
Targeting the CXCR4/CXCL12 Axis in Cancer Therapy: Analysis of Recent Advances in the Development of Potential Anticancer Agents

 , , , , ,
, , , , ,  and
and 
Abstract

1. Introduction
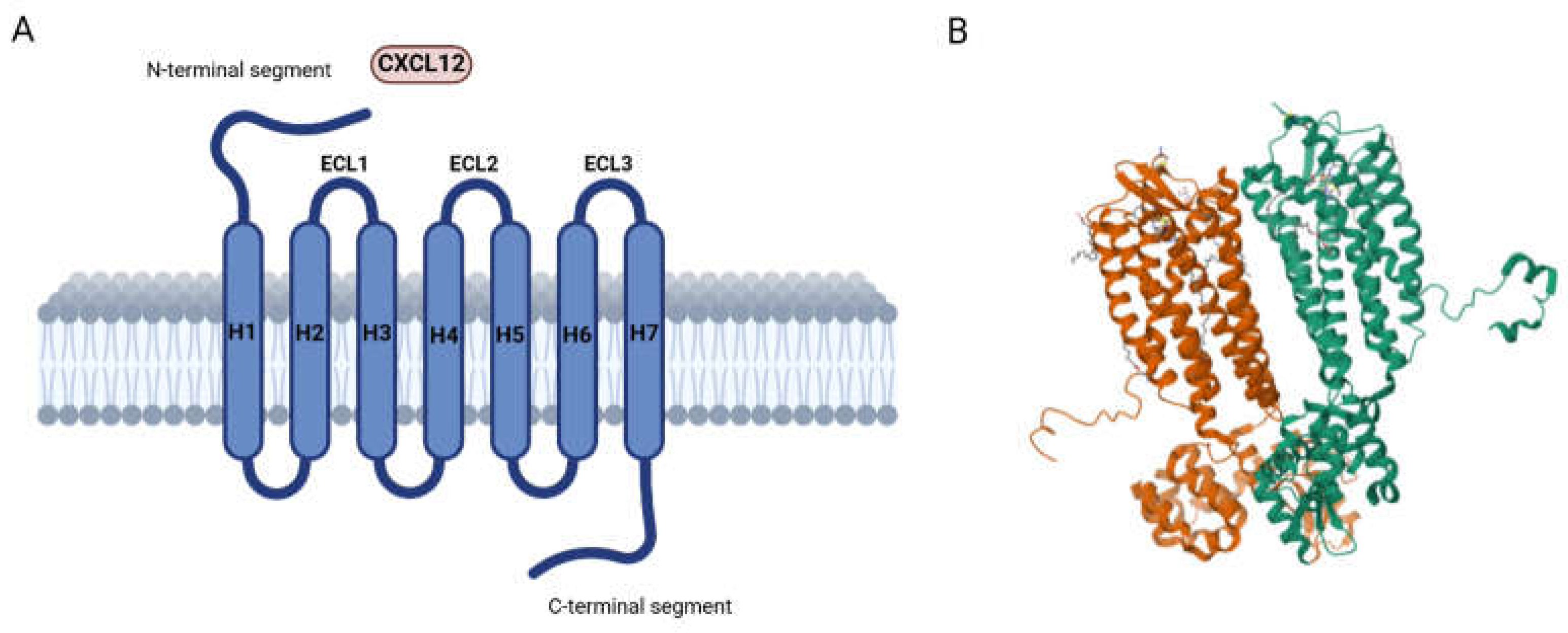
2. CXCR4: Localization and Physio-Pathological Role
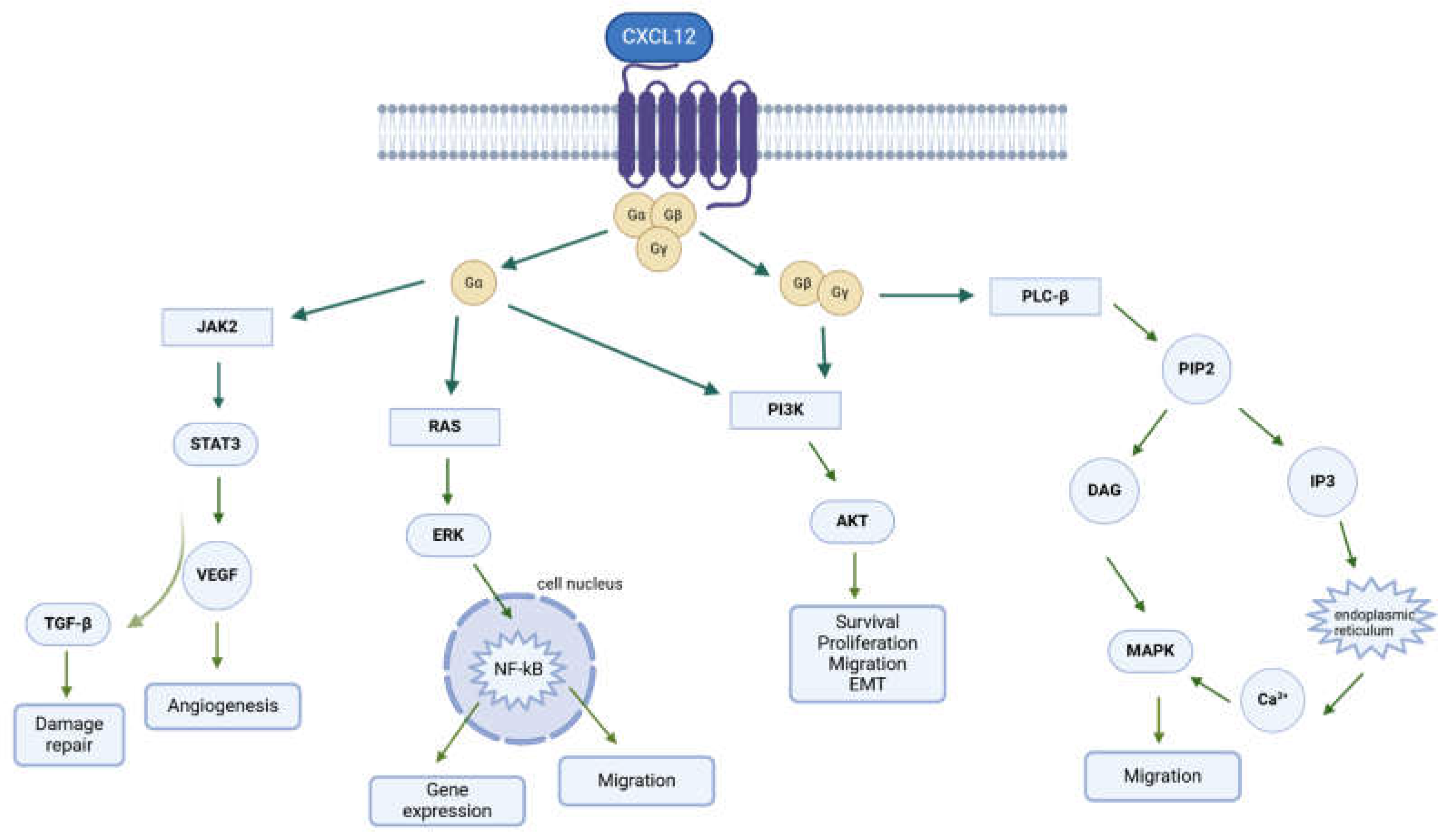
3. CXCR4/CXCL12 Axis in Cancer
- (i)
- The upregulation of vascular endothelial growth factor (VEGF) expression in tumor tissue;
- (ii)
- Reductions in the expression of glycolytic enzyme phosphoglycerate kinase 1 (PGK1), which in turn suppresses the secretion of VEGF;
- (iii)
- The upregulation of several angiogenesis-associated genes in cancer cells;
- (iv)
- Routing the recruitment of endothelial progenitor cells to the vicinity of new vessels.
