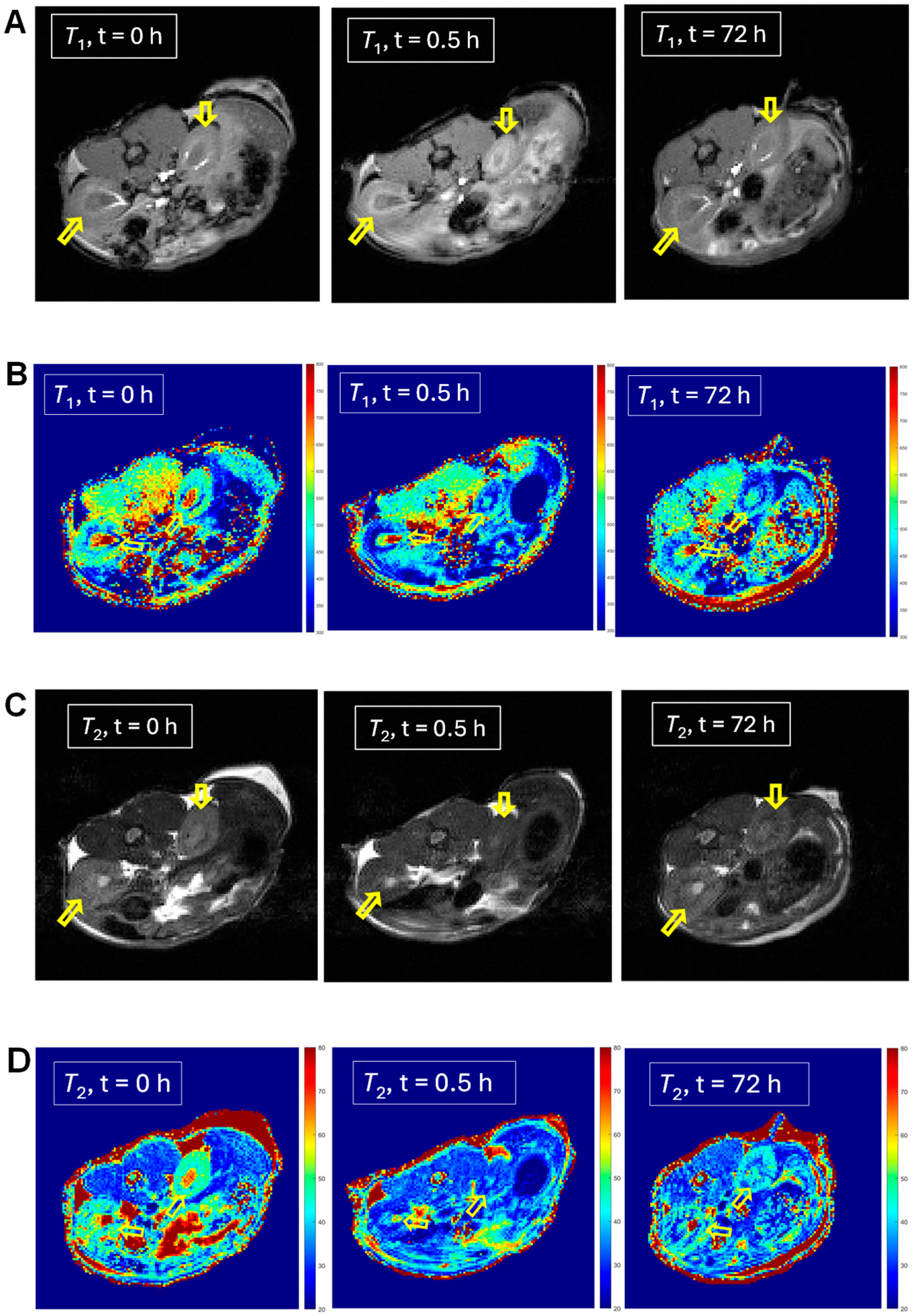
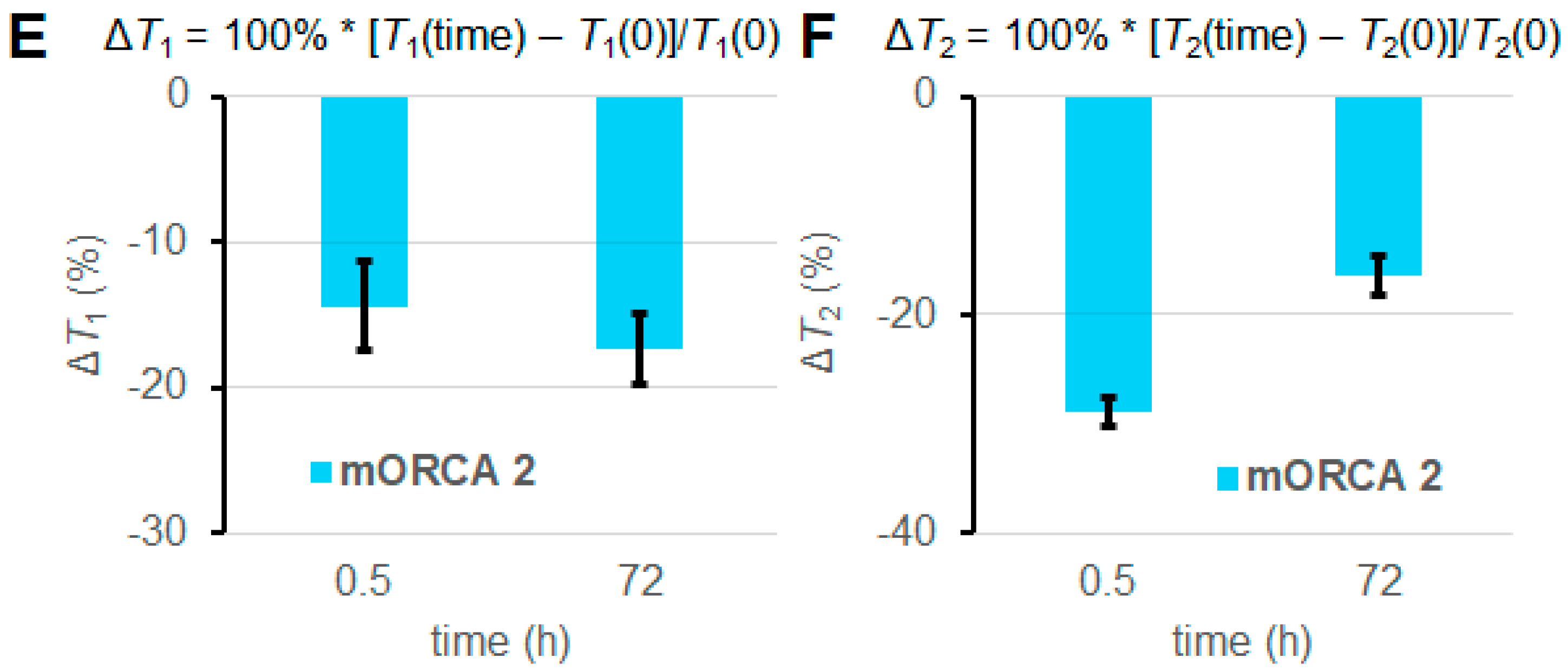
2.3. In Vivo MRI of 1 and 2
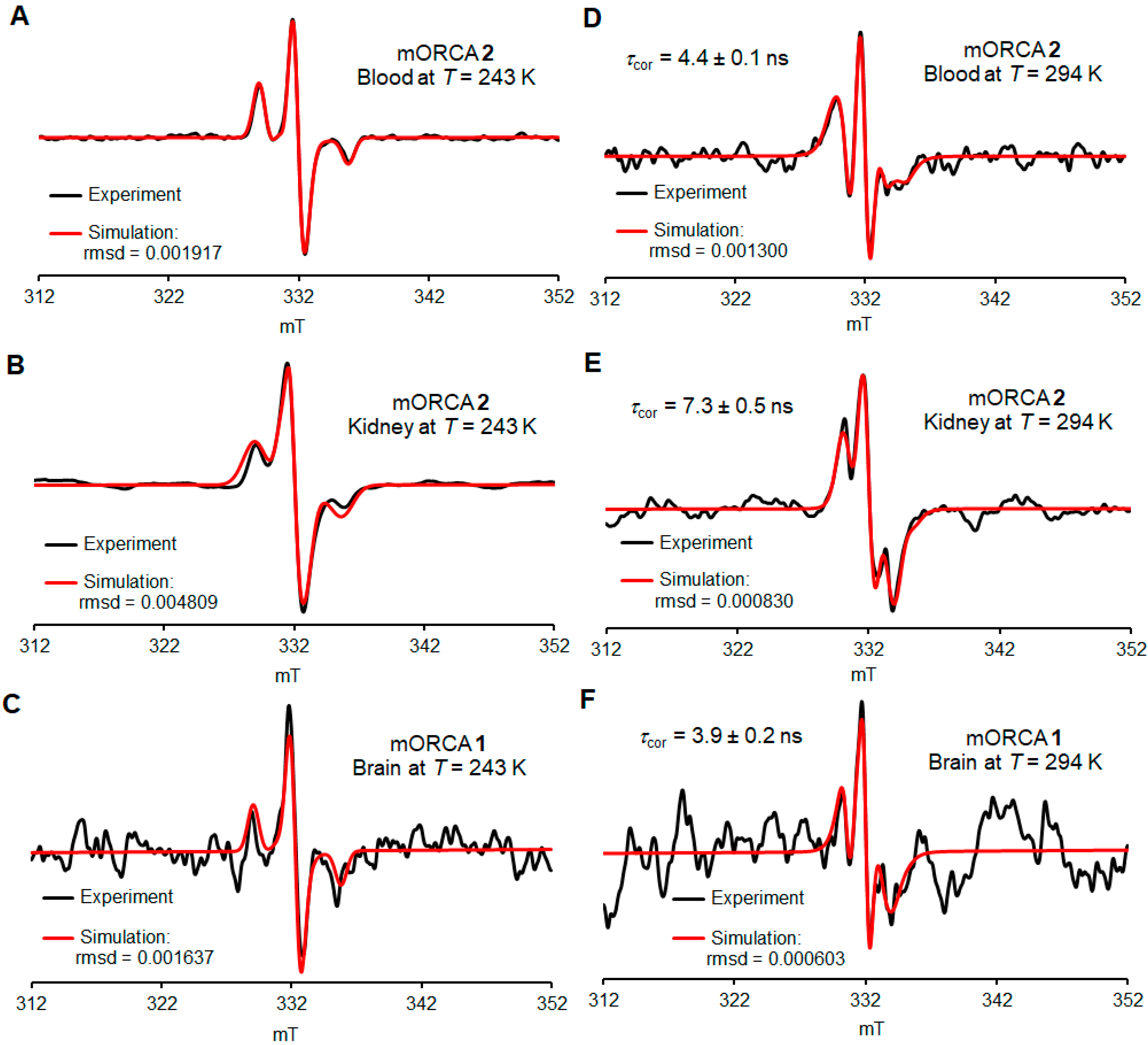
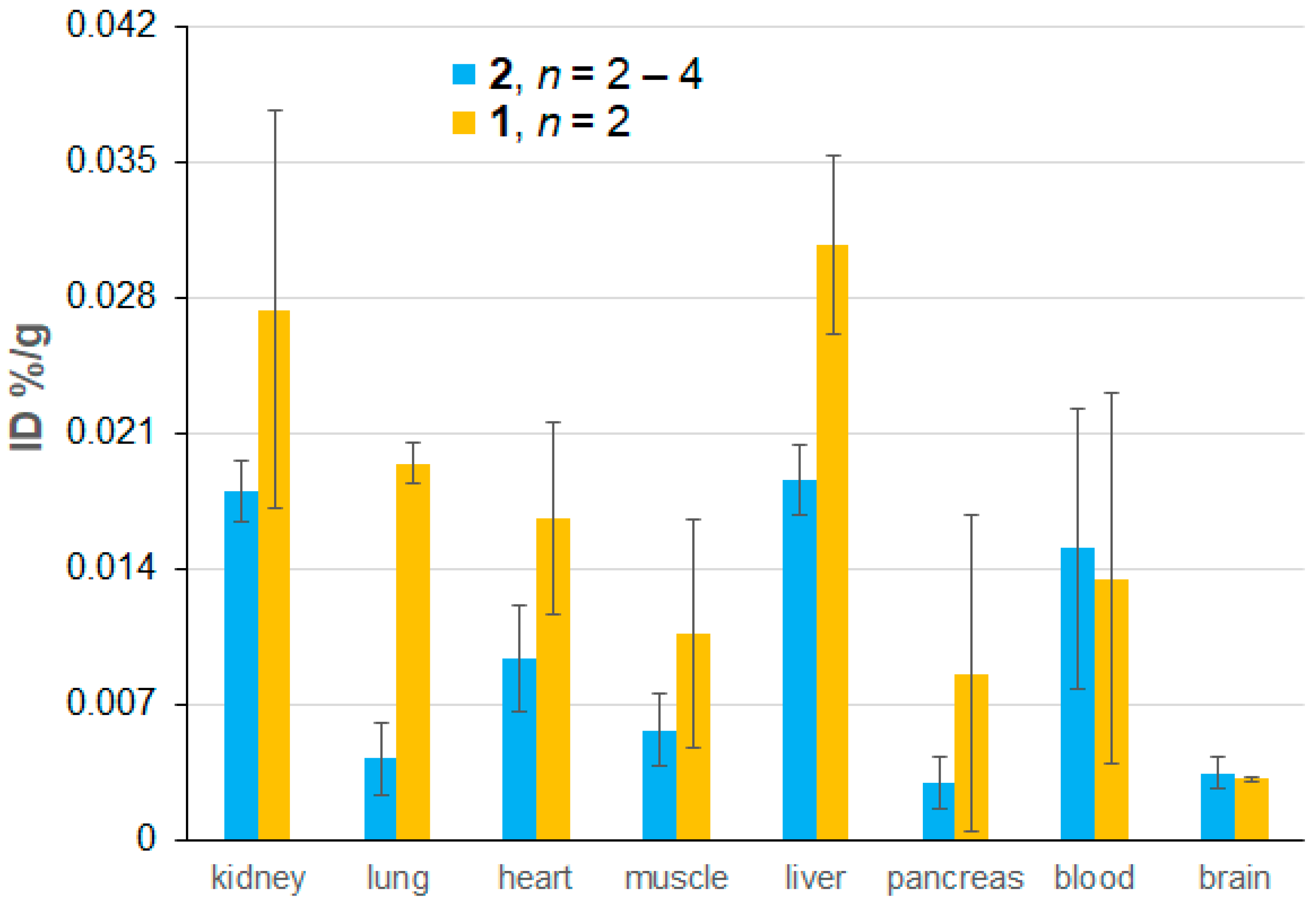
2.4. Ex Vivo EPR Spectroscopy of 1 and 2
3. Materials and Methods
3.1. Materials
3.2. Instruments
3.3. Synthesis
3.4. In-Cell EPR Spectroscopy
3.5. In Vivo MRI
3.6. Ex Vivo EPR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brasch, R.C.; London, D.A.; Wesbey, G.E.; Tozer, T.N.; Nitecki, D.E.; Williams, R.D.; Doemeny, J.; Tuck, L.D.; Lallemand, D.P. Work in Progress: Nuclear Magnetic Resonance Study of a Paramagnetic Nitroxide Contrast Agent for Enhancement of Renal Structures in Experimental Animals. Radiology 1983, 147, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Hyodo, F.; Matsumoto, A.; Koretsky, A.P.; Sowers, A.L.; Mitchell, J.B.; Krishna, M.C. High-Resolution Mapping of Tumor Redox Status by Magnetic Resonance Imaging Using Nitroxides as Redox-Sensitive Contrast Agents. Clin. Cancer Res. 2006, 12, 2455–2462. [Google Scholar] [CrossRef] [PubMed]
- Hyodo, F.; Matsumoto, K.; Matsumoto, A.; Mitchell, J.B.; Krishna, M.C. Probing the Intracellular Redox Status of Tumors with Magnetic Resonance Imaging and Redox-Sensitive Contrast Agents. Cancer Res. 2006, 66, 9921–9928. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Yamasaki, T.; Nakamura, M.; Ishikawa, J.; Ueno, M.; Nakanishi, I.; Sekita, A.; Ozawa, Y.; Kamada, T.; Aoki, I.; et al. Brain Contrasting Ability of Blood-Brain-Barrier–Permeable Nitroxyl Contrast Agents for Magnetic Resonance Redox Imaging. Magn. Reson. Med. 2016, 76, 935–945. [Google Scholar] [CrossRef]
- Matsumoto, K.; Mitchell, J.B.; Krishna, M.C. Multimodal Functional Imaging for Cancer/Tumor Microenvironments Based on MRI, EPRI, and PET. Molecules 2021, 26, 1614. [Google Scholar] [CrossRef]
- Rajca, A.; Wang, Y.; Boska, M.; Paletta, J.T.; Olankitwanit, A.; Swanson, M.A.; Mitchell, D.G.; Eaton, S.S.; Eaton, G.R.; Rajca, S. Organic Radical Contrast Agents for Magnetic Resonance Imaging. J. Am. Chem. Soc. 2012, 134, 15724–15727. [Google Scholar] [CrossRef]
