

Total Synthesis and Stereochemical Assignment of Alternapyrone


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
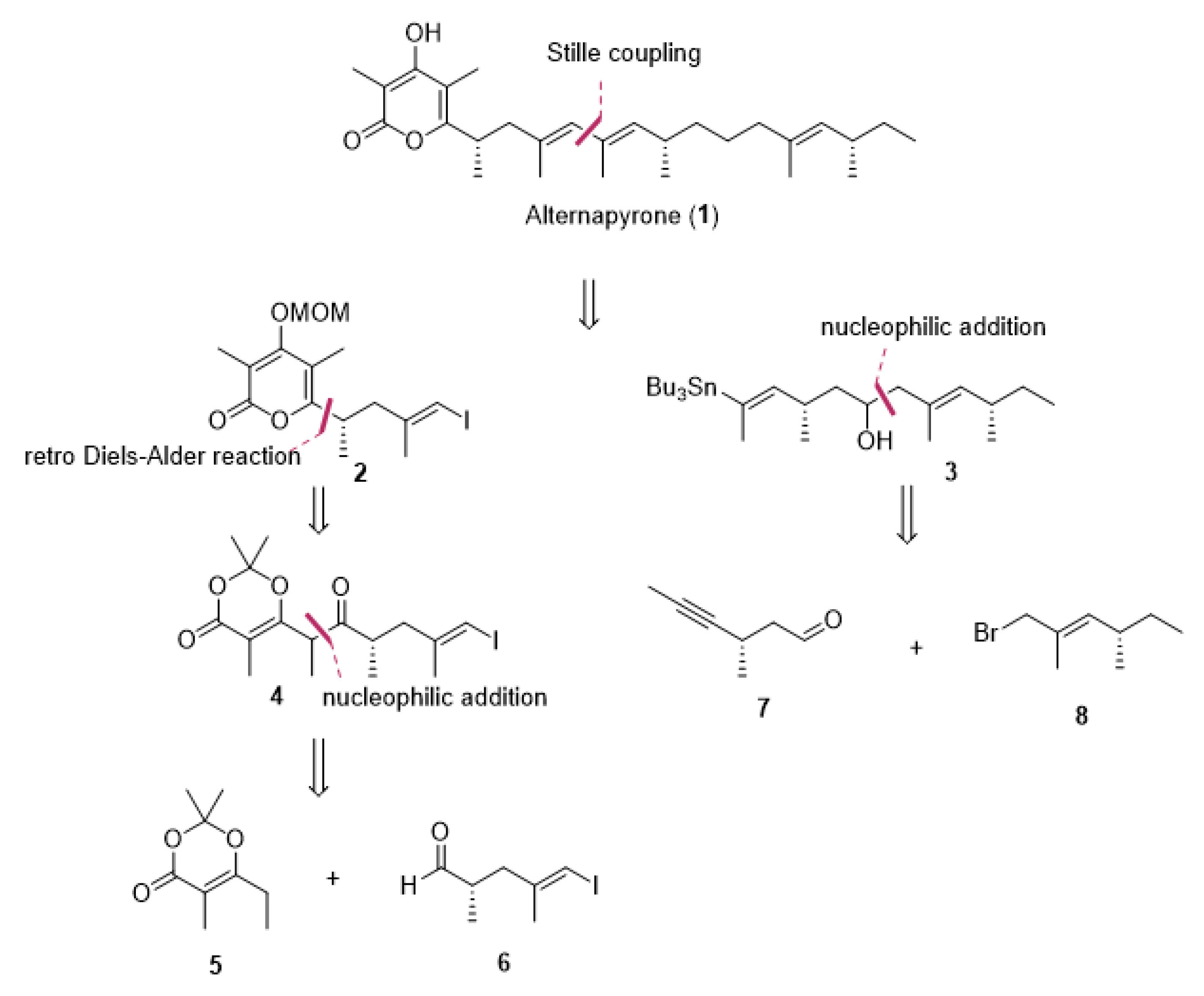
:1. Introduction
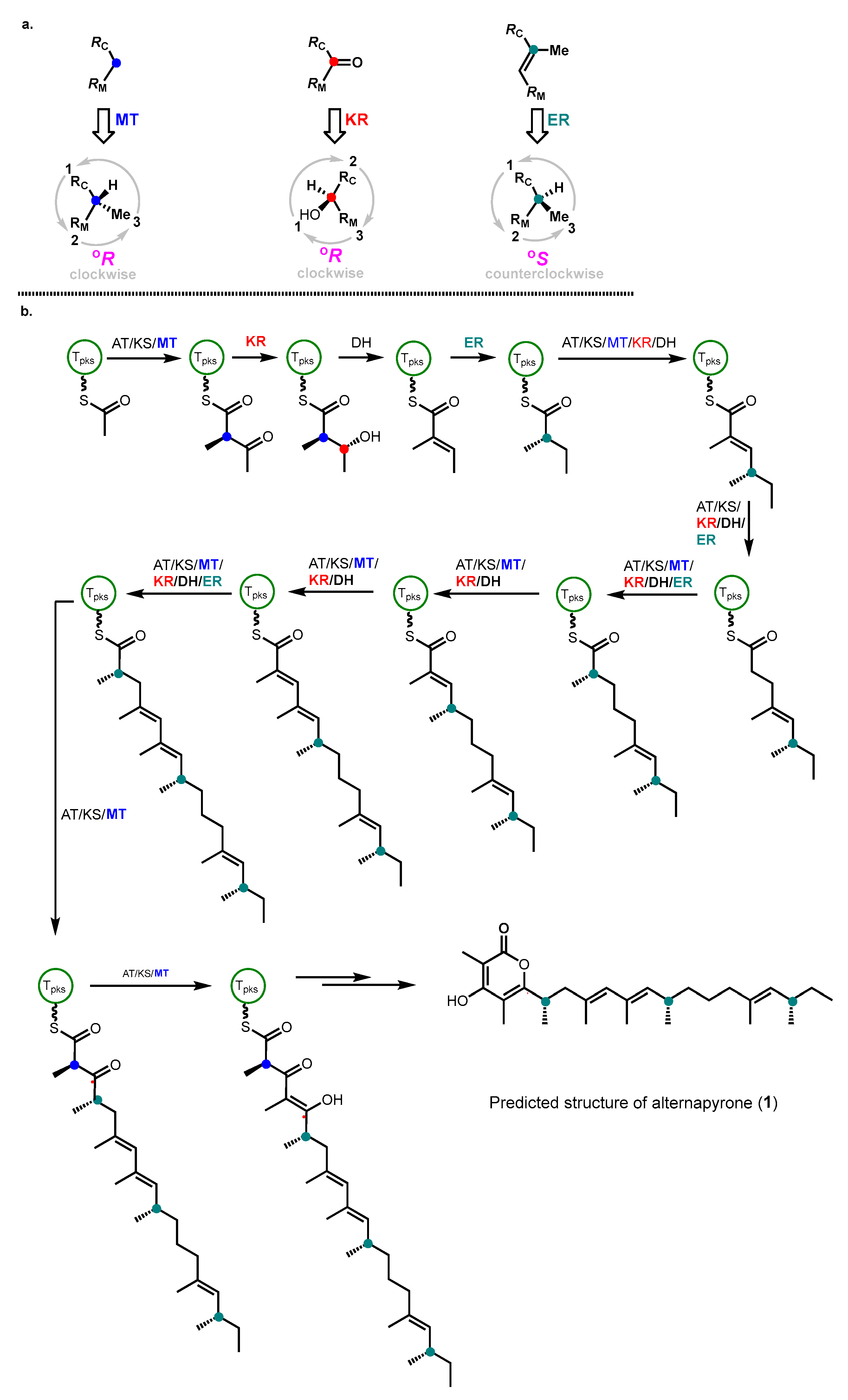
