

Synthesis and Evaluation of the Antiproliferative Activity of the Derivatives of 3,5-Diaryl-3,4-dihydro-2H-pyrrole-2-carboxylic Acids
 , , , ,
, , , ,
Abstract

1. Introduction
2. Results and Discussion
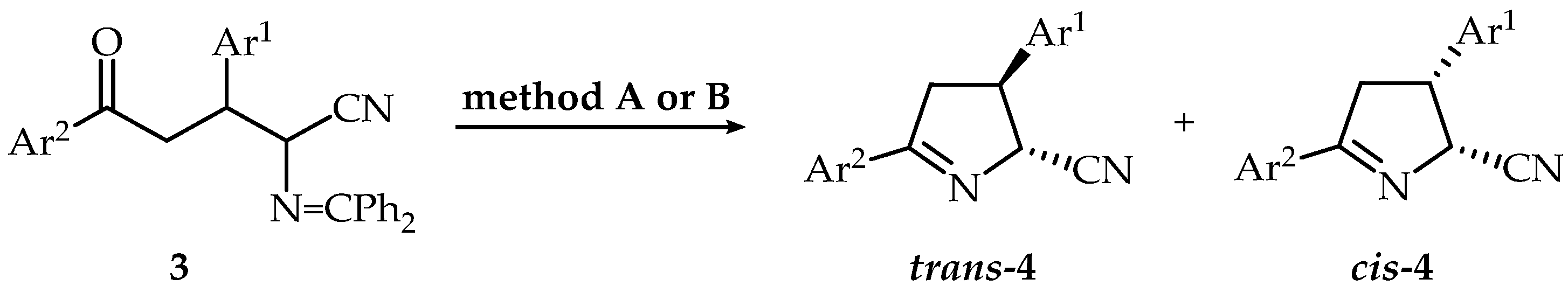
2.1. Synthesis
2.2. Antiproliferative Activity
3. Materials and Methods
3.1. General Information
3.1.1. LC-HRMS Analyses
3.1.2. In Vitro Antiproliferative Activity
3.2. Synthesis and Characterization
3.2.1. Synthesis of 3,5-Diaryl-2-[(diphenylmethylene)amino]-5-oxonitriles (3a–o), General Procedure
3.2.2. Synthesis of 3,5-Diaryl-3,4-dihydro-2H-pyrrole-2-carbonitriles (Δ1-Pyrroline-5-carbonitriles) (4a–h, 4m–o), General Procedure
3.2.3. Reaction of 3,5-Diaryl-3,4-dihydro-2H-pyrrole-2-carbonitriles with 4 M HCl/Dioxane in Dry Methanol, General Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carins, R.A.; Harris, S.I.; Mak, W.T. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Heiden Vander, G.M.; Lunt, Y.S.; Dayton, L.T.; Fiske, P.B.; Israelsen, J.W.; Mattaini, R.K.; Vokes, I.N.; Stephanopoulos, L.C.; Metallo, M.C.; Locasale, W.J. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.J.; Fendt, S.-M.; Becker, D.F. The proline cycle as a potential cancer therapy target. Biochemistry 2018, 57, 3433–3444. [Google Scholar] [CrossRef]
- Geng, P.; Qin, W.; Xu, G. Proline metabolism in cancer. Amino Acids 2021, 53, 1769–1777. [Google Scholar] [CrossRef]
- Wang, D.; Duan, J.-j.; Guo, Y.-f.; Chen, J.-j.; Chen, T.-q.; Wang, J.; Yu, S.-c. Targeting the glutamine-arginine-proline metabolism axis in cancer. J. Enzym. Inhib. Med. Chem. 2024, 39, 2367129. [Google Scholar] [CrossRef]
- Cai, F.; Miao, Y.; Liu, C.; Wu, T.; Shen, S.; Su, X.; Shi, Y. Pyrroline-5-carboxylate reductase 1 promotes proliferation and inhibits apoptosis in non-small cell lung cancer. Oncol. Lett. 2018, 15, 731–740. [Google Scholar] [CrossRef]
- Bogner, A.N.; Stiers, K.M.; Tanner, J.J. Structure, biochemistry, and gene expression patterns of the proline biosynthetic enzyme pyrroline-5-carboxylate reductase (PYCR), an emerging cancer therapy target. Amino Acids 2021, 53, 1817–1834. [Google Scholar] [CrossRef]
- Shvekhgeimer, M.G.A. Methods for the Synthesis of 3,4-2H-Dihydropyrroles (Δ1-Pyrrolines) and Their Chemical Transformations. (Review). Chem. Heterocycl. Compd. 2003, 39, 405–448. [Google Scholar] [CrossRef]
- Chandan, N.; Thompson, A.L.; Moloney, M.G. Rapid synthesis of substituted pyrrolines and pyrrolidines by nucleophilic ring closure at activated oximes. Org. Biomol. Chem. 2012, 10, 7863–7868. [Google Scholar] [CrossRef]