- Luo, T.; Wang, B.; Chen, R.; Qi, Q.; Wu, R.; Xie, S.; Chen, H.; Han, J.; Wu, D.; Cao, S. Research Progress of Nitroxide Radical-Based MRI Contrast Agents: From Structure Design to Application. J. Mater. Chem. B 2025, 13, 372–398. [Google Scholar] [CrossRef]
- Yuan, X.; Yu, H.; Wang, L.; Uddin, M.A.; Ouyang, C. Nitroxide Radical Contrast Agents for Safe Magnetic Resonance Imaging: Progress, Challenges, and Perspectives. Mater. Horiz. 2025, 12, 1726–1756. [Google Scholar] [CrossRef]
- Wahab, A.; Suhail, M.; Eggers, T.; Shehzad, K.; Akakuru, O.U.; Ahmad, Z.; Sun, Z.; Iqbal, M.Z.; Kong, X. Innovative Perspectives on Metal-Free Contrast Agents for MRI: Enhancing Imaging Efficacy and AI-Driven Future Diagnostics. Acta Biomater. 2025, 193, 83–106. [Google Scholar] [CrossRef]
- Sowers, M.A.; McCombs, J.R.; Wang, Y.; Paletta, J.T.; Morton, S.W.; Dreaden, E.C.; Boska, M.D.; Ottaviani, M.F.; Hammond, P.T.; Rajca, A. Redox-Responsive Branched-Bottlebrush Polymers for in-vivo MRI and Fluorescence Imaging. Nat. Commun. 2014, 5, 5460. [Google Scholar] [CrossRef]
- Nguyen, H.V.T.; Chen, Q.; Paletta, J.T.; Harvey, P.; Jiang, Y.; Zhang, H.; Boska, M.D.; Ottaviani, M.F.; Jasanoff, A.; Rajca, A. Nitroxide-Based Macromolecular Contrast Agents with Unprecedented Transverse Relaxivity and Stability for Magnetic Resonance Imaging of Tumors. ACS Cent. Sci. 2017, 3, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.V.T.; Detappe, A.; Gallagher, N.M.; Zhang, H.; Harvey, P.; Yan, C.; Mathieu, C.; Golder, M.R.; Jiang, Y.; Ottaviani, M.F. Triply Loaded Nitroxide Brush-Arm Star Polymers Enable Metal-Free Millimetric Tumor Detection by Magnetic Resonance Imaging. ACS Nano 2018, 12, 11343–11354. [Google Scholar] [CrossRef] [PubMed]
- Marx, L.; Chiarelli, R.; Guiberteau, T.; Rassat, A. A comparative study of the reduction by ascorbate of 1,1,3,3-tetraethylisoindolin-2-yloxyl and of 1,1,3,3-tetramethylisoindolin-2-yloxyl. J. Chem. Soc. Perkin Trans. 1 2000, 1181–1182. [Google Scholar] [CrossRef]