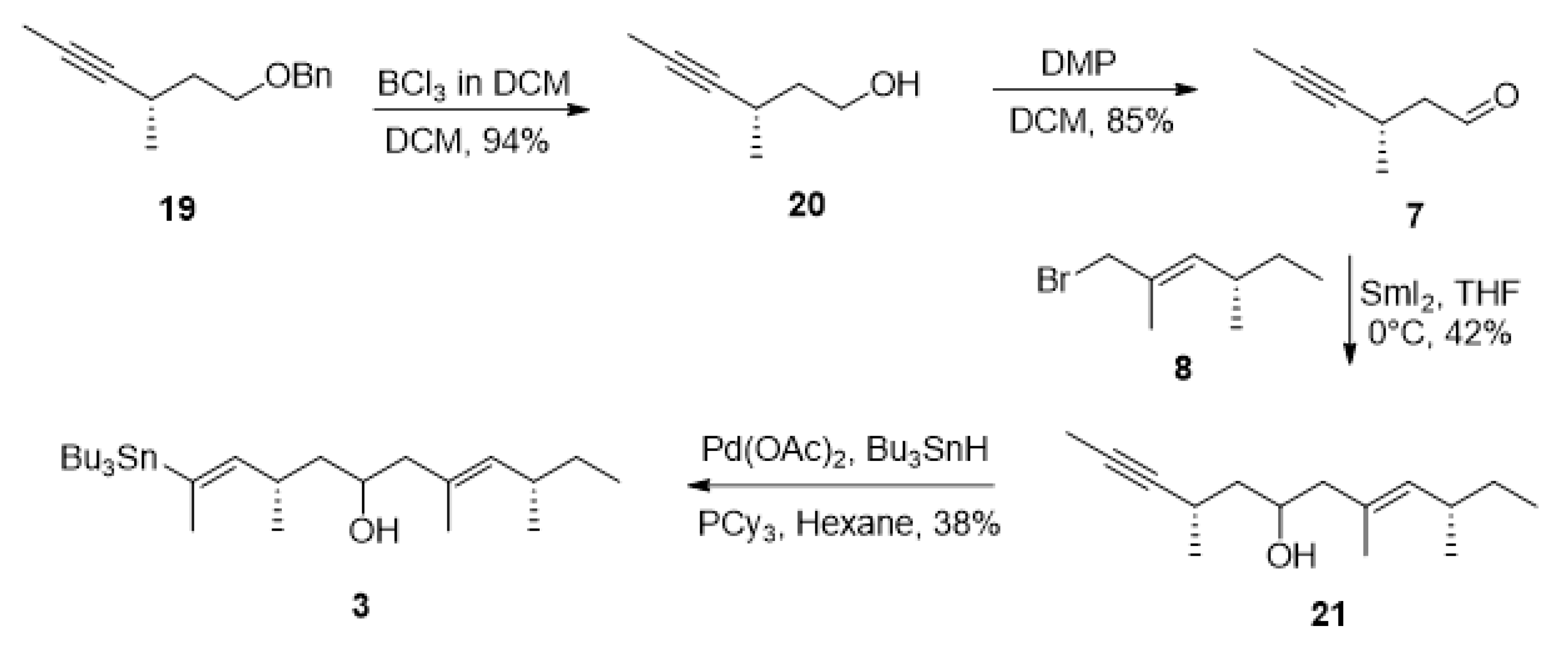
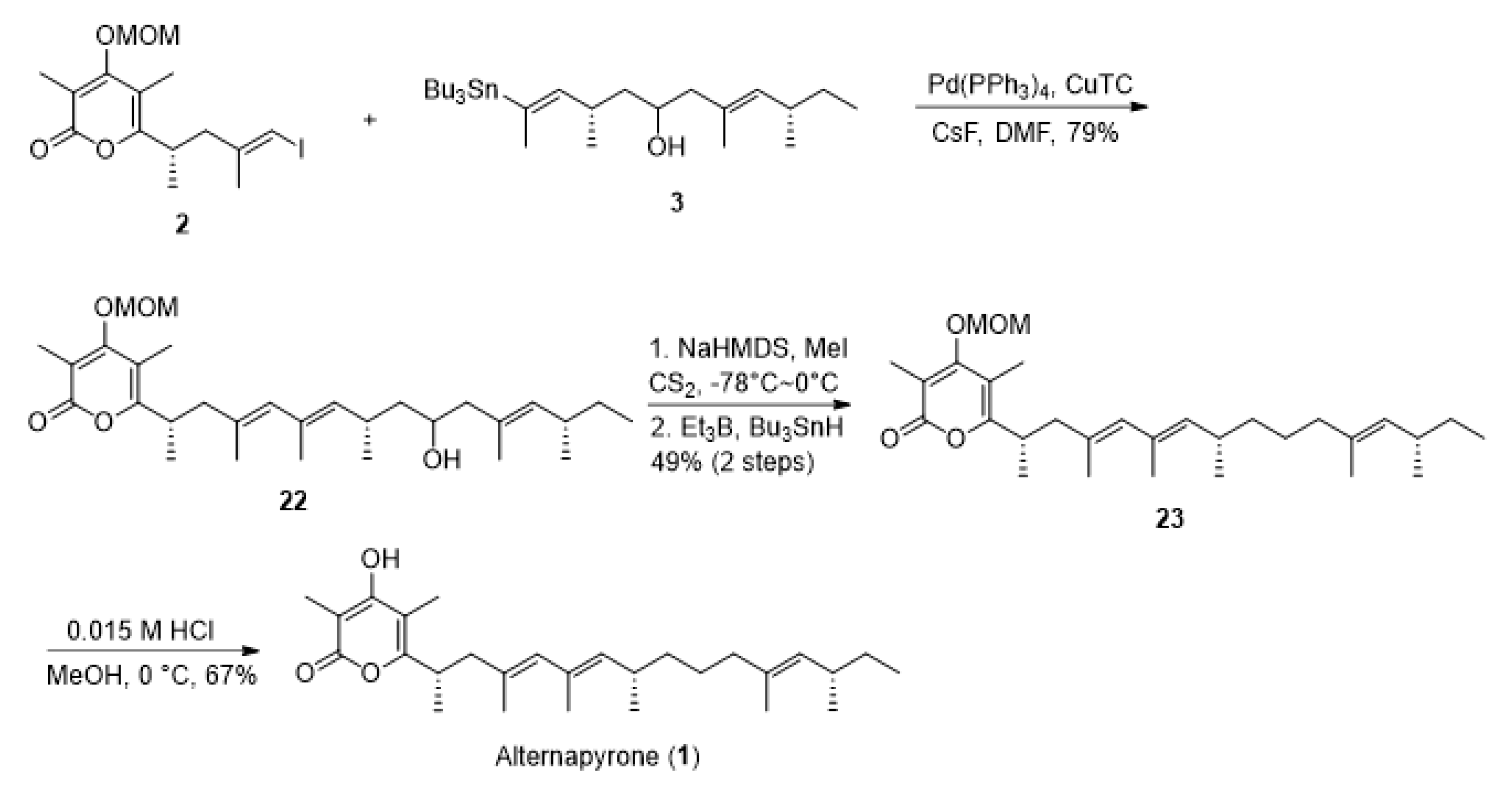
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Experimental Procedures
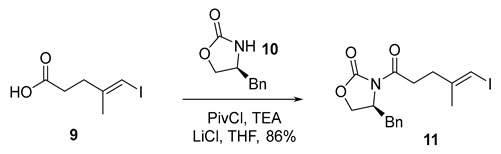
- Rf = 0.40 (20% EtOAc/hexanes), UV and PMA stain.