4. Small Molecules Targeting the CXCR4/CXCL12 Axis
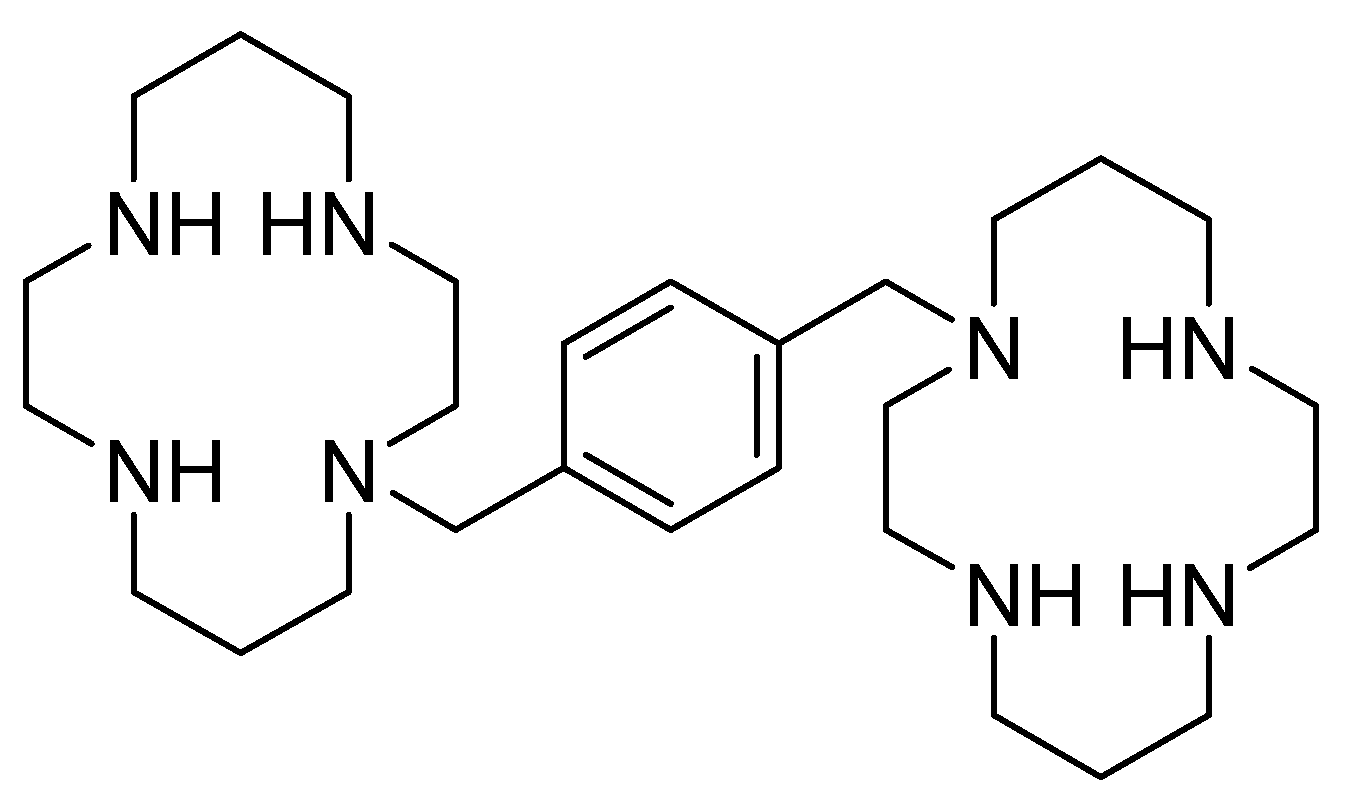
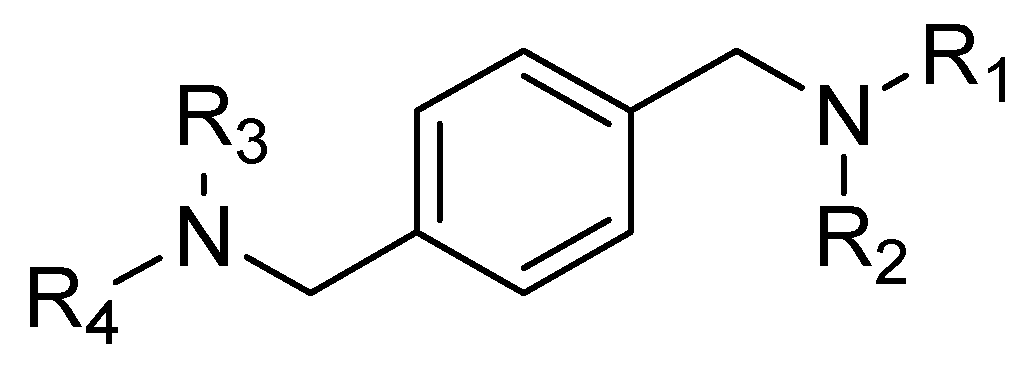
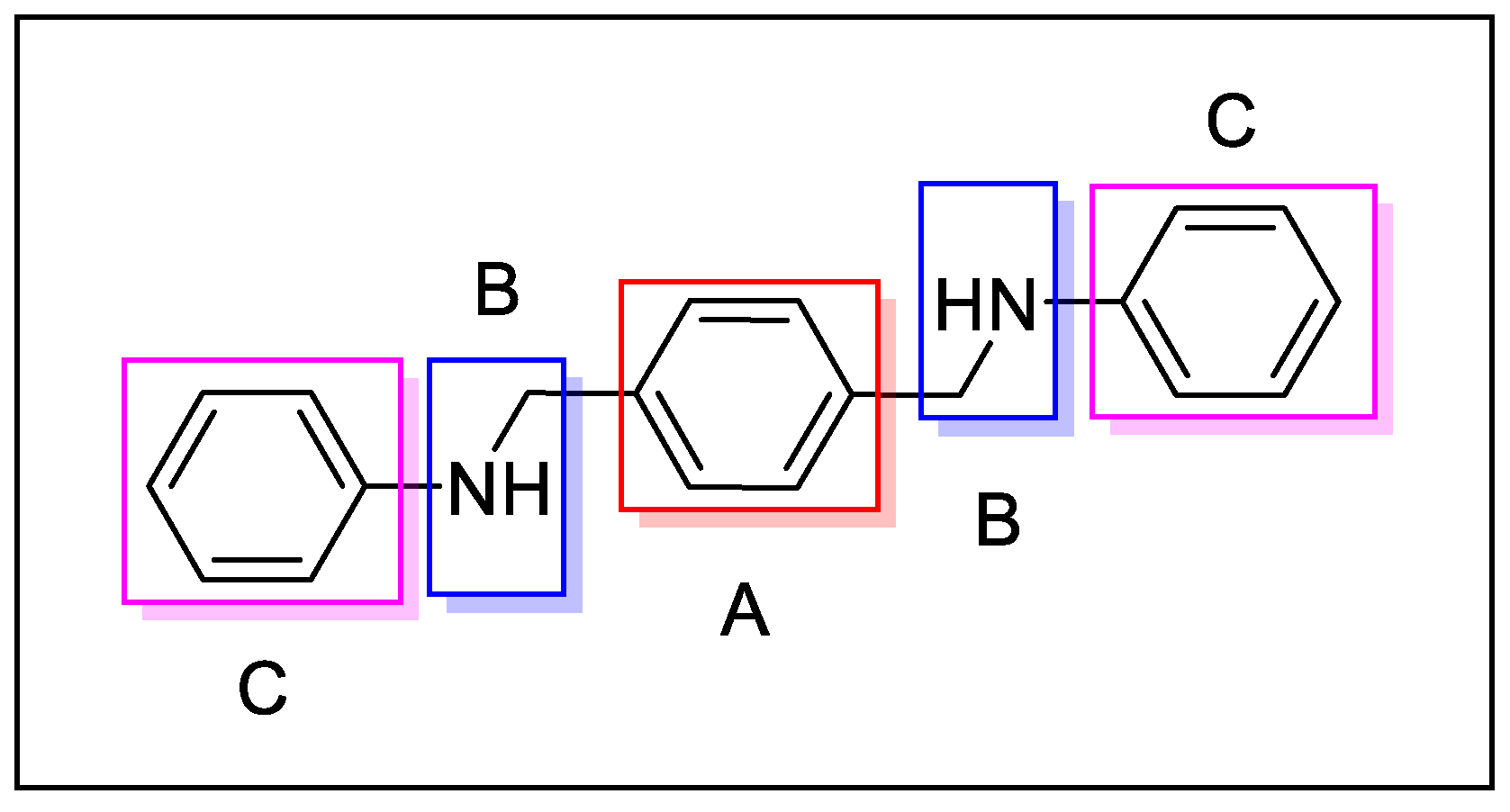
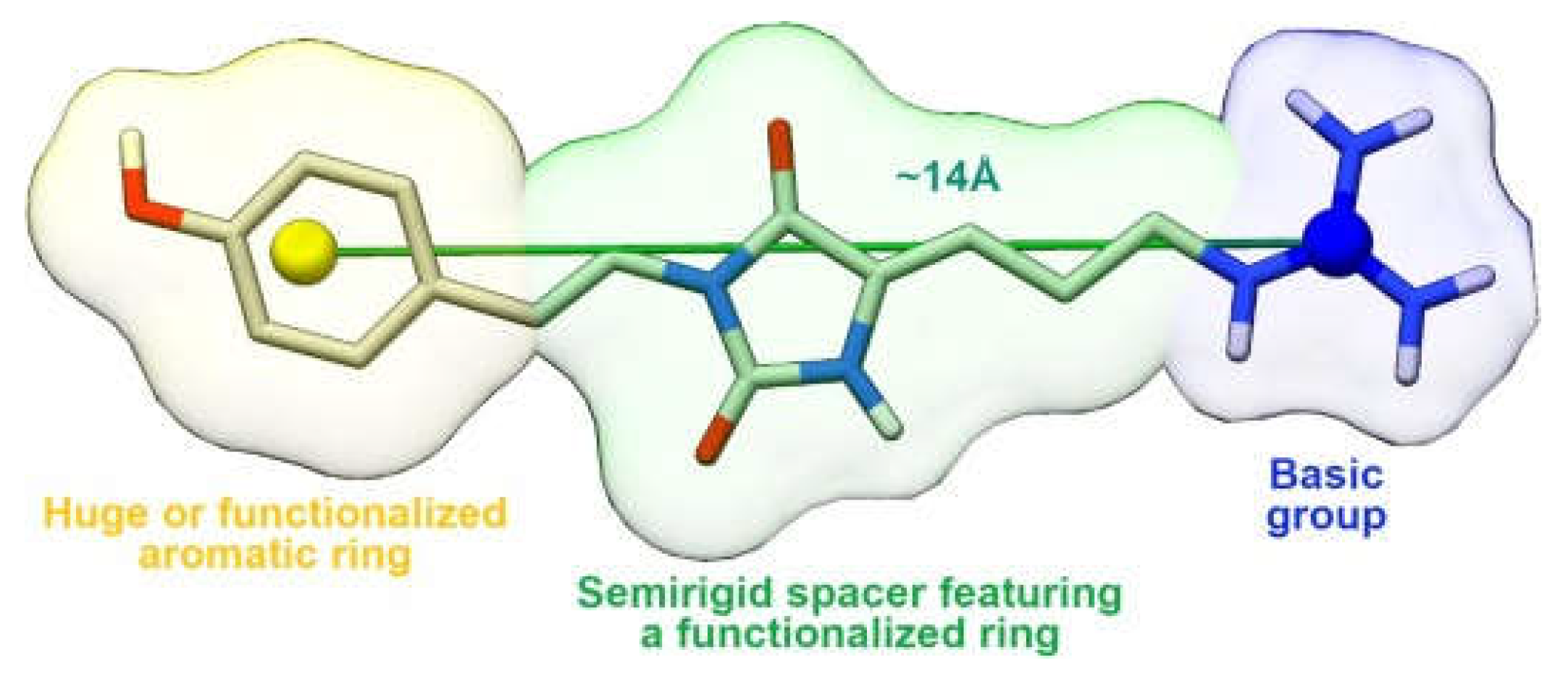
4.1. AMD3100 Derivatives
- The central aromatic ring is critical for high CXCR4 affinity. The substitution of the central phenyl ring (Figure 5, A) with a cyclohexane ring leads to a complete loss of activity. In the same way, the removal of the central ring yields unactive compounds. In addition, the replacement of the central ring with a bicycle or tricycle ring decreases the activity, as well as the ortho-substitution of sections B and C (Figure 5).
- One carbon separation between the central phenyl ring and the nitrogen of the acyclic linker is essential for high potency. The insertion of a methyl group on benzyl carbon or nitrogen leads to a significant decrease of in. Moreover, the elongation of the aliphatic chain between segments A and C leads to a loss of activity.
- Anti-CXCR4 activity is much more sensitive to para substitution on the terminal aromatic rings compared to meta and ortho substitution; in particular, electron-donating groups in the para positions of the terminal phenyl rings (Figure 5, C) increase the activity. On the other hand, the introduction of electron-withdrawing groups leads to a reduction in activity [60].