- Bobko, A.A.; Kirilyuk, I.A.; Grigor’ev, I.A.; Zweier, J.L.; Khramtsov, V.V. Reversible reduction of nitroxides to hydroxylamines: Roles for ascorbate and glutathione. Free Radic. Biol. Med. 2007, 42, 404–412. [Google Scholar] [CrossRef]
- Wang, Y.; Paletta, J.T.; Berg, K.; Reinhart, E.; Rajca, S.; Rajca, A. Synthesis of Unnatural Amino Acids Functionalized with Sterically Shielded Pyrroline Nitroxides. Org. Lett. 2014, 16, 5298–5300. [Google Scholar] [CrossRef]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and Reduction Kinetics of Sterically Shielded Pyrrolidine Nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically Shielded Spin Labels for in-cell EPR Spectroscopy: Analysis of Stability in Reducing Environment. Free Radic. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, H.; Paletta, J.T.; Rajca, S.; Rajca, A. Reduction Kinetics and Electrochemistry of Tetracarboxylate Nitroxides. Free Radic. Res. 2018, 52, 327–334. [Google Scholar] [CrossRef]
- Bleicken, S.; Assafa, T.E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Enrica Bordignon, E. gem-Diethyl Pyrroline Nitroxide Spin Labels: Synthesis, EPR Characterization, Rotamer Libraries and Biocompatibility. ChemistryOpen 2019, 8, 1057–1065. [Google Scholar] [CrossRef]
- Rančić, A.; Babić, N.; Orio, M.; Peyrot, F. Structural Features Governing the Metabolic Stability of Tetraethyl-Substituted Nitroxides in Rat Liver Microsomes. Antioxidants 2023, 12, 402. [Google Scholar] [CrossRef]
- Jana, S.; Evans, E.G.B.; Jang, H.S.; Zhang, S.; Zhang, H.; Rajca, A.; Gordon, S.E.; Zagotta, W.N.; Stoll, S.; Mehl, R.A. Ultra-Fast Bioorthogonal Spin-Labeling and Distance Measurements in Mammalian Cells Using Small, Genetically Encoded Tetrazine Amino Acids. J. Am. Chem. Soc. 2023, 145, 14608–14620. [Google Scholar] [CrossRef] [PubMed]