- = +25.5 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C16H18INO3Na+ [M + Na]+: calcd. 422.0224; found: 422.0225.
- 1H NMR (400 MHz, CDCl3) δ 7.37–7.24 (m, 3H), 7.23–7.17 (m, 2H), 6.03 (q, J = 1.1 Hz, 1H), 4.67 (ddt, J = 12.8, 7.1, 3.3 Hz, 1H), 4.26–4.14 (m, 2H), 3.28 (dd, J = 13.4, 3.4 Hz, 1H), 3.22–2.98 (m, 2H), 2.77 (dd, J = 13.4, 9.6 Hz, 1H), 2.68–2.52 (m, 2H), 1.90 (d, J = 1.0 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 172.1, 153.5, 146.2, 135.3, 129.5, 129.1, 127.5, 76.4, 66.4, 55.3, 38.0, 33.9, 33.8, 24.1.
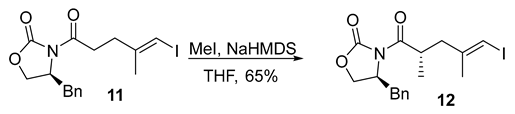
- Rf = 0.40 (20% EtOAc/hexanes), UV and PMA stain.
- = +35.3 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C17H20INO3Na+ [M + Na]+: calcd. 436.0380; found: 436.0380.
- 1H NMR (500 MHz, CDCl3) δ 7.36–7.25 (m, 3H), 7.23–7.18 (m, 2H), 5.97 (s, 1H), 4.64 (tt, J = 10.5, 3.1 Hz, 1H), 4.26–4.14 (m, 2H), 4.05–3.95 (m, 1H), 3.25 (dd, J = 13.4, 3.3 Hz, 1H), 2.77 (dd, J = 13.4, 9.6 Hz, 1H), 2.66 (dd, J = 13.5, 7.2 Hz, 1H), 2.29 (dd, J = 13.6, 7.4 Hz, 1H), 1.86 (s, 3H), 1.19 (d, J = 6.8 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 176.2, 153.2, 145.3, 135.3, 129.6, 129.1, 127.5, 77.3, 66.3, 55.5, 43.2, 38.0, 36.0, 23.9, 17.1.
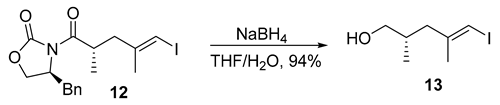
- Rf = 0.40 (25% EtOAc/hexanes), UV and PMA stain.
- = −9.6 (c 1.25, CHCl3);
- 1H NMR (500 MHz, CDCl3) δ 5.92–5.86 (m, 1H), 3.52–3.38 (m, 2H), 2.35 (ddd, J = 13.5, 6.1, 1.2 Hz, 1H), 2.01 (ddd, J = 13.5, 8.4, 0.9 Hz, 1H), 1.92–1.81 (m, 1H), 1.83 (d, J = 1.1 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 146.7, 75.8, 67.8, 43.7, 33.9, 23.9, 16.4.
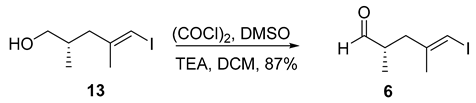
- Rf = 0.50 (20% EtOAc/hexanes), UV and PMA stain.
- = −22.7 (c 0.72, CHCl3);
- 1H NMR (500 MHz, CDCl3) δ 9.62 (d, J = 1.5 Hz, 1H), 5.99–5.95 (m, 1H), 2.64 (ddd, J = 13.9, 6.0, 1.3 Hz, 1H), 2.59–2.48 (m, 1H), 2.19 (ddd, J = 13.9, 8.4, 0.8 Hz, 1H), 1.82 (d, J = 1.1 Hz, 3H), 1.06 (d, J = 7.0 Hz, 3H).
- 13C NMR (126 MHz, CDCl3) δ 203.8, 144.6, 77.2, 44.3, 40.2, 23.8, 13.2.
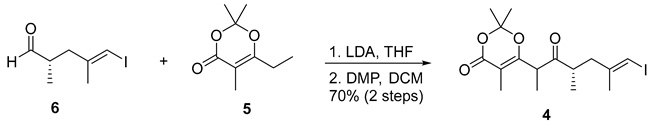
- Rf = 0.40 (20% EtOAc/hexanes), UV and PMA stain.
- HRMS (ESI, m/z) for C16H23IO4Na+ [M + Na]+: calcd. 429.0533; found: 429.0521.