- Ma, T.; Fu, X.; Kee, C.W.; Zong, L.; Pan, Y.; Huang, K.-W.; Tan, C.-H. Pentanidium-Catalyzed Enantioselective Phase-Transfer Conjugate Addition Reactions. J. Am. Chem. Soc. 2011, 133, 2828–2831. [Google Scholar] [CrossRef] [PubMed]
- Halland, N.; Hazell, R.G.; Jørgensen, K.A. Organocatalytic Asymmetric Conjugate Addition of Nitroalkanes to α,β-Unsaturated Enones Using Novel Imidazoline Catalysts. J. Org. Chem. 2002, 67, 8331–8338. [Google Scholar] [CrossRef] [PubMed]
- Pandit, P.; Chatterjee, N.; Maiti, D.K. First synthesis of fused-Δ1-pyrrolines via intramolecular 1,3-dipolar cycloaddition of ketoimine: A complete diastereoselective approach. Chem. Commun. 2011, 47, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Ren, J.; Wang, Z. TfOH-Catalyzed Formal [3 + 2] Cycloaddition of Cyclopropane 1,1-Diesters with Nitriles. J. Org. Chem. 2014, 79, 790–796. [Google Scholar] [CrossRef]
- Medran, N.S.; La-Venia, A.; Testero, S.A. Metal-mediated synthesis of pyrrolines. RSC Adv. 2019, 9, 6804–6844. [Google Scholar] [CrossRef]
- Lee, H.; Nam, H.; Lee, S.Y. Enantio- and Diastereoselective Variations on α-Iminonitriles: Harnessing Chiral Cyclopropenimine-Thiourea Organocatalysts. J. Am. Chem. Soc. 2024, 146, 3065–3074. [Google Scholar] [CrossRef]
- Tsuge, O.; Ueno, K.; Kanemasa, S.; Yorozu, K. Michael Addition and Alkylation of 2-Azaallyl Anions Derived from N-(1-Cyanoalkyl)imines, and Stereoselective Cyclization of Imine Esters or Ketones Leading to 1-Pyrrolines. Bull. Chem. Soc. Jpn. 1987, 60, 3347–3358. [Google Scholar] [CrossRef]
- Meyer, N.; Opatz, T. A Short Synthesis of Polysubstituted Pyrrolidines via α-(Alkylideneamino)nitriles. Synlett 2004, 5, 787–790. [Google Scholar] [CrossRef]
- Tasheva, D.; Petrova, A.; Simova, S. Convenient Synthesis of Some Substituted 5-Oxonitriles under Aqueous Conditions: Synthesis of 3,4-Dihydro-2H-pyrrole-2-carbonitriles. Synth. Commun. 2007, 37, 3971–3979. [Google Scholar] [CrossRef]
- Bai, X.-F.; Li, L.; Xu, Z.; Zheng, Z.-J.; Xia, C.-G.; Cui, Y.-M.; Xu, L.-W. Asymmetric Michael Addition of Aldimino Esters with Chalcones Catalyzed by Silver/Xing-Phos: Mechanism-Oriented Divergent Synthesis of Chiral Pyrrolines. Chem. Eur. J. 2016, 22, 10399–10404. [Google Scholar] [CrossRef]