4.2. Amide and Sulfonamide Derivatives
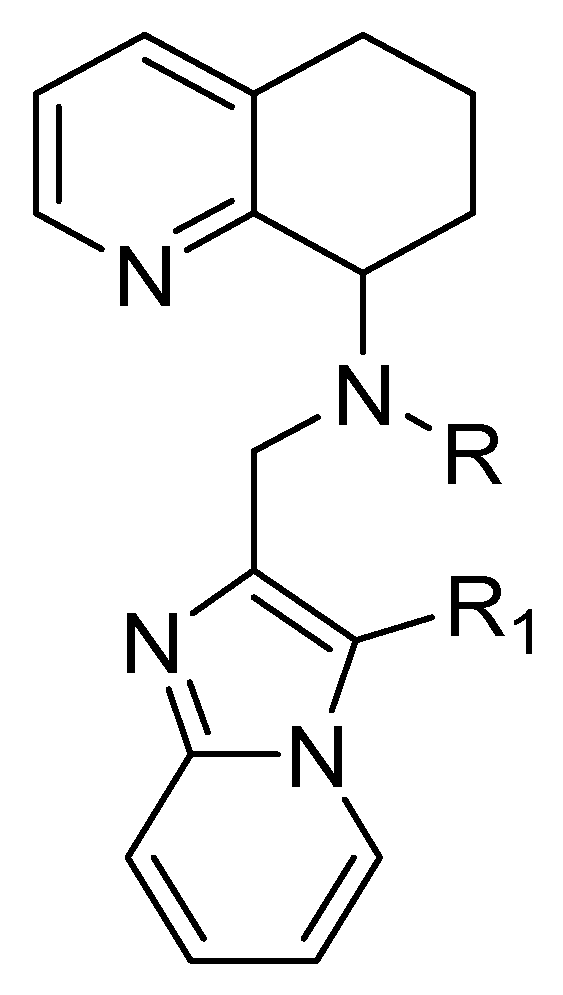
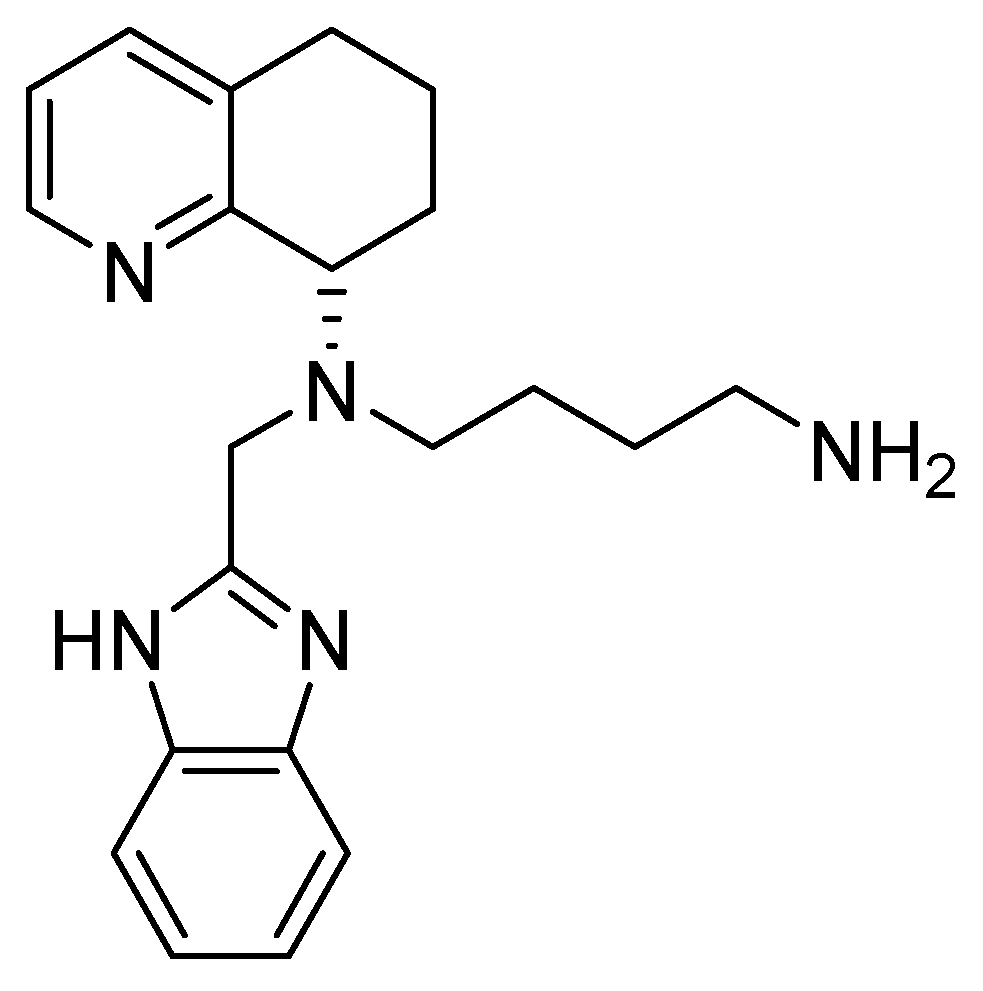
4.3. Tetrahydroquinoline–Benzimidazole-Based Scaffold
- A chiral tetrahydroquinoline (THQ);
- A basic heterocycle;
- A butyl amine chain [68].
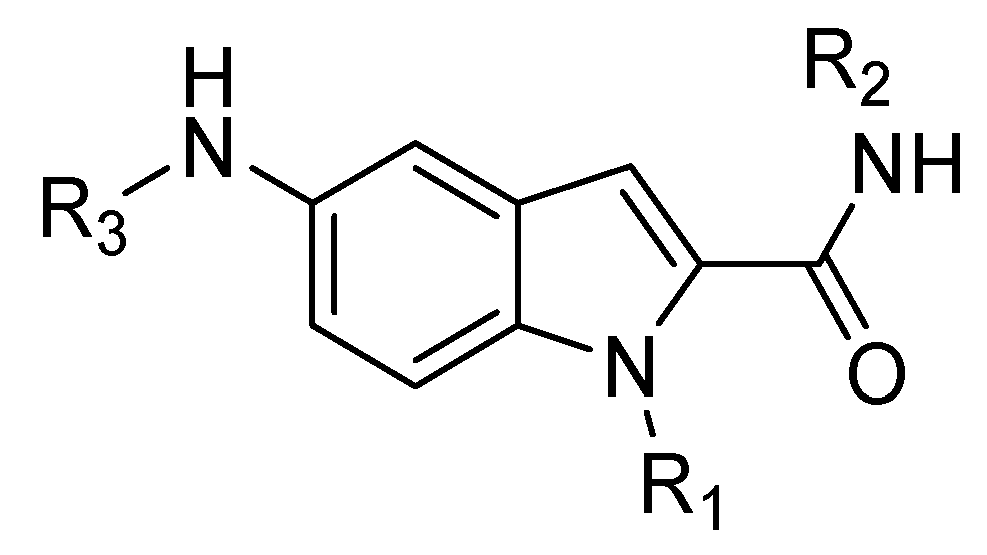
4.4. Indole Scaffold Derivatives
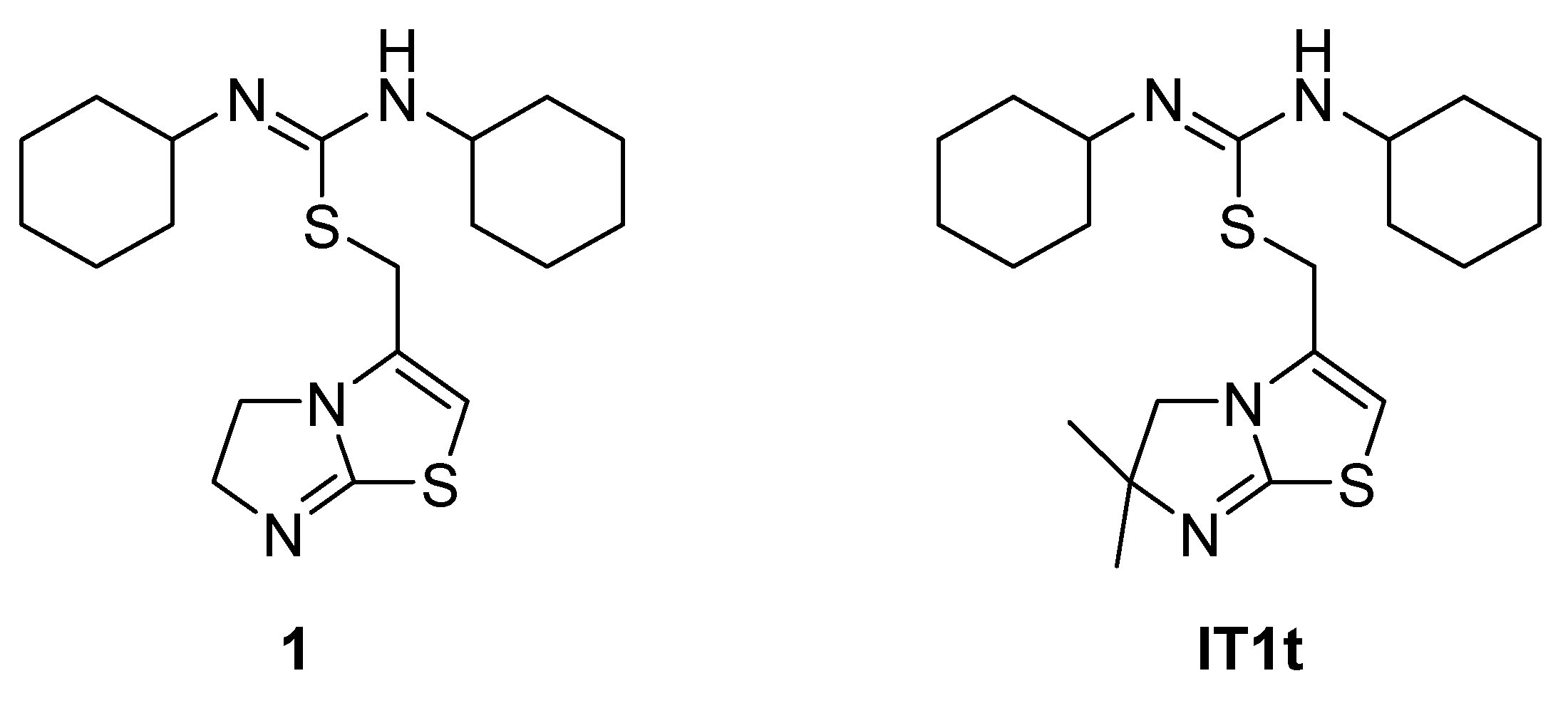
4.5. Isothioureas Derivatives
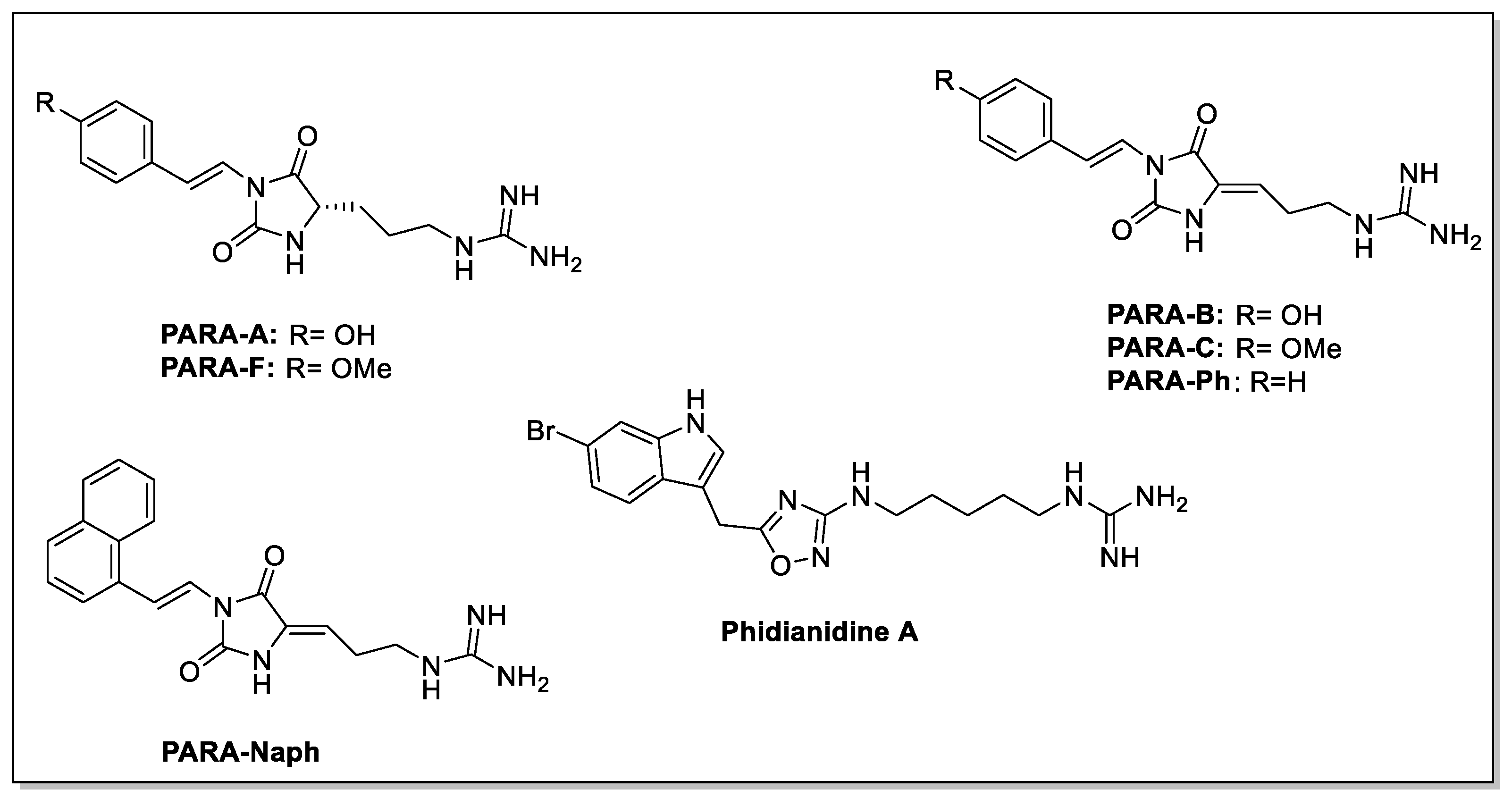
4.6. Guanidine Derivatives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Bouyahya, A.; Mechchate, H.; Oumeslakht, L.; Zeouk, I.; Aboulaghras, S.; Balahbib, A.; Zengin, G.; Kamal, M.A.; Gallo, M.; Montesano, D.; et al. The Role of Epigenetic Modifications in Human Cancers and the Use of Natural Compounds as Epidrugs: Mechanistic Pathways and Pharmacodynamic Actions. Biomolecules 2022, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.; Groom, A.; MacDonald, I. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.; Sokol, C.; Luster, A. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Shi, Y.; Riese, D.J., 2nd; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 574667. [Google Scholar] [CrossRef]
- Teixidó, J.; Martínez-Moreno, M.; Díaz-Martínez, M.; Sevilla-Movilla, S. The good and bad faces of the CXCR4 chemokine receptor. Int. J. Biochem. Cell Biol. 2018, 95, 121–131. [Google Scholar] [CrossRef]