- 1H NMR (400 MHz, CDCl3) δ 5.95–5.90 (m, 2.5H), 3.76 (q, J = 6.9 Hz, 1H), 3.54 (q, J = 6.9 Hz, 1.5H), 2.94–2.78 (m, 2.5H), 2.56 (ddd, J = 14.0, 6.4, 1.1 Hz, 1H), 2.47 (ddd, J = 13.4, 7.8, 1.0 Hz, 1.5H), 2.23–2.13 (m, 2.5H), 1.91 (s, 3H), 1.90 (s, 4.5H), 1.82 (d, J = 1.1 Hz, 4.5H), 1.76 (d, J = 1.1 Hz, 3H), 1.64 (s, 3H), 1.63 (s, 4.5H), 1.63–1.59 (m, 7.5H), 1.26 (d, J = 6.9 Hz, 3H), 1.23 (d, J = 6.9 Hz, 4.5H), 1.05 (d, J = 7.1 Hz, 3H), 1.02 (d, J = 6.7 Hz, 4.5H).
- 13C NMR (101 MHz, CDCl3) δ 209.1, 208.2, 162.7, 162.6, 162.4, 144.7, 144.7, 105.6, 105.5, 102.5, 102.3, 77.9, 77.6, 48.8, 46.8, 44.2, 42.8, 42.3, 41.9, 26.3, 26.0, 24.0, 24.0, 23.9, 23.8, 17.4, 16.7, 12.4, 12.3, 10.4, 10.4.
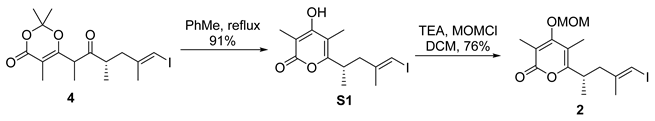
- Rf = 0.50 (100% EtOAc/hexanes), UV and PMA stain.
- = +88.1 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C13H17IO3Na+ [M + Na]+: calcd. 371.0115; found: 371.0109.
- 1H NMR (400 MHz, CDCl3) δ 5.90 (q, J = 1.1 Hz, 1H), 3.16–3.02 (m, 1H), 2.57 (ddd, J = 13.7, 8.4, 1.0 Hz, 1H), 2.37 (ddd, J = 13.7, 6.7, 1.0 Hz, 1H), 2.01 (s, 3H), 1.96 (s, 3H), 1.79 (d, J = 1.1 Hz, 3H), 1.17 (d, J = 6.9 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 166.3, 165.0, 160.5, 145.1, 106.9, 98.5, 77.5, 43.9, 33.2, 24.1, 18.1, 9.7, 8.8.
- Rf = 0.40 (33% EtOAc/hexanes), UV and PMA stain.
- = +109.1 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C15H21IO4Na+ [M + Na]+: calcd. 415.0377; found: 415.0370.
- 1H NMR (500 MHz, CDCl3) δ 5.88 (q, J = 1.1 Hz, 1H), 5.04 (s, 2H), 3.56 (s, 3H), 3.11–3.00 (m, 1H), 2.58 (ddd, J = 13.7, 8.3, 1.0 Hz, 1H), 2.39 (ddd, J = 13.7, 6.7, 1.0 Hz, 1H), 2.01 (s, 3H), 1.92 (s, 3H), 1.80 (d, J = 1.1 Hz, 3H), 1.18 (d, J = 6.8 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 166.3, 165.7, 160.2, 145.2, 110.1, 109.1, 99.2, 77.4, 58.0, 44.0, 33.5, 24.1, 17.9, 11.0, 10.5.
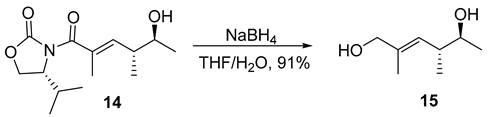
- Rf = 0.50 (150% EtOAc/hexanes), UV and PMA stain.
- = +20.0 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C8H16O2Na+ [M + Na]+: calcd. 167.1043; found: 167.1043.
- 1H NMR (400 MHz, CDCl3) δ 5.32–5.23 (m, 1H), 4.02 (d, J = 1.5 Hz, 2H), 3.59–3.48 (m, 1H), 2.45–2.31 (m, 1H), 2.03–1.82 (m, 2H), 1.69 (d, J = 1.5 Hz, 3H), 1.18 (d, J = 6.2 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 137.1, 127.9, 72.0, 68.6, 40.3, 20.5, 17.1, 14.3.

- Rf = 0.50 (12.5% EtOAc/hexanes), UV and PMA stain.
- = +26.0 (c 1.00, CHCl3);
- 1H NMR (400 MHz, CDCl3) δ 7.71–7.63 (m, 4H), 7.46–7.33 (m, 6H), 5.27 (dq, J = 10.0, 1.5 Hz, 1H), 4.08 (d, J = 1.5 Hz, 2H), 3.53–3.42 (m, 1H), 2.45–2.31 (m, 1H), 1.64 (d, J = 1.4 Hz, 3H), 1.17 (d, J = 6.2 Hz, 3H), 1.06 (s, 9H), 0.94 (d, J = 6.8 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 137.0, 135.7, 133.9, 129.8, 127.8, 126.5, 71.9, 68.9, 40.3, 27.0, 20.1, 19.4, 17.0, 14.2.
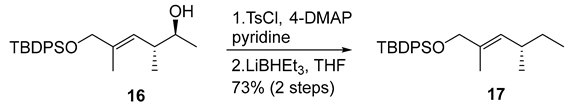
- Rf = 0.80 (10% EtOAc/hexanes), UV and PMA stain.
- = +15.0 (c 0.10, CHCl3);
- HRMS (ESI, m/z) for C24H34OSiNa+ [M + Na]+: calcd. 389.2271; found: 389.2273
- 1H NMR (500 MHz, CDCl3) δ 7.74–7.66 (m, 4H), 7.45–7.33 (m, 6H), 5.22–5.15 (m, 1H), 4.06 (d, J = 1.5 Hz, 2H), 2.36–2.24 (m, 1H), 1.62 (d, J = 1.4 Hz, 3H), 1.39–1.17 (m, 2H), 1.06 (s, 9H), 0.93 (d, J = 6.6 Hz, 3H), 0.85 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 135.7, 134.2, 132.8, 131.3, 129.6, 127.7, 69.4, 33.7, 30.5, 27.0, 20.9, 19.5, 13.9, 12.1.

- Rf = 0.40 (20% Et2O/pentane), PMA stain.
- = +34.3 (c 1.05, CHCl3);
- 1H NMR (500 MHz, CDCl3) δ 5.17 (dq, J = 9.5, 1.3 Hz, 1H), 4.00 (d, J = 1.2 Hz, 2H), 2.36–2.21 (m, 1H), 1.67 (d, J = 1.4 Hz, 3H), 1.41—1.15 (m, 2H), 0.93 (d, J = 6.7 Hz, 3H), 0.83 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 133.5, 132.9, 69.3, 33.9, 30.4, 20.8, 14.0, 12.1.