- Usatov, M.S.; Dobrynin, S.A.; Polienko, Y.F.; Morozov, D.A.; Glazachev, Y.I.; An’kov, S.V.; Tolstikova, T.G.; Gatilov, Y.V.; Bagryanskaya, I.Y.; Raizvikh, A.E.; et al. Hydrophilic Reduction-Resistant Spin Labels of Pyrrolidine and Pyrroline Series from 3,4-Bis-Hydroxymethyl-2,2,5,5-Tetraethylpyrrolidine-1-Oxyl. Int. J. Mol. Sci. 2024, 25, 1550. [Google Scholar] [CrossRef] [PubMed]
- Sowiński, M.P.; Warnke, A.; Lund, B.A.; Skagseth, S.; Cordes, D.B.; Lovett, J.E.; Haugland, M.M. Spirocyclic Pyrrolidinyl Nitroxides with Exo -Methylene Substituents. ChemPlusChem 2024, 89, e202400387. [Google Scholar] [CrossRef] [PubMed]
- Mitin, D.; Bullinger, F.; Dobrynin, S.; Engelmann, J.; Scheffler, K.; Kolokolov, M.; Krumkacheva, O.; Buckenmaier, K.; Kirilyuk, I.; Chubarov, A. Contrast Agents Based on Human Serum Albumin and Nitroxides for 1H-MRI and Overhauser-Enhanced MRI. Int. J. Mol. Sci. 2024, 25, 4041. [Google Scholar] [CrossRef]
- Sowiński, M.P.; Gahlawat, S.; Lund, B.A.; Warnke, A.-L.; Hopmann, K.H.; Lovett, J.E.; Haugland, M.M. Conformational Tuning Improves the Stability of Spirocyclic Nitroxides with Long Paramagnetic Relaxation Times. Commun. Chem. 2023, 6, 111. [Google Scholar] [CrossRef]
- Asanbaeva, N.B.; Dobrynin, S.A.; Morozov, D.A.; Haro-Mares, N.; Gutmann, T.; Buntkowsky, G.; Bagryanskaya, E.G. An EPR Study on Highly Stable Nitroxyl-Nitroxyl Biradicals for Dynamic Nuclear Polarization Applications at High Magnetic Fields. Molecules 2023, 28, 1926. [Google Scholar] [CrossRef]
- Schauenburg, D.; Weil, T. Not So Bioorthogonal Chemistry. J. Am. Chem. Soc. 2025, 147, 8049–8062. [Google Scholar] [CrossRef]
- Shajahan, A.; Parashar, S.; Goswami, S.; Ahmed, S.M.; Nagarajan, P.; Sampathkumar, S.G. Carbohydrate-Neuroactive Hybrid Strategy for Metabolic Glycan Engineering of the Central Nervous System in Vivo. J. Am. Chem. Soc. 2017, 139, 693–700. [Google Scholar] [CrossRef]
- Kayser, H.; Zeitler, R.; Kannicht, C.; Grunow, D.; Nuck, R.; Reutter, W. Biosynthesis of a Nonphysiological Sialic Acid in Different Rat Organs, Using N-Propanoyl-D-Hexosamines as Precursors. J. Biol. Chem. 1992, 267, 16934–16938. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef]