- Nie, J.; Hua, M.-Q.; Xiong, H.-Y.; Zheng, Y.; Ma, J.-A. Asymmetric Phase-Transfer-Catalyzed Conjugate Addition of Glycine Imine to Exocyclic α,β-Unsaturated Ketones: Construction of Polycyclic Imines Containing Three Stereocenters. J. Org. Chem. 2012, 77, 4209–4216. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Watanabe, S.; Takahashi, T.; Tokoro, Y.; Fukuzawa, S. Silver/ThioClickFerrophos Complex as an Effective Catalyst for Asymmetric Conjugate Addition of Glycine Imino Ester to Unsaturated Malonates and α-Enones. Org. Lett. 2013, 15, 4418–4421. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, D.S.; Ofial, A.R.; Mayr, H. Nucleophilic reactivities of Schiff base derivatives of amino acids. Tetrahedron 2019, 75, 459–463. [Google Scholar] [CrossRef]
- Tasheva, D.; Petkova, N.; Dryanska, V. A Convenient Synthesis of Esters of β-Phenylglutamic Acid Under Aqueous Conditions. Synth. Commun. 2003, 3, 3661–3670. [Google Scholar] [CrossRef]
- Poonia, N.S.; Yadav, B.; Kumar, C.; Bhagwat, V. Coordinative role of alkali cations in organic reactions. 1. Selective methylation of the alcoholic group of kojic acid. J. Org. Chem. 1977, 42, 2030–2031. [Google Scholar] [CrossRef]
- Poonia, N.S.; Chhabra, K.; Kumar, C.; Sharma, T.C.; Bhagwat, V.W. Coordinative role of alkali cations in organic synthesis. 2. The chalcone-flavanone system. J. Org. Chem. 1977, 42, 3311–3313. [Google Scholar] [CrossRef]
- Bukhari, N.A.S.; Jasamai, M.; Jantan, I.; Ahmad, W. Review of Methods and Various Catalysts Used for Chalcone Synthesis. Mini-Rev. Org. Chem. 2013, 10, 73–83. [Google Scholar]
- Kulkarni, P. Calcium oxide catalyzed synthesis of chalcone under microwave condition. Curr. Microw. Chem. 2015, 2, 144–149. [Google Scholar] [CrossRef]
- van der Werf, A.; Kellogg, R.M. Synthesis of some proline derivatives by means of Michael additions of glycine esters. Tetrahedron Lett. 1991, 32, 3727–3730. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; Polt, R.L. A mild and efficient route to Schiff base derivatives of amino acids. J. Org. Chem. 1982, 47, 2663–2666. [Google Scholar] [CrossRef]
- Kanemasa, S.; Uchida, O.; Wada, E. Stereoselective Michael addition of α-amino esters in the presence of lithium bromide/1,8-diazabicyclo[5.4.0]undec-7-ene. J. Org. Chem. 1990, 55, 4411–4417. [Google Scholar] [CrossRef]
- Opio, J.O.; Labidalle, S.; Galons, H.; Miocque, M.; Zaparucha, A.; Loupy, A. Preparation of 2-Substituted Vinylamino Acid Derivatives. Synth. Commun. 1991, 21, 1743–1754. [Google Scholar] [CrossRef]
- Pfaff, D.; Nemecek, G.; Podlech, J. A Lewis acid-promoted Pinner reaction. Beilstein J. Org. Chem. 2013, 9, 1572–1577. [Google Scholar] [CrossRef] [PubMed]