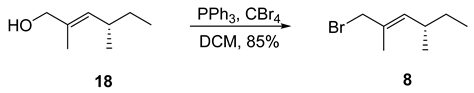
- Rf = 0.90 (100% pentane), UV and PMA stain.
- = +9.7 (c 0.50, CHCl3);
- 1H NMR (400 MHz, CDCl3) δ 5.39–5.32 (m, 1H), 3.99–3.95 (m, 2H), 2.32–2.17 (m, 1H), 1.76 (d, J = 1.4 Hz, 3H), 1.43–1.15 (m, 2H), 0.93 (d, J = 6.6 Hz, 3H), 0.83 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 137.9, 130.9, 42.3, 34.6, 30.2, 20.3, 15.0, 12.0.
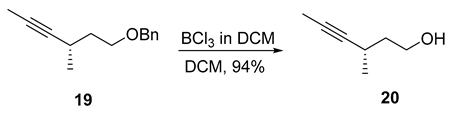
- Rf = 0.30 (33.3% Et2O/pentane), PMA stain.
- = +21.7 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C7H12ONa+ [M + Na]+: calcd. 135.0780; found: 135.0783.
- 1H NMR (500 MHz, CDCl3) δ 3.83–3.71 (m, 2H), 2.61–2.48 (m, 1H), 1.85 (s, 1H), 1.77 (d, J = 2.4 Hz, 3H), 1.72–1.54 (m, 2H), 1.15 (dd, J = 6.9, 3H).
- 13C NMR (101 MHz, CDCl3) δ 83.3, 76.7, 61.5, 39.8, 23.1, 21.7, 3.6.

- Rf = 0.50 (25% Et2O/pentane), PMA stain.
- = +12.0 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C7H10ONa+ [M + Na]+: calcd. 133.0624; found: 133.0624.
- 1H NMR (500 MHz, CDCl3) δ 9.79 (t, J = 2.1 Hz, 1H), 2.98—2.86 (m, 1H), 2.53 (ddd, J = 16.5, 7.6, 2.2 Hz, 1H), 2.45 (ddd, J = 16.5, 6.3, 2.0 Hz, 1H), 1.77 (d, J = 2.4 Hz, 3H), 1.20 (d, J = 6.9 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 201.8, 81.7, 77.2, 50.4, 21.4, 20.9, 3.5.
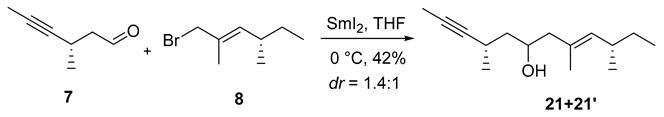
- 21: Rf = 0.40 (10% EtOAc/hexanes), PMA stain.
- = +45.3 (c 0.10, CHCl3);
- HRMS (ESI, m/z) for C15H26ONa+ [M+Na]+: calcd. 245.1876; found: 245.1876.
- 1H NMR (500 MHz, CDCl3) δ 5.01 (dq, J = 9.5, 1.3 Hz, 1H), 3.94 (tt, J = 8.6, 4.1 Hz, 1H), 2.76–2.65 (m, 1H), 2.33–2.23 (m, 1H), 2.18 (ddd, J = 13.2, 4.4, 1.1 Hz, 1H), 2.06 (ddd, J = 13.2, 9.0, 0.9 Hz, 1H), 1.91 (s, 1H), 1.80 (d, J = 2.4 Hz, 3H), 1.66 (d, J = 1.4 Hz, 3H), 1.51–1.39 (m, 2H), 1.40–1.28 (m, 1H), 1.27–1.18 (m, 1H), 1.16 (d, J = 7.0 Hz, 3H), 0.93 (d, J = 6.7 Hz, 3H), 0.84 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 135.7, 130.6, 83.4, 76.4, 66.8, 48.5, 44.4, 34.3, 30.5, 23.0, 22.1, 21.3, 16.5, 12.1, 3.7.
- 21′: Rf = 0.50 (10% EtOAc/hexanes), PMA stain.
- = +42.7 (c 0.10, CHCl3);
- HRMS (ESI, m/z) for C15H26ONa+ [M+Na]+: calcd. 245.1876; found: 245.1876.
- 1H NMR (500 MHz, CDCl3) δ5.00 (dq, J = 9.5, 1.3 Hz, 1H), 3.84 (ddt, J = 8.9, 8.0, 4.4 Hz, 1H), 2.60–2.49 (m, 1H), 2.35–2.22 (m, 1H), 2.19 (ddd, J = 13.4, 4.1, 1.2 Hz, 1H), 2.02 (ddd, J = 13.3, 8.8, 0.9 Hz, 1H), 1.79 (d, J = 2.4 Hz, 3H), 1.65 (d, J = 1.4 Hz, 3H), 1.68–1.58 (m, 1H), 1.49 (ddd, J = 13.6, 6.5, 4.7 Hz, 1H), 1.39–1.28 (m, 1H), 1.26–1.15 (m, 1H), 1.17 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.7 Hz, 3H), 0.84 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 135.5, 130.6, 83.8, 76.6, 67.5, 48.0, 44.0, 34.4, 30.5, 23.3, 21.6, 20.9, 16.5, 12.3, 3.6.
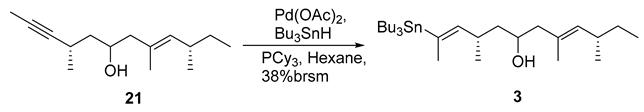
- Rf = 0.70 (10% EtOAc/hexanes), UV and PMA stain.
- = +25.8 (c 0.10, CHCl3);
- HRMS (ESI, m/z) for C27H54OSnNa+ [M+Na]+: calcd. 537.3089; found: 537.3093.
- 1H NMR (400 MHz, CDCl3) δ 5.21 (dq, J = 9.1, 1.9 Hz, 1H), 5.03–4.96 (m, 1H), 3.67–3.56 (m, 1H), 2.99–2.85 (m, 1H), 2.34–2.19 (m, 1H), 2.13 (dd, J = 13.1, 4.4 Hz, 1H), 2.02 (ddd, J = 13.2, 8.8, 0.9 Hz, 1H), 1.87 (d, J = 1.8 Hz, 3H), 1.66–1.55 (m, 2H),1.60 (d, J = 1.4 Hz, 3H), 1.54–1.38 (m, 7H), 1.37–1.16 (m, 10H), 0.98–0.75 (m, 21H).
- 13C NMR (101 MHz, CDCl3) δ 147.2, 136.6, 135.6, 130.7, 67.0, 48.9, 44.9, 34.3, 30.5, 29.3, 29.1, 27.5, 21.9, 21.3, 19.4, 16.4, 13.9, 12.1, 9.2.