- Manetti, M.; Liakouli, V.; Fatini, C.; Cipriani, P.; Bonino, C.; Vettori, S.; Guiducci, S.; Montecucco, C.; Abbate, R.; Valentini, G.; et al. Association between a stromal cell-derived factor 1 (SDF-1/CXCL12) gene polymorphism and microvascular disease in systemic sclerosis. Ann. Rheum. Dis. 2009, 68, 408–411. [Google Scholar] [CrossRef]
- Yu, L.; Cecil, J.; Peng, S.B.; Schrementi, J.; Kovacevic, S.; Paul, D.; Su, E.W.; Wang, J. Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene 2006, 374, 174–179. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Gupta, S.K. Cloning and relative expression analysis of rat stromal cell derived factor-1 (SDF-1)1: SDF-1 alpha mRNA is selectively induced in rat model of myocardial infarction. Inflammation 2001, 25, 293–300. [Google Scholar] [CrossRef]
- Yang, Y.; Li, J.; Lei, W.; Wang, H.; Ni, Y.; Liu, Y.; Yan, H.; Tian, Y.; Wang, Z.; Yang, Z.; et al. CXCL12-CXCR4/CXCR7 Axis in Cancer: From Mechanisms to Clinical Applications. Int. J. Biol. Sci. 2023, 19, 3341–3359. [Google Scholar] [CrossRef]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [PubMed]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Pawig, L.; Klasen, C.; Weber, C.; Bernhagen, J.; Noels, H. Diversity and Inter-Connections in the CXCR4 Chemokine Receptor/Ligand Family: Molecular Perspectives. Front. Immunol. 2015, 6, 429. [Google Scholar]
- RCSB PDB. Available online: https://www.wwpdb.org/pdb?id=pdb_00003odu (accessed on 10 January 2025).