- Roger, R.; Neilson, D.G. The Chemistry of Imidates. Chem. Rev. 1961, 61, 179–211. [Google Scholar] [CrossRef]
- Zil‘berman, E.N. Reactions of nitriles with hydrogen halides and nucleophilic reagents. Russ. Chem. Rev. 1962, 31, 615–633. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Pereira, A.M.; Cidade, H.; Tiritan, M.E. Stereoselective Synthesis of Flavonoids: A Brief Overview. Molecules 2023, 28, 426. [Google Scholar] [CrossRef]
- Pickard, P.L.; Tolbert, T.L. Organic Syntheses, Collective Volume V; John Wiley: Hoboken, NJ, USA, 1973; pp. 520–522. [Google Scholar]
- Kohler, E.P.; Chadwell, H.M. Organic Syntheses, Collective Volume I; John Wiley: Hoboken, NJ, USA, 1941; pp. 78–80. [Google Scholar]
- Roman, B.I.; De Ryck, T.; Patronov, A.; Slavov, S.H.; Vanhoecke, B.W.; Katritzky, A.R.; Bracke, M.E.; Stevens, C.V. 4-Fluoro-3′,4′,5′-trimethoxychalcone as a new anti-invasive agent. From discovery to initial validation in an in vivo metastasis model. Eur. J. Med. Chem. 2015, 101, 627–639. [Google Scholar] [CrossRef]
- Li, J.-T.; Yang, W.-Z.; Wang, S.-X.; Li, S.-H.; Li, T.-S. Improved synthesis of chalcones under ultrasound irradiation. Ultrason. Sonochem. 2002, 9, 237–239. [Google Scholar] [CrossRef]
- Mateeva, N.; Kode, R.; Redda, K. Synthesis of Novel Flavonoid Derivatives as Potential HIV-Integrase Inhibitors. J. Het. Chem. 2002, 39, 1251–1258. [Google Scholar] [CrossRef]

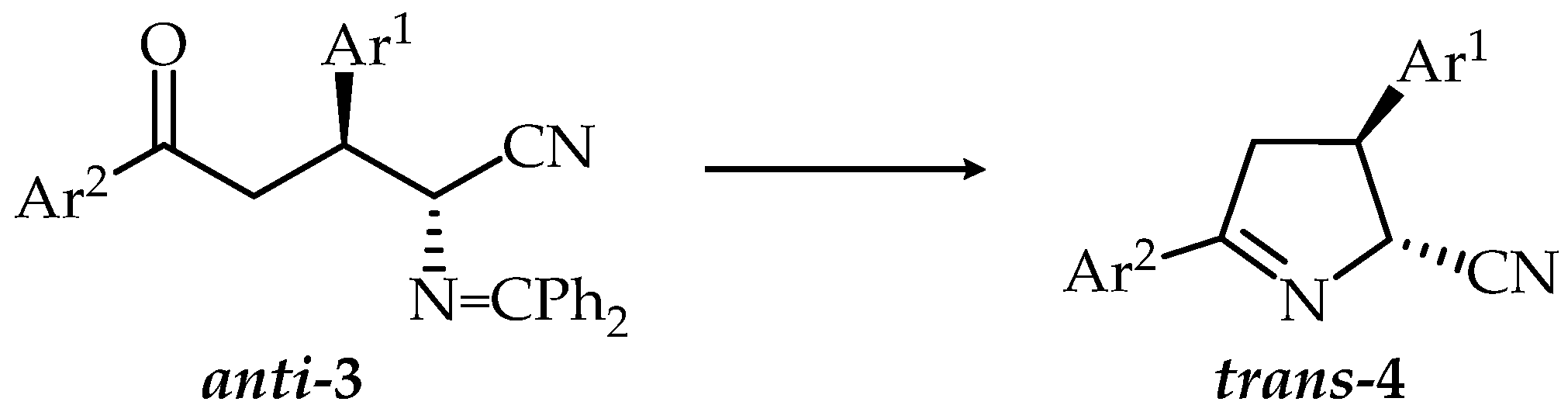


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method A a | Method B b | ||||||
|---|---|---|---|---|---|---|---|
| 3 | Ar1 | Ar2 | Reaction Time (min) | Yield c (%) | D.R. d anti:syn | Yield e (%) | D.R. d anti:syn |
| a | C6H5 | C6H5 | 30 | 84 f | 99:1 | 92 | 40:60 |
| b | 4-CH3OC6H4 | C6H5 | 30 | 84 | 92:8 | 92 | 39:61 |
| c | 4-(CH3)2NC6H4 | C6H5 | 30 | 75 | 88:12 | - | - |
| d | 4-NO2C6H4 | C6H5 | 10 | 94 e | 82:18 | - | - |
| e | C6H5 | 4-CH3OC6H4 | 60 | 87 | 93:7 | 89 | 44:56 |
| f | 4-ClC6H4 | 4-CH3OC6H4 | 30 | 85 | 94:6 | 91 | 42:58 |
| g | 4-FC6H4 | 3,4,5-(CH3O)3C6H2 | 240 | 87 e | 49:51 | - | - |
| h | pyridine-3-yl | C6H5 | 20 | 67 | 82:18 | 84 | 40:60 |
| i | 4-CH3C6H4 | C6H5 | 30 | 85 f | 91 | 44:56 | |
| j | 4-ClC6H4 | C6H5 | 30 | 85 f | 95 | 40:60 | |
| k | C6H5 | 4-CH3C6H4 | 30 | 85 f | 85 | 53:47 | |
| l | C6H5 | 4-ClC6H4 | 30 | 89 f | 84 | 37:63 | |
| Method C a | ||||
|---|---|---|---|---|
| 3 | Ar1 | Reaction Time (min) | Yield b (%) | D.R. c (anti:syn) |