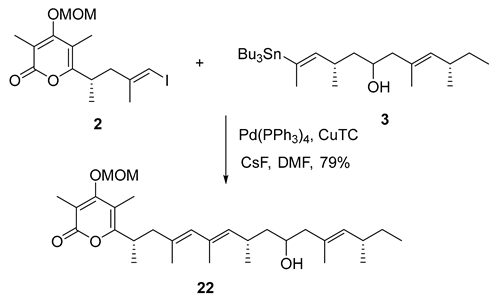
- Rf = 0.40 (33% EtOAc/hexanes), UV and PMA stain.
- = +85.4 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C30H48O5Na+ [M+Na]+: calcd. 511.3394; found: 511.3379.
- 1H NMR (400 MHz, CDCl3) δ 5.56 (s, 1H), 5.02 (s, 2H), 4.98 (dd, J = 9.4, 1.7 Hz, 1H), 4.90–4.82 (m, 1H), 3.65–3.55 (m, 1H), 3.56 (s, 3H), 3.13–3.00 (m, 1H), 2.79–2.63 (m, 1H), 2.37 (ddd, J = 13.3, 8.2, 1.0 Hz, 1H), 2.32–2.15 (m, 2H), 2.12 (dd, J = 13.1, 4.1 Hz, 1H), 2.03–1.97 (m,1H), 2.01 (s, 3H), 1.94 (s, 3H), 1.70 (d, J = 1.4 Hz, 3H), 1.65 (d, J = 1.4 Hz, 3H), 1.59 (d, J = 1.4 Hz, 3H), 1.49–1.21 (m, 4H), 1.20 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.82 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 166.5, 166.0, 161.7, 135.7, 135.3, 132.2, 131.9, 131.7, 130.6, 109.8, 108.7, 99.1, 66.8, 57.9, 48.9, 45.2, 45.1, 34.3, 33.8, 30.5, 29.6, 21.9, 21.3, 17.9, 17.2, 16.5, 12.1, 11.0, 10.5.

- Rf = 0.50 (25% EtOAc/hexanes), UV and PMA stain.
- = +61.2 (c 0.50, CHCl3);
- HRMS (ESI, m/z) for C32H50O5S2Na+ [M+Na]+: calcd. 601.2992; found: 601.2990.
- 1H NMR (400 MHz, CDCl3) δ 5.74–5.63 (m, 1H), 5.52 (s, 1H), 5.02 (s, 2H), 4.92 (d, J = 9.3 Hz, 1H), 4.82–4.74 (m, 1H), 3.56 (s, 3H), 3.12–2.98 (m, 1H), 2.61–2.43 (m, 2H), 2.51 (s, 3H), 2.35 (dd, J = 13.3, 8.0 Hz, 1H), 2.28–2.11 (m, 3H), 2.01 (s, 3H), 1.93 (s, 3H), 1.81–1.70 (m, 1H), 1.69 (d, J = 1.4 Hz, 3H), 1.61 (d, J = 1.4 Hz, 3H), 1.59–1.47 (m, 1H), 1.54 (d, J = 1.4 Hz, 3H), 1.40–1.09 (m, 2H), 1.18 (d, J = 6.8 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H), 0.81 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 215.2, 166.5, 166.0, 161.7, 135.6, 134.2, 132.4, 132.4, 131.5, 129.2, 109.8, 108.7, 99.1, 82.1, 57.9, 45.2, 44.5, 41.1, 34.3, 33.8, 30.5, 29.5, 21.6, 20.7, 18.9, 18.0, 17.9, 17.2, 16.7, 12.2, 11.0, 10.5.
- Rf = 0.30 (12.5% EtOAc/hexanes), UV and PMA stain.
- = +78.0 (c 0.10, CHCl3);
- HRMS (ESI, m/z) for C30H48O4Na+ [M+Na]+: calcd. 495.3445; found: 495.3456.
- 1H NMR (400 MHz, CDCl3) δ 5.56 (s, 1H), 5.03 (s, 2H), 4.93–4.80 (m, 2H), 3.56 (s, 3H), 3.15–2.97 (m, 1H), 2.42–2.29 (m, 2H), 2.28–2.14 (m, 2H), 2.01 (s, 3H), 1.97–1.87 (m, 2H), 1.94 (s, 3H), 1.70 (d, J = 1.4 Hz, 3H), 1.61 (d, J = 1.4 Hz, 3H), 1.56 (d, J = 1.4 Hz, 3H), 1.42–1.08 (m, 6H), 1.20 (d, J = 6.8 Hz, 3H), 0.97–0.85 (m, 6H), 0.82 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, CDCl3) δ 166.5, 166.0, 161.8, 136.3, 133.8, 132.0, 131.8, 131.4, 130.9, 109.8, 108.7, 99.1, 57.9, 45.3, 40.0, 37.2, 34.1, 33.8, 32.5, 30.7, 26.0, 21.2, 21.2, 17.9, 17.3, 16.2, 12.1, 11.0, 10.5.
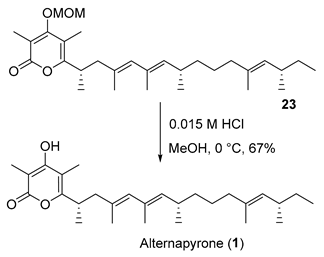
- Rf = 0.40 (50% EtOAc/hexanes), UV and PMA stain.
- = +32.0 (c 0.10, CHCl3);
- HRMS (ESI, m/z) for C28H44O3H+ [M+H]+: calcd. 429.3363; found: 429.3364.
- 1H NMR (400 MHz, Acetone-d6) δ 5.58 (s, 1H), 4.94–4.83 (m, 2H), 3.28–3.15 (m, 1H), 2.46–2.37 (m, 1H), 2.33 (ddd, J = 13.2, 8.6, 1.0 Hz, 1H), 2.29–2.23 (m, 1H), 2.19 (dd, J = 12.9, 6.5 Hz, 1H), 1.96 (s, 3H), 1.92–1.99 (m, 2H), 1.91 (s, 3H), 1.74 (d, J = 1.4 Hz, 3H), 1.63 (d, J = 1.4 Hz, 3H), 1.57 (d, J = 1.4 Hz, 3H), 1.44–1.10 (m, 6H), 1.17 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H), 0.82 (t, J = 7.4 Hz, 3H).
- 13C NMR (101 MHz, Acetone-d6) δ 165.3, 165.1, 161.5, 136.6, 134.5, 133.3, 132.2, 132.1, 131.9, 106.9, 98.4, 45.9, 40.5, 37.9, 34.8, 34.0, 33.1, 31.3, 26.6, 21.6, 21.4, 18.3, 18.0, 17.4, 16.3, 12.3, 10.0, 9.2.