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef]
- Hadad, I.; Veithen, A.; Springael, J.Y.; Sotiropoulou, P.A.; Mendes Da Costa, A.; Miot, F.; Naeije, R.; De Deken, X.; Entee, K.M. Stroma cell-derived factor-1α signaling enhances calcium transients and beating frequency in rat neonatal cardiomyocytes. PLoS ONE 2013, 8, e56007. [Google Scholar]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar]
- Gutkind, J.S. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J. Biol. Chem. 1998, 273, 1839–1842. [Google Scholar] [CrossRef]
- Vlahakis, S.R.; Villasis-Keever, A.; Gomez, T.; Vanegas, M.; Vlahakis, N.; Paya, C.V. G protein-coupled chemokine receptors induce both survival and apoptotic signaling pathways. J. Immunol. 2002, 169, 5546–5554. [Google Scholar]
- Vila-Coro, A.J.; Rodríguez-Frade, J.M.; Martín De Ana, A.; Moreno-Ortíz, M.C.; Martínez, A.C.; Mellado, M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999, 13, 1699–1710. [Google Scholar]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Cao, Z.; Lis, R.; Siempos, I.I.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Ding, B.S. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat. Cell Biol. 2015, 17, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Mezzapelle, R. The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front. Immunol. 2020, 11, 2109. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef]
- Zanetti, G.; Negro, S.; Megighian, A.; Mattarei, A.; Lista, F.; Fillo, S.; Rigoni, M.; Pirazzini, M.; Montecucco, C. A CXCR4 receptor agonist strongly stimulates axonal regeneration after damage. Ann. Clin. Transl. Neurol. 2019, 6, 2395–2402. [Google Scholar] [CrossRef]
- Li, M.; Chang, C.J.; Lathia, J.D.; Wang, L.; Pacenta, H.L.; Cotleur, A.; Ransohoff, R.M. Chemokine receptor CXCR4 signaling modulates the growth factor-induced cell cycle of self-renewing and multipotent neural progenitor cells. Glia 2011, 59, 108–118. [Google Scholar] [CrossRef]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galun, E.; et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef]
- Yin, X.; Liu, Z.; Zhu, P.; Wang, Y.; Ren, Q.; Chen, H.; Xu, J. CXCL12/CXCR4 promotes proliferation, migration, and invasion of adamantinomatous craniopharyngiomas via PI3K/AKT signal pathway. J. Cell Biochem. 2019, 120, 9724–9736. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, Q.; Bai, X.; Yu, Z.; Sun, M.; Zhao, H.; Mi, X.; Wang, E.; Yao, W.; Jin, F.; et al. Activation of STAT3 is involved in malignancy mediated by CXCL12-CXCR4 signaling in human breast cancer. Oncol. Rep. 2014, 32, 2760–2768. [Google Scholar] [PubMed]
- Klein, S.; Abraham, M.; Bulvik, B.; Dery, E.; Weiss, I.D.; Barashi, N.; Abramovitch, R.; Wald, H.; Harel, Y.; Olam, D.; et al. CXCR4 Promotes Neuroblastoma Growth and Therapeutic Resistance through miR-15a/16-1-Mediated ERK and BCL2/Cyclin D1 Pathways. Cancer Res. 2018, 78, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Kim, J.; Revelo-Penafiel, M.P.; Angel, R.; Dawson, D.W.; Lowy, A.M. The chemokine receptor CXCR4 is expressed in pancreatic intraepithelial neoplasia. Gut 2008, 57, 1555–1560. [Google Scholar] [CrossRef]
- Furusato, B.; Mohamed, A.; Uhlén, M.; Rhim, J.S. CXCR4 and cancer. Pathol. Int. 2010, 60, 497–505. [Google Scholar]
- Liang, Z.; Brooks, J.; Willard, M.; Liang, K.; Yoon, Y.; Kang, S.; Shim, H. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signalling pathway. Biochem. Biophys. Res. Commun. 2007, 359, 716–722. [Google Scholar]
- Chu, C.Y.; Cha, S.T.; Lin, W.C.; Lu, P.H.; Tan, C.T.; Chang, C.C.; Lin, B.R.; Jee, S.H.; Kuo, M.L. Stromal cell-derived factor-1α (SDF-1α/CXCL12)-enhanced angiogenesis of human basal cell carcinoma cells involves ERK1/2-NFkB/interleukin-6 pathway. Carcinogenesis 2009, 30, 205–213. [Google Scholar]
- Novi, S.; Vestuto, V.; Campiglia, P.; Tecce, N.; Bertamino, A.; Tecce, M.F. Anti-Angiogenic Effects of Natural Compounds in Diet-Associated Hepatic Inflammation. Nutrients 2023, 15, 2748. [Google Scholar] [CrossRef]
- Kukreja, P.; Abdel-Mageed, A.B.; Mondal, D.; Liu, K.; Agrawal, K.C. Up-regulation of CXCR4 expression in PC-3 cells by stromal-derived factor-1alpha (CXCL12) increases endothelial adhesion and transendothelial migration: Role of MEK/ERK signaling pathway-dependent NF-kappaB activation. Cancer Res. 2005, 65, 9891–9898. [Google Scholar]
- Hartmann, T.N.; Burger, J.A.; Glodek, A.; Fujii, N.; Burger, M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene 2005, 24, 4462–4471. [Google Scholar]