- Prescher, J.A.; Dube, D.H.; Bertozzi, C.R. Chemical Remodelling of Cell Surfaces in Living Animals. Nature 2004, 430, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in Living Systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Changa, P.V.; Preschera, J.A.; Sletten, E.M.; Baskin, J.M.; Miller, I.A.; Agard, N.J.; Lo, A.; Bertozzi, C.R. Copper-Free Click Chemistry in Living Animals. Proc. Natl. Acad. Sci. USA 2010, 107, 1821–1826. [Google Scholar] [CrossRef]
- Jacobs, C.L.; Goon, S.; Yarema, K.J.; Hinderlich, S.; Hang, H.C.; Chai, D.H.; Bertozzi, C.R. Substrate Specificity of the Sialic Acid Biosynthetic Pathway. Biochemistry 2001, 40, 12864–12874. [Google Scholar] [CrossRef]
- Jones, M.B.; Teng, H.; Rhee, J.K.; Lahar, N.; Baskaran, G.; Yarema, K.J. Characterization of the Cellular Uptake and Metabolic Conversion of Acetylated N-Acetylmannosamine (ManNAc) Analogues to Sialic Acids. Biotechnol. Bioeng. 2004, 85, 394–405. [Google Scholar] [CrossRef]
- Palaniappan, K.K.; Bertozzi, C.R. Chemical Glycoproteomics. Chem. Rev. 2016, 116, 14277–14306. [Google Scholar] [CrossRef]
- Mitry, M.M.A.; Greco, F.; Osborn, H.M.I. In Vivo Applications of Bioorthogonal Reactions: Chemistry and Targeting Mechanisms. Chem.–Eur. J. 2023, 29, e202203942. [Google Scholar] [CrossRef]
- Neves, A.A.; Wainman, Y.A.; Wright, A.; Kettunen, M.I.; Rodrigues, T.B.; McGuire, S.; Hu, D.-E.; Bulat, F.; Geninatti Crich, S.; Stöckmann, H.; et al. Imaging Glycosylation In Vivo by Metabolic Labeling and Magnetic Resonance Imaging. Angew. Chem. Int. Ed. 2016, 55, 1286–1290. [Google Scholar] [CrossRef]
- Agatemor, C.; Buettner, M.J.; Ariss, R.; Muthiah, K.; Saeui, C.T.; Yarema, K.J. Exploiting Metabolic Glycoengineering to Advance Healthcare. Nat. Rev. Chem. 2019, 3, 605–620. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Cai, K.; He, H.; Liu, Y.; Yen, J.; Wang, Z.; Xu, M.; Sun, Y.; Zhou, X.; et al. Selective in Vivo Metabolic Cell-Labeling-Mediated Cancer Targeting. Nat. Chem. Biol. 2017, 13, 415–424. [Google Scholar] [CrossRef]