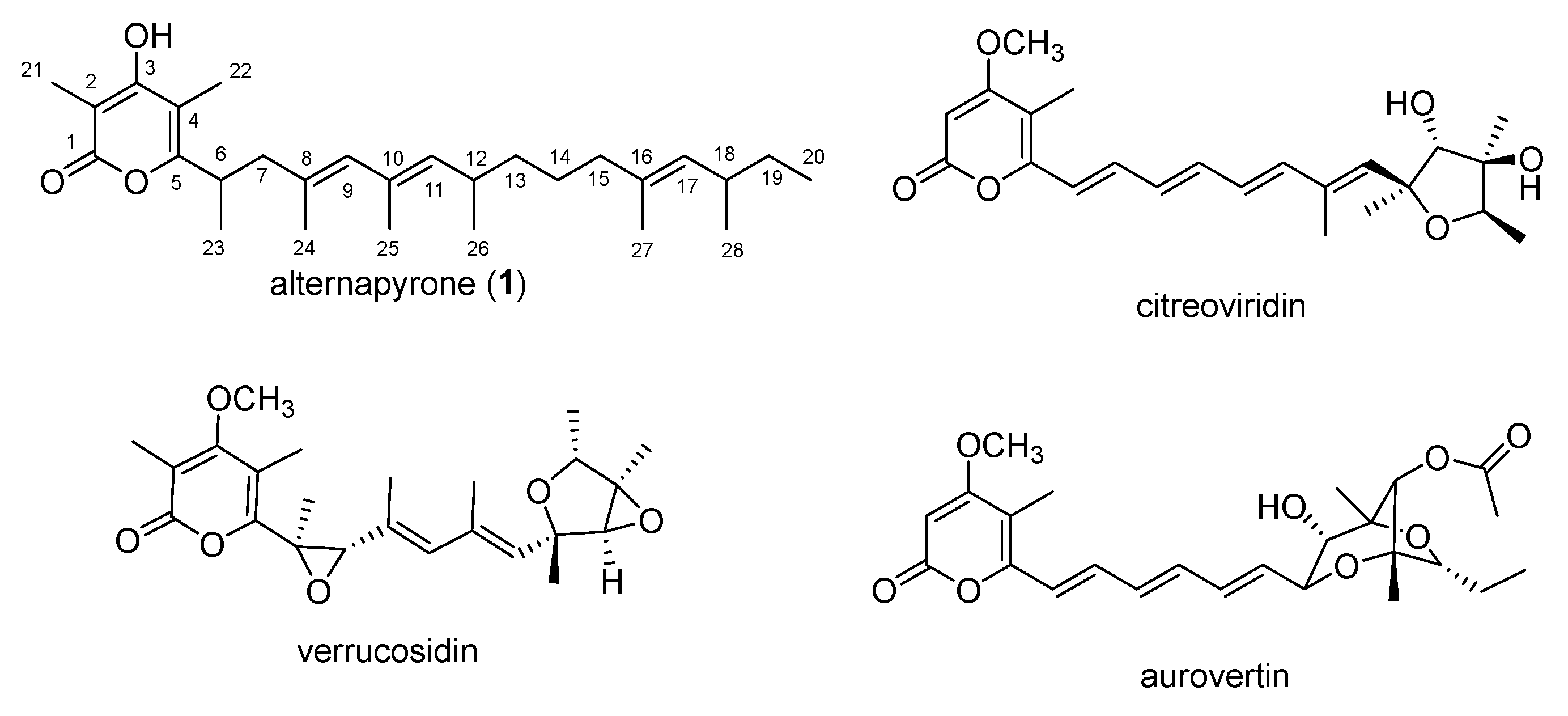
- Natural alternapyrone1: a colorless oil; 1H NMR and 13C NMR data (see Table S1); APCI-TOFMS m/z: calculated for C28H44O3H+ [M + H]+ 429.3363, found 429.3374
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cox, R.J. Polyketides, proteins and genes in fungi: Programmed nano-machines begin to reveal their secrets. Org. Biomol. Chem. 2007, 5, 2010–2026. [Google Scholar] [CrossRef]
- Lin, T.-S.; Chiang, Y.-M.; Wang, C.C.C. Biosynthetic Pathway of the Reduced Polyketide Product Citreoviridin in Aspergillus terreus var. aureus Revealed by Heterologous Expression in Aspergillus nidulans. Org. Lett. 2016, 18, 1366–1369. [Google Scholar] [CrossRef]
- Park, H.R.; Ryoo, I.J.; Choo, S.J.; Hwang, J.H.; Kim, J.Y.; Cha, M.R.; Shin Ya, K.; Yoo, I.D. Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 down-regulator. Toxicology 2007, 229, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.C.; Chang, H.Y.; Hsu, C.H.; Kuo, W.H.; Chang, K.J.; Juan, H.F. Targeting therapy for breast carcinoma by ATP synthase inhibitor aurovertin B. J. Proteome Res. 2008, 7, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Simpson, T.J. Chapter 3 Fungal Type I Polyketide Synthases. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2009; Volume 459, pp. 49–78. [Google Scholar] [CrossRef]
- Takino, J.; Kotani, A.; Ozaki, T.; Peng, W.; Yu, J.; Guo, Y.; Mochizuki, S.; Akimitsu, K.; Hashimoto, M.; Ye, T.; et al. Biochemistry-Guided Prediction of the Absolute Configuration of Fungal Reduced Polyketides. Angew. Chem. Int. Ed. 2021, 60, 23403–23411. [Google Scholar] [CrossRef]
- Fujii, I.; Yoshida, N.; Shimomaki, S.; Oikawa, H.; Ebizuka, Y. An Iterative Type I Polyketide Synthase PKSN Catalyzes Synthesis of the Decaketide Alternapyrone with Regio-Specific Octa-Methylation. Chem. Biol. 2005, 12, 1301–1309. [Google Scholar] [CrossRef]
- Li, H.; Hu, J.; Wei, H.; Solomon, P.S.; Vuong, D.; Lacey, E.; Stubbs, K.A.; Piggott, A.M.; Chooi, Y.-H. Chemical Ecogenomics-Guided Discovery of Phytotoxic α-Pyrones from the Fungal Wheat Pathogen Parastagonospora nodorum. Org. Lett. 2018, 20, 6148–6152. [Google Scholar] [CrossRef]
- Hu, Y.; Zhao, X.; Song, Y.; Jiang, J.; Long, T.; Cong, M.; Miao, Y.; Liu, Y.; Yang, Z.; Zhu, Y.; et al. Anti-inflammatory and Neuroprotective α-Pyrones from a Marine-Derived Strain of the Fungus Arthrinium arundinis and Their Heterologous Expression. J. Nat. Prod. 2024, 87, 1975–1982. [Google Scholar] [CrossRef]
- Phakeovilay, J.; Imaram, W.; Vuttipongchaikij, S.; Bunnak, W.; Lazarus, C.M.; Wattana-Amorn, P. C-Methylation controls the biosynthetic programming of alternapyrone. Org. Biomol. Chem. 2022, 20, 5050–5054. [Google Scholar] [CrossRef]
- Tufano, E.; Lee, E.; Barilli, M.; Casali, E.; Oštrek, A.; Jung, H.; Morana, M.; Kang, J.; Kim, D.; Chang, S.; et al. Iridium Acylnitrenoid-Initiated Biomimetic Cascade Cyclizations: Stereodefined Access to Polycyclic δ-Lactams. J. Am. Chem. Soc. 2023, 87, 24724–24735. [Google Scholar] [CrossRef]
- Preindl, J.; Schulthoff, S.; Wirtz, C.; Lingnau, J.; Fürstner, A. Polyunsaturated C-Glycosidic 4-Hydroxy-2-pyrone Derivatives: Total Synthesis Shows that Putative Orevactaene Is Likely Identical with Epipyrone A. Angew. Chem. Int. Ed. 2017, 56, 7525–7530. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Wu, Y.; Liu, B. Enantioselective Synthesis of the Proposed Structure of Santinol D. Eur. J. Org. Chem. 2020, 2020, 948–960. [Google Scholar] [CrossRef]
- Shimamura, H.; Sunazuka, T.; Izuhara, T.; Hirose, T.; Shiomi, K.; Omura, S. Total synthesis and biological evaluation of verticipyrone and analogues. Org. Lett. 2007, 9, 65–67. [Google Scholar] [CrossRef]
- Morris, C.L.; Hu, Y.L.; Head, G.D.; Brown, L.J.; Whittingham, W.G.; Brown, R.C.D. Oxidative Cyclization Reactions of Trienes and Dienynes: Total Synthesis of Membrarollin. J. Org. Chem. 2009, 74, 981–988. [Google Scholar] [CrossRef]