| m | 3-F-4-CH3-C6H3 | 125 | 88 | 44:56 |
| n | 3-Cl-4-CH3-C6H3 | 130 | 91 | 44:56 |
| o | 3-Br-4-CH3-C6H3 | 335 | 90 | 45:55 |
| Method A a | Method B b | |||||
|---|---|---|---|---|---|---|
| 4 | Ar1 | Ar2 | Reaction Time (min) | Yield c (%) | Reaction Time (min) | Yield d (%) trans, cis |
| a | -C6H5 | -C6H5 | 120 | 96 | 150 | 39, 46 |
| b | -C6H4(4-OCH3) | -C6H5 | 180 | 91 | 150 | 30, 43 |
| c | -C6H4(4-(N(CH3)2) | -C6H5 | 30 | 75 | - | - |
| d | -C6H4(4-NO2) | -C6H5 | 210 | 86 e | - | - |
| e | -C6H5 | -C6H4(4-OCH3) | 300 | 78 | 180 | 41, 49 |
| f | -C6H4(4-Cl) | -C6H4(4-OCH3) | 300 | 92 | 360 | 38, 49 |
| g | -C6H4(4-F) | -C6H2(3,4,5-OCH3)3 | 1440 | 28, 38 d | 360 | 51, 41 |
| h | pyridine-3-yl | -C6H5 | 210 | 86 | 400 | 51, 30 |
| i | -C6H4(4-CH3) | -C6H5 | - | - | 270 | 36, 43 |
| j | -C6H4(4-Cl) | -C6H5 | - | - | 180 | 45, 43 |
| k | -C6H5 | -C6H4(4-CH3) | - | - | 180 | 50, 36 |
| l | -C6H5 | -C6H4(4-Cl) | - | - | 205 | 34, 51 |
| m | -C6H3(3-F-4-CH3) | -C6H2(2-OH-4,6-(OCH3)2) | 120 | 37, 55 d | - | - |
| n | -C6H3(3-Cl-4-CH3) | -C6H2(2-OH-4,6-(OCH3)2) | 120 | 45, 51 d | - | - |
| o | -C6H3(3-Br-4-CH3) | -C6H2(2-OH-4,6-(OCH3)2) | 120 | 35, 51 d | - | - |
| 5 | Ar1 | Ar2 | Reaction Time (min) | Yield a (%) |
|---|---|---|---|---|
| a | -C6H5 | -C6H5 | 300 1440 | 59 64 b |
| b | -C6H4(4-OCH3) | -C6H5 | 270 | 68 |
| c | -C6H4(4-(N(CH3)2) | -C6H5 | 150 | 73 |
| d | -C6H4(4-NO2) | -C6H5 | 150 | 39 c |
| e | -C6H5 | -C6H4(4-OCH3) | 270 | 80 |
| f | -C6H4(4-Cl) | -C6H4(4-OCH3) | 240 | 71 |
| g | -C6H4(4-F) | -C6H2(3,4,5-OCH3)3 | 170 1620 | - 53 b |
| i | -C6H4(4-CH3) | -C6H5 | 240 | 64 |
| j | -C6H4(4-Cl) | -C6H5 | 240 | 63 |
| k | -C6H5 | -C6H4(4-CH3) | 420 | 64 |
| l | -C6H5 | -C6H4(4-Cl) | 240 | 62 |
| m | -C6H3(3-F-4-CH3) | -C6H2(2-OH-4,6-(OCH3)2) | 120 | 68 |
| 6m | -C6H3(3-F-4-CH3) | -C6H2(2-OH-4,6-(OCH3)2) | 1620 | 56 d |
| Cell Line | Cisplatin * | trans-4k | cis-4k | cis-4e | cis-4h |
|---|---|---|---|---|---|
| IC50 [µM] ± SD (SI) | IC50 [µM] ± SD (SI) | IC50 [µM] ± SD (SI) | IC50 [µM] ± SD (SI) | IC50 [µM] ± SD (SI) | |
| MCF-10A | 23.2 ± 1.4 (1) | 182 ± 5.8 (1) | 104 ± 6 (1) | 427 ± 12 (1) | 2277 ± 178 (1) |
| MCF-7 | 45.8 ± 3.2 (0.50) | 568 ± 57 (0.32) | 374 ± 6 (0.28) | 1766 ± 232 (0.24) | 1377 ± 255 (1.65) |
| MDA-MB-231 | 1.8 ± 0.1 (12.7) | 313 ± 20 (0.58) | 2562 ± 107 (0.04) | >3619 (<0.12) | >4110 (<0.55) |
| H1299 | 5.0 ± 0.9 (4.62) | 354 ± 15 (0.51) | 205 ± 6 (0.51) | 840 ± 43 (0.51) | 2984 ± 62 (0.76) |