- Wang, H.; Mooney, D.J. Metabolic Glycan Labelling for Cancer-Targeted Therapy. Nat. Chem. 2020, 12, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Tran, T.T.; Li, Q.; Yan, X.; Zhou, M.; Kundu, K.; Fanucci, G.E.; Guo, Z. A metabolically engineered spin-labeling approach for studying glycans on cells. Chem. Sci. 2020, 11, 12522–12532. [Google Scholar] [CrossRef] [PubMed]
- Sampathkumar, S.-G.; Li, A.V.; Yarema, K.J. Synthesis of Non-Natural ManNAc Analogs for the Expression of Thiols on Cell-Surface Sialic Acids. Nat. Protoc. 2006, 1, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Shahabi, D. Synthesis of Amide Derivatives for Electron Deficient Amines and Functionalized Carboxylic Acids Using EDC and DMAP and a Catalytic Amount of HOBt as the Coupling Reagents. Tetrahedron Lett. 2021, 63, 152719. [Google Scholar] [CrossRef]
- Hattie, M.; Stubbs, K.A. Generalising a Simple Methodology for the Regioselective Anomeric Deacetylation of Carbohydrates. ChemistrySelect 2020, 5, 875–877. [Google Scholar] [CrossRef]
- Kim, E.J.; Sampathkumar, S.-G.; Jones, M.B.; Rhee, J.K.; Baskaran, G.; Ramya, T.N.C.; Yarema, K.J. Characterization of the Metabolic Flux and Apoptotic Effects of O-Hydroxyl- and N-Acyl-Modified N-Acetylmannosamine Analogs in Jurkat Cells. J. Biol. Chem. 2004, 279, 18342–18352. [Google Scholar] [CrossRef]
- Yang, Z.; Stein, R.A.; Pink, M.; Madzelan, P.; Ngendahimana, T.; Rajca, S.; Wilson, M.A.; Eaton, S.S.; Eaton, G.R.; Mchaourab, H.S.; et al. Cucurbit [7] uril Enhances Distance Measurements of Spin-Labeled Proteins. J. Am. Chem. Soc. 2023, 145, 25726–25736. [Google Scholar] [CrossRef]
- Liu, H.; Jensen, L.; Hansen, S.; Petersen, S.V.; Takahashi, K.; Ezekowitz, A.B.; Hansen, F.D.; Jensenius, J.C.; Thiel, S. Lectin blood mannose binding: Characterization and Quantification of Mouse Mannan-Binding Lectins (MBL-A and MBL-C) and Study of Acute Phase Responses. Scand. J. Immunol. 2001, 53, 489–497. [Google Scholar] [CrossRef]