- Li, X.; Ma, Q.; Xu, Q.; Liu, H.; Lei, J.; Duan, W.; Bhat, K.; Wang, F.; Wu, E.; Wang, Z. SDF-1/CXCR4 signalling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012, 322, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Wang, G.; Huo, H.; Li, Q.; Zhao, Y.; Liu, Y. SDF-1/CXCR4 signalling up-regulates survivin to regulate human sacral chondrosarcoma cell cycle and epithelial-mesenchymal transition via ERK and PI3K/AKT pathway. Med. Oncol. 2015, 32, 377. [Google Scholar] [CrossRef] [PubMed]
- Liao, A.; Shi, R.; Jiang, Y.; Tian, S.; Li, P.; Song, F.; Qu, Y.; Li, J.; Yun, H.; Yang, X. SDF-1/CXCR4 Axis Regulates Cell Cycle Progression and Epithelial-Mesenchymal Transition via Up-regulation of Survivin in Glioblastoma. Mol. Neurobiol. 2017, 54, 7550. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, P.; Chang, Y.; Xu, Q.; Wu, Z.; Ma, Q.; Wang, Z. The SDF-1/CXCR4 axis induces epithelial-mesenchymal transition in hepatocellular carcinoma. Mol. Cell. Biochem. 2014, 392, 77–84. [Google Scholar] [CrossRef]
- Hu, T.H.; Yao, Y.; Yu, S.; Han, L.L.; Wang, W.J.; Guo, H.; Tian, T.; Ruan, Z.P.; Kang, X.M.; Wang, J.; et al. SDF-1/CXCR4 promotes epithelial-mesenchymal transition and progression of colorectal cancer by activation of the Wnt/β-catenin signalling pathway. Cancer Lett. 2014, 354, 417–426. [Google Scholar] [CrossRef]
- Parmo-Cabañas, M.; Molina-Ortiz, I.; Matías-Román, S.; García-Bernal, D.; Carvajal-Vergara, X.; Valle, I.; Pandiella, A.; Arroyo, A.G.; Teixidó, J. Role of metalloproteinases MMP-9 and MT1-MMP in CXCL12-promoted myeloma cell invasion across basement membranes. J. Pathol. 2006, 208, 108–118. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Zuba-Surma, E.; Kucia, M.; Reca, R.; Wojakowski, W.; Ratajczak, J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia 2006, 20, 1915–1924. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget 2014, 5, 11283–11296. [Google Scholar] [CrossRef]
- Rosenkilde, M.M.; Gerlach, L.O.; Jakobsen, J.S.; Skerlj, R.T.; Bridger, G.J.; Schwartz, T.W. Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: Transfer of binding site to the CXCR3 receptor. J. Biol. Chem. 2004, 279, 3033–3041. [Google Scholar] [CrossRef] [PubMed]
- Fricker, S.P.; Anastassov, V.; Cox, J.; Darkes, M.C.; Grujic, O.; Idzan, S.R.; Labrecque, J.; Lau, G.; Mosi, R.M.; Nelson, K.L.; et al. Characterization of the molecular pharmacology of AMD3100: A specific antagonist of the G-protein coupled chemokine receptor, CXCR4. Biochem. Pharmacol. 2006, 72, 588–596. [Google Scholar] [CrossRef]
- Rios, A.; Hsu, S.H.; Blanco, A.; Buryanek, J.; Day, A.L.; McGuire, M.F.; Brown, R.E. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 together with lapatinib, metformin and niacinamide. Oncoscience 2016, 3, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Zhai, B.; Yu, Y.; Kiyotsugu, Y.; Raschle, T.; Etzkorn, M.; Seo, H.C.; Nagiec, M.; Luna, R.E.; Reinherz, E.L.; et al. Quantitative phosphoproteomic analysis reveals system-wide signalling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2182-90. [Google Scholar] [CrossRef]
- Chetram, M.A.; Odero-Marah, V.; Hinton, C.V. Loss of PTEN permits CXCR4-mediated tumorigenesis through ERK1/2 in prostate cancer cells. Mol. Cancer Res. 2011, 9, 90–102. [Google Scholar] [CrossRef]
- Rubin, J.B.; Kung, A.L.; Klein, R.S.; Chan, J.A.; Sun, Y.; Schmidt, K.; Kieran, M.W.; Luster, A.D.; Segal, R.A. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13513–13518. [Google Scholar] [CrossRef]
- Zhan, W.; Liang, Z.; Zhu, A.; Kurtkaya, S.; Shim, H.; Snyder, J.P.; Liotta, D.C. Discovery of small molecule CXCR4 antagonists. J. Med. Chem. 2007, 50, 5655–5664. [Google Scholar] [CrossRef]
- Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Non-peptidic chemokine receptors antagonists as emerging anti-HIV agents. J. Enzyme. Inhib. Med. Chem. 2002, 17, 69–76. [Google Scholar] [CrossRef]
- Liang, Z.; Zhan, W.; Zhu, A.; Yoon, Y.; Lin, S.; Sasaki, M.; Klapproth, J.M.; Yang, H.; Grossniklaus, H.E.; Xu, J.; et al. Development of a unique small molecule modulator of CXCR4. PLoS ONE 2012, 7, e34038. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Wang, B.; Chen, Y.; Liu, H.; Shi, L. Novel CXCR4 Inhibitor CPZ1344 Inhibits the Proliferation, Migration and Angiogenesis of Glioblastoma. Pathol. Oncol. Res. 2020, 26, 2597–2604. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bai, S.; Cheng, Q.; Zeng, Y.; Xu, X.; Guan, G. Naringenin promotes SDF-1/CXCR4 signaling pathway in BMSCs osteogenic differentiation. Folia Histochem. Cytobiol. 2021, 59, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Shi, Q.; Liang, Z.; Yoon, Y.; Han, Y.; Feng, A.; Liu, S.; Oum, Y.; Yun, C.C.; Shim, H. Development of CXCR4 modulators by virtual HTS of a novel amide-sulfamide compound library. Eur. J. Med. Chem. 2017, 126, 464–475. [Google Scholar] [CrossRef]