- Rentsch, A.; Kalesse, M. The Total Synthesis of Corallopyronin A and Myxopyronin B. Angew. Chem. Int. Ed. 2012, 51, 11381–11384. [Google Scholar] [CrossRef]
- Fujita, K.; Matsui, R.; Suzuki, T.; Kobayashi, S. Concise Total Synthesis of (−)-Myxalamide A. Angew. Chem. Int. Ed. 2012, 51, 7271–7274. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, S.; Kuroda, S.; Imamura, K.; Tatsuta, K. The first total synthesis and structural determination of lagunamycin. Tetrahedron Lett. 2006, 47, 6183–6186. [Google Scholar] [CrossRef]
- Grassi, D.; Alexakis, A. Copper-Free Asymmetric Allylic Alkylation Using Grignard Reagents on Bifunctional Allylic Bromides. Org. Lett. 2012, 14, 1568–1571. [Google Scholar] [CrossRef]
- Adamo, M.F.A.; Pergoli, R.; Moccia, M. Alkynyl-2-deoxy-d-riboses, a cornucopia for the generation of families of C-nucleosides. Tetrahedron 2010, 66, 9242–9251. [Google Scholar] [CrossRef]
- Tanabe, Y.; Sato, E.; Nakajima, N.; Ohkubo, A.; Ohno, O.; Suenaga, K. Total Synthesis of Biselyngbyolide A. Org. Lett. 2014, 16, 2858–2861. [Google Scholar] [CrossRef]
- Takamura, H.; Kikuchi, T.; Iwamoto, K.; Nakao, E.; Harada, N.; Otsu, T.; Endo, N.; Fukuda, Y.; Ohno, O.; Suenaga, K.; et al. Unified Total Synthesis, Stereostructural Elucidation, and Biological Evaluation of Sarcophytonolides. J. Org. Chem. 2018, 83, 11028–11056. [Google Scholar] [CrossRef] [PubMed]
- Cornil, J.; Echeverria, P.G.; Reymond, S.; Phansavath, P.; Ratovelomanana-Vidal, V.; Guerinot, A.; Cossy, J. Synthetic Studies toward the C14-C29 Fragment of Mirabalin. Org. Lett. 2016, 18, 4534–4537. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Hsu, S.C.; Hsu, F.L.; Uang, B.J. Asymmetric Synthesis of (–)-Pterosin N from a Chiral 1,3-Dioxolanone. Eur. J. Org. Chem. 2014, 2014, 4351–4355. [Google Scholar] [CrossRef]
- Jeso, V.; Cherry, L.; Macklin, T.K.; Pan, S.C.; LoGrasso, P.V.; Micalizio, G.C. Convergent synthesis and discovery of a natural product-inspired paralog-selective Hsp90 inhibitor. Org. Lett. 2011, 13, 5108–5111. [Google Scholar] [CrossRef]
- Kikuchi, H.; Hoshi, T.; Kitayama, M.; Sekiya, M.; Katou, Y.; Ueda, K.; Kubohara, Y.; Sato, H.; Shimazu, M.; Kurata, S.; et al. New diterpene pyrone-type compounds, metarhizins A and B, isolated from entomopathogenic fungus, Metarhizium flavoviride and their inhibitory effects on cellular proliferation. Tetrahedron 2009, 65, 469–477. [Google Scholar] [CrossRef]
- Frisch, F.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Kleinbeck, F.; Carreira, E.M. Total Synthesis of Bafilomycin A1. Angew. Chem. Int. Ed. 2008, 48, 578–581. [Google Scholar] [CrossRef]
- Hosokawa, S.; Sato, H. Synthesis of the C1–C17 Segment of Bafilomycin N. Synlett 2019, 30, 577–580. [Google Scholar] [CrossRef]
- Toyoshima, A.; Sasaki, M. Toward a total synthesis of amphidinolide N: Convergent synthesis of the C1–C13 segment. Tetrahedron Lett. 2016, 57, 3532–3534. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Feng, J.; Wang, D.; Tang, B.; Xu, C.; Ye, T. Total Synthesis and Stereochemical Assignment of Alternapyrone. Molecules 2025, 30, 1597. https://doi.org/10.3390/molecules30071597
Zhang H, Feng J, Wang D, Tang B, Xu C, Ye T. Total Synthesis and Stereochemical Assignment of Alternapyrone. Molecules. 2025; 30(7):1597. https://doi.org/10.3390/molecules30071597
Chicago/Turabian StyleZhang, Hui, Jiaxuan Feng, Di Wang, Bencan Tang, Chao Xu, and Tao Ye. 2025. "Total Synthesis and Stereochemical Assignment of Alternapyrone" Molecules 30, no. 7: 1597. https://doi.org/10.3390/molecules30071597
APA StyleZhang, H., Feng, J., Wang, D., Tang, B., Xu, C., & Ye, T. (2025). Total Synthesis and Stereochemical Assignment of Alternapyrone. Molecules, 30(7), 1597. https://doi.org/10.3390/molecules30071597