- Miller, A.; Phillips, A.; Gor, J.; Wallis, R.; Perkins, S.J. Near-planar Solution Structures of Mannose-binding Lectin Oligomers Provide Insight on Activation of Lectin Pathway of Complement. J. Biol. Chem. 2012, 287, 3930–3945. [Google Scholar] [CrossRef]
- Ikeda, Y.; Long, D.M. Comparative Effects of Direct and Indirect Hydroxyl Radical Scavengers on Traumatic Brain Oedema. Acta Neurochir. Suppl. 1990, 51, 74–76. [Google Scholar] [CrossRef]
- Cyran, C.C.; Sennino, B.; Chaopathomkul, B.; Fu, Y.; Rogut, V.; Shames, D.M.; Wendland, M.F.; McDonald, D.M.; Brasch, R.C. Magnetic Resonance Imaging Assays for Dimethyl Sulfoxide Effect on Cancer Vasculature. Investig. Radiol. 2008, 43, 298–305. [Google Scholar] [CrossRef]
- Delgado-Goñi, T.; Martin-Sitjar, J.M.; Simões, R.V.; Acosta, M.; Lope-Piedrafita, S.; Arúsa, C. Dimethyl sulfoxide (DMSO) as a potential contrast agent for brain tumors. NMR Biomed. 2013, 26, 173–184. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | 2 | 1 | ||||
|---|---|---|---|---|---|---|
| ΔT1 (%) | ΔT2 (%) | ΔT1 (%) | ΔT2 (%) | |||
| Kidneys | n = 1 | −17 ± 2 | −16 ± 2 | n = 1 | −21 ± 2 | −17 ± 1 |
| Kidneys | n = 9 | +2 ± 5 | −6.5 ± 1.6 | n = 2 | −8 ± 13 | −7 ± 10 |
| Kidneys | n = 7 | −3.4 ± 4.2 | −7.4 ± 1.9 | |||
| Kidneys | n = 4 | −10.5 ± 3.7 | −10.2 ± 2.4 | |||
| Liver | n = 1 | −41 ± 3 | −7.5 ± 2.6 | n = 1 | −1 ± 4 | 0 ± 3 |
| Liver | n = 7 | −6 ± 8 | +6 ± 4 | n = 2 | +9 ± 10 | +7 ± 7 |
| Liver | n = 4 | −19.7 ± 8.5 | −2.9 ± 1.7 | |||
| Liver | n = 3 | −26.0 ± 8.1 | −3.8 ± 2.1 | |||
| Heart | n = 1 | −26 ± 7 | −28 ± 5 | n = 1 | ||
| Heart | n = 5 | +7 ± 10 | −8 ± 7 | n = 2 | ||
| Heart | n = 3 | 0 ± 15 | −19 ± 5 | |||
| Brain | n = 1 | −16 ± 1 | −4.0 ± 1.1 | n = 1 | +8 ± 1 | −1.3 ± 0.8 |
| Brain | n = 2 | −11.9 ± 4.5 | −1.7 ± 2.2 | n = 2 | +10 ± 2 | −0.5 ± 0.9 |
| Tissue | n 1 | τcor (ns) 2 | Fast Component 2,3 |
|---|---|---|---|
| Blood | 7 | 4.0 ± 0.3 | 0.00 |
| Kidney | 10 | 6.0 ± 0.5 | 0.40 ± 0.06 |
| Liver | 4 | 9.2 ± 0.4 | 0.37 ± 0.05 |