| A549 | 38.6 ± 4.7 (0.60) | 11.7 ± 0.8 (15.5) | 82.2 ± 5.8 (1.27) | 341 ± 26 (1.25) | 311 ± 19 (7.33) |
| HeLa | 19.6 ± 3.2 (1.18) | 84.1 ± 2.3 (2.16) | 169 ± 10 (0.61) | 158 ± 6.5 (2.69) | 2219 ± 136 (1.03) |
| HepG2 | 19.1 ± 0.4 (1.22) | 159 ± 4.6 (1.14) | 235 ± 15 (0.44) | 554 ± 34 (0.77) | 2996 ± 107 (0.76) |
| HT-29 | 8.2 ± 0.3 (2.84) | 136 ± 7.7 (1.33) | 853 ± 46 (0.12) | 308 ± 8 (1.39) | 1640 ± 132 (1.39) |
| PC3 | 3.4 ± 0.5 (6.93) | 121 ± 13 (1.50) | 297 ± 25 (0.35) | 478 ± 58 (0.80) | 1915 ± 70 (1.19) |
| Cell Line | trans-4m | cis-4m | cis-6m |
|---|---|---|---|
| IC50 [µM] ± SD (SI) | IC50 [µM] ± SD (SI) | IC50 [µM] ± SD (SI) | |
| MCF-10A | 79.3 ± 2.5 (1) | 170 ± 12 (1) | 429 ± 28 (1) |
| MCF-7 | 149 ± 12 (0.53) | 185 ± 26 (0.92) | 923 ± 37 (0.46) |
| MDA-MB-231 | 717 ± 73 (0.11) | 16.0 ± 0.7 (10.5) | 1989 ± 40 (0.22) |
| H1299 | 36.4 ± 2.0 (2.18) | 25.4 ± 2.3 (6.68) | 1876 ± 51 (0.23) |
| A549 | 32.2 ± 1.1 (2.46) | 97.6 ± 13 (1.74) | 136 ± 5.4 (3.16) |
| HeLa | 1168 ± 42 (0.07) | 224 ± 15 (0.75) | 1180 ± 37 (0.36) |
| HepG2 | 44.6 ± 2.8 (1.78) | 117 ± 4.8 (1.45) | 117 ± 6.2 (3.67) |
| HT-29 | 28.1 ± 2.0 (2.82) | 19.6 ± 1.8 (8.66) | 64.3 ± 4.8 (6.67) |
| PC3 | 44.3 ± 5.4 (1.79) | 115 ± 4.8 (1.47) | 125 ± 7.6 (3.44) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihaylova, V.; Iliev, I.; Vasileva, A.; Mazzio, E.; Mochona, B.; Mateeva, N.; Tasheva, D. Synthesis and Evaluation of the Antiproliferative Activity of the Derivatives of 3,5-Diaryl-3,4-dihydro-2H-pyrrole-2-carboxylic Acids. Molecules 2025, 30, 1602. https://doi.org/10.3390/molecules30071602
Mihaylova V, Iliev I, Vasileva A, Mazzio E, Mochona B, Mateeva N, Tasheva D. Synthesis and Evaluation of the Antiproliferative Activity of the Derivatives of 3,5-Diaryl-3,4-dihydro-2H-pyrrole-2-carboxylic Acids. Molecules. 2025; 30(7):1602. https://doi.org/10.3390/molecules30071602
Chicago/Turabian StyleMihaylova, Vesela, Ivan Iliev, Anelia Vasileva, Elizabeth Mazzio, Bereket Mochona, Nelly Mateeva, and Donka Tasheva. 2025. "Synthesis and Evaluation of the Antiproliferative Activity of the Derivatives of 3,5-Diaryl-3,4-dihydro-2H-pyrrole-2-carboxylic Acids" Molecules 30, no. 7: 1602. https://doi.org/10.3390/molecules30071602
APA StyleMihaylova, V., Iliev, I., Vasileva, A., Mazzio, E., Mochona, B., Mateeva, N., & Tasheva, D. (2025). Synthesis and Evaluation of the Antiproliferative Activity of the Derivatives of 3,5-Diaryl-3,4-dihydro-2H-pyrrole-2-carboxylic Acids. Molecules, 30(7), 1602. https://doi.org/10.3390/molecules30071602