- Wu, R.; Yu, W.; Yao, C.; Liang, Z.; Yoon, Y.; Xie, Y.; Shim, H.; Bai, R. Amide-sulfamide modulators as effective anti-tumor metastatic agents targeting CXCR4/CXCL12 axis. Eur. J. Med. Chem. 2020, 185, 111823. [Google Scholar] [CrossRef]
- Bai, R.; Liang, Z.; Yoon, Y.; Salgado, E.; Feng, A.; Gurbani, S.; Shim, H. Novel anti-inflammatory agents targeting CXCR4: Design, synthesis, biological evaluation and preliminary pharmacokinetic study. Eur. J. Med. Chem. 2017, 136, 360–371. [Google Scholar] [CrossRef]
- Truax, V.M.; Zhao, H.; Katzman, B.M.; Prosser, A.R.; Alcaraz, A.A.; Saindane, M.T.; Howard, R.B.; Culver, D.; Arrendale, R.F.; Gruddanti, P.R.; et al. Discovery of tetrahydroisoquinoline-based CXCR4 antagonists. ACS Med. Chem. Lett. 2013, 4, 1025–1030. [Google Scholar] [CrossRef]
- O’Boyle, G.; Swidenbank, I.; Marshall, H.; Barker, C.E.; Armstrong, J.; White, S.A.; Fricker, S.P.; Plummer, R.; Wright, M.; Lovat, P.E. Inhibition of CXCR4-CXCL12 chemotaxis in melanoma by AMD11070. Br. J. Cancer 2013, 108, 1634–1640. [Google Scholar] [CrossRef]
- Ueda, S.; Kato, M.; Inuki, S.; Ohno, H.; Evans, B.; Wang, Z.X.; Peiper, S.C.; Izumi, K.; Kodama, E.; Matsuoka, M.; et al. Identification of novel non-peptide CXCR4 antagonists by ligand-based design approach. Bioorg. Med. Chem. Lett. 2008, 18, 4124–4129. [Google Scholar] [CrossRef]
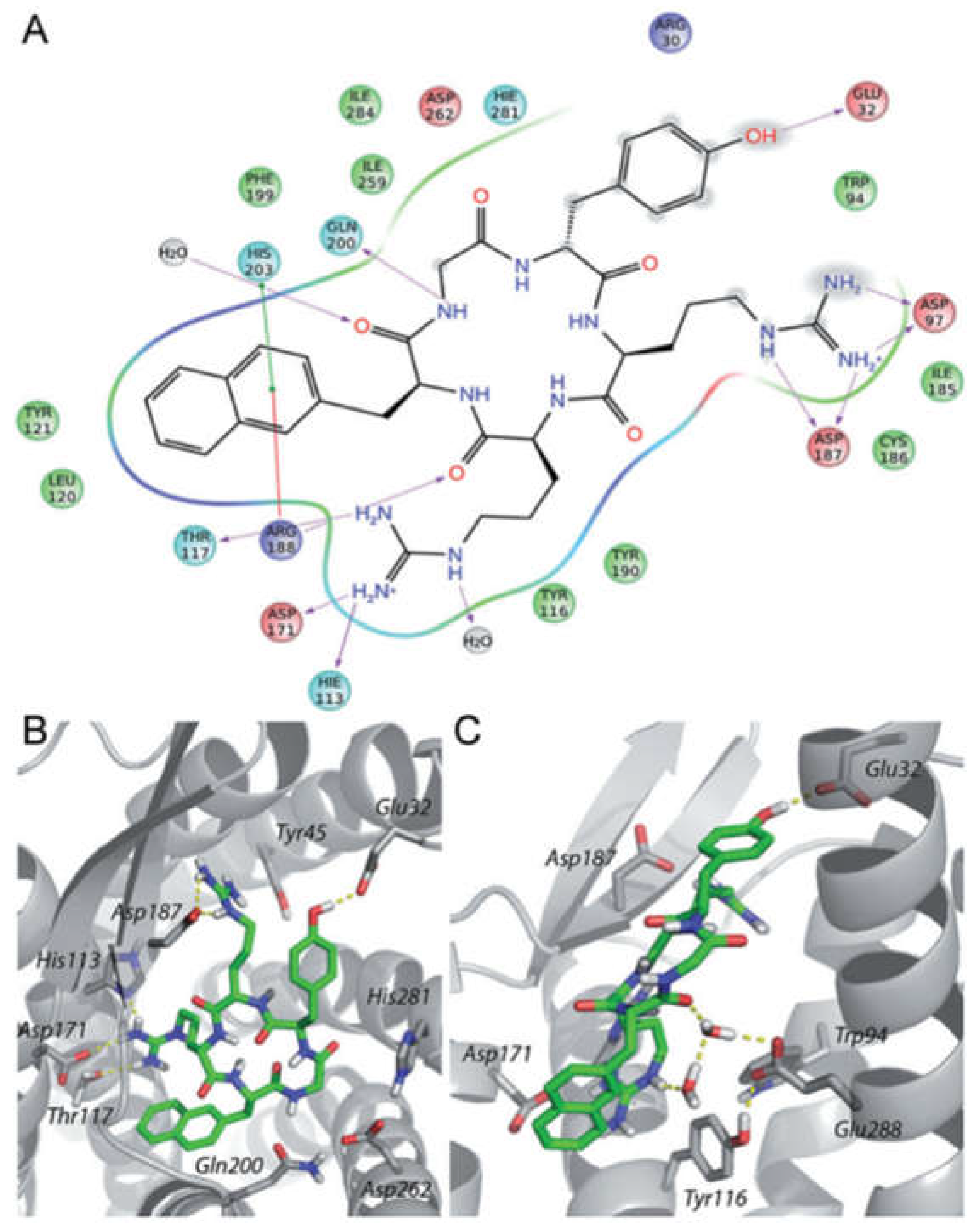
- Thiele, S.; Mungalpara, J.; Steen, A.; Rosenkilde, M.M.; Våbenø, J. Determination of the binding mode for the cyclopentapeptide CXCR4 antagonist FC131 using a dual approach of ligand modifications and receptor mutagenesis. Br. J. Pharmacol. 2014, 171, 5313–5329. [Google Scholar] [CrossRef]
- Ostacolo, C.; Di Sarno, V.; Lauro, G.; Pepe, G.; Musella, S.; Ciaglia, T.; Vestuto, V.; Autore, G.; Bifulco, G.; Marzocco, S.; et al. Identification of an indol-based multi-target kinase inhibitor through phenotype screening and target fishing using inverse virtual screening approach. Eur. J. Med. Chem. 2019, 1, 61–75. [Google Scholar]
- Ciaglia, T.; Vestuto, V.; Di Sarno, V.; Musella, S.; Smaldone, G.; Di Matteo, F.; Napolitano, V.; Miranda, M.R.; Pepe, G.; Basilicata, M.G.; et al. Peptidomimetics as potent dual SARS-CoV-2 cathepsin-L and main protease inhibitors: In silico design, synthesis and pharmacological characterization. Eur. J. Med. Chem. 2024, 266, 116128. [Google Scholar] [CrossRef] [PubMed]
- Thoma, G.; Streiff, M.B.; Kovarik, J.; Glickman, F.; Wagner, T.; Beerli, C.; Zerwes, H.G. Orally bioavailable isothioureas block function of the chemokine receptor CXCR4 in vitro and in vivo. J. Med. Chem. 2008, 51, 7915–7920. [Google Scholar] [PubMed]
- Van Hout, A.; D’huys, T.; Oeyen, M.; Schols, D.; Van Loy, T. Comparison of cell-based assays for the identification and evaluation of competitive CXCR4 inhibitors. PLoS ONE 2017, 12, e0176057. [Google Scholar] [CrossRef]
- Tulotta, C.; Stefanescu, C.; Beletkaia, E.; Bussmann, J.; Tarbashevich, K.; Schmidt, T.; Snaar-Jagalska, B.E. Inhibition of signaling between human CXCR4 and zebrafish ligands by the small molecule IT1t impairs the formation of triple-negative breast cancer early metastases in a zebrafish xenograft model. Dis. Model. Mech. 2016, 9, 141–153. [Google Scholar]
- Vitale, R.M.; Gatti, M.; Carbone, M.; Barbieri, F.; Felicità, V.; Gavagnin, M.; Florio, T.; Amodeo, P. Minimalist hybrid ligand/receptor-based pharmacophore model for CXCR4 applied to a small-library of marine natural products led to the identification of phidianidine a as a new CXCR4 ligand exhibiting antagonist activity. ACS Chem. Biol. 2013, 8, 2762–2770. [Google Scholar]
- Carbone, M.; Li, Y.; Irace, C.; Mollo, E.; Castelluccio, F.; Di Pascale, A.; Cimino, G.; Santamaria, R.; Guo, Y.W.; Gavagnin, M. Structure and cytotoxicity of phidianidines A and B: First finding of 1,2,4-oxadiazole system in a marine natural product. Org. Lett. 2011, 13, 2516–2519. [Google Scholar] [CrossRef]
- Vitale, R.M.; Thellung, S.; Tinto, F.; Solari, A.; Gatti, M.; Nuzzo, G.; Ioannou, E.; Roussis, V.; Ciavatta, M.L.; Manzo, E.; et al. Identification of the hydantoin alkaloids parazoanthines as novel CXCR4 antagonists by computational and in vitro functional characterization. Bioorg. Chem. 2020, 105, 104337. [Google Scholar]