| Lung | 4 | 5.5 ± 0.6 | 0.51 ± 0.03 |
| Brain | 1 | 3.9 | 0.73 |
| Tissue | mORCA 1 | mORCA 2 | ORCAFluor | ||
|---|---|---|---|---|---|
| %ID (n) | % 1 | %ID (n) | % 1 | %ID | |
| Blood | 0.014 ± 0.010 (2) | 0.24 | 0.015 ± 0.007 (4) | 0.27 | 5.6 ± 0.7 |
| Kidney | 0.027 ± 0.010 (2) | 5.7 | 0.018 ± 0.002 (4) | 3.8 | 0.48 ± 0.06 |
| Liver | 0.031 ± 0.005 (2) | 10.8 | 0.019 ± 0.002 (4) | 6.6 | 0.28 ± 0.08 |
| Lung | 0.020 ± 0.001 (2) | 1.7 | 0.004 ± 0.002 (4) | 0.36 | 1.2 ± 0.2 |
| Heart | 0.017 ± 0.005 (2) | 2.5 | 0.009 ± 0.003 (4) | 1.4 | 0.67 ± 0.10 |
| Pancreas | 0.009 ± 0.008 (2) | na | 0.003 ± 0.001 (4) | na | na |
| Muscle | 0.011 ± 0.006 (2) | na | 0.006 ± 0.002 (3) | na | na |
| Spleen | 0.019 ± 0.004 (2) | 2.6 | 0.023 ± 0.007 (2) | 3.2 | 0.72 ± 0.14 |
| Brain | 0.0032 ± 0.0001 (2) | na | 0.0035 ± 0.0008 (2) | na | <<0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Dhakal, S.; Curtis, E.; Miller, H.; Paletta, J.T.; Gee, C.; Rajca, S.; Kievit, F.; Rajca, A. Towards Metabolic Organic Radical Contrast Agents (mORCAs) for Magnetic Resonance Imaging. Molecules 2025, 30, 1581. https://doi.org/10.3390/molecules30071581
Zhang S, Dhakal S, Curtis E, Miller H, Paletta JT, Gee C, Rajca S, Kievit F, Rajca A. Towards Metabolic Organic Radical Contrast Agents (mORCAs) for Magnetic Resonance Imaging. Molecules. 2025; 30(7):1581. https://doi.org/10.3390/molecules30071581
Chicago/Turabian StyleZhang, Shuyang, Sabina Dhakal, Evan Curtis, Hunter Miller, Joseph T. Paletta, Connor Gee, Suchada Rajca, Forrest Kievit, and Andrzej Rajca. 2025. "Towards Metabolic Organic Radical Contrast Agents (mORCAs) for Magnetic Resonance Imaging" Molecules 30, no. 7: 1581. https://doi.org/10.3390/molecules30071581
APA StyleZhang, S., Dhakal, S., Curtis, E., Miller, H., Paletta, J. T., Gee, C., Rajca, S., Kievit, F., & Rajca, A. (2025). Towards Metabolic Organic Radical Contrast Agents (mORCAs) for Magnetic Resonance Imaging. Molecules, 30(7), 1581. https://doi.org/10.3390/molecules30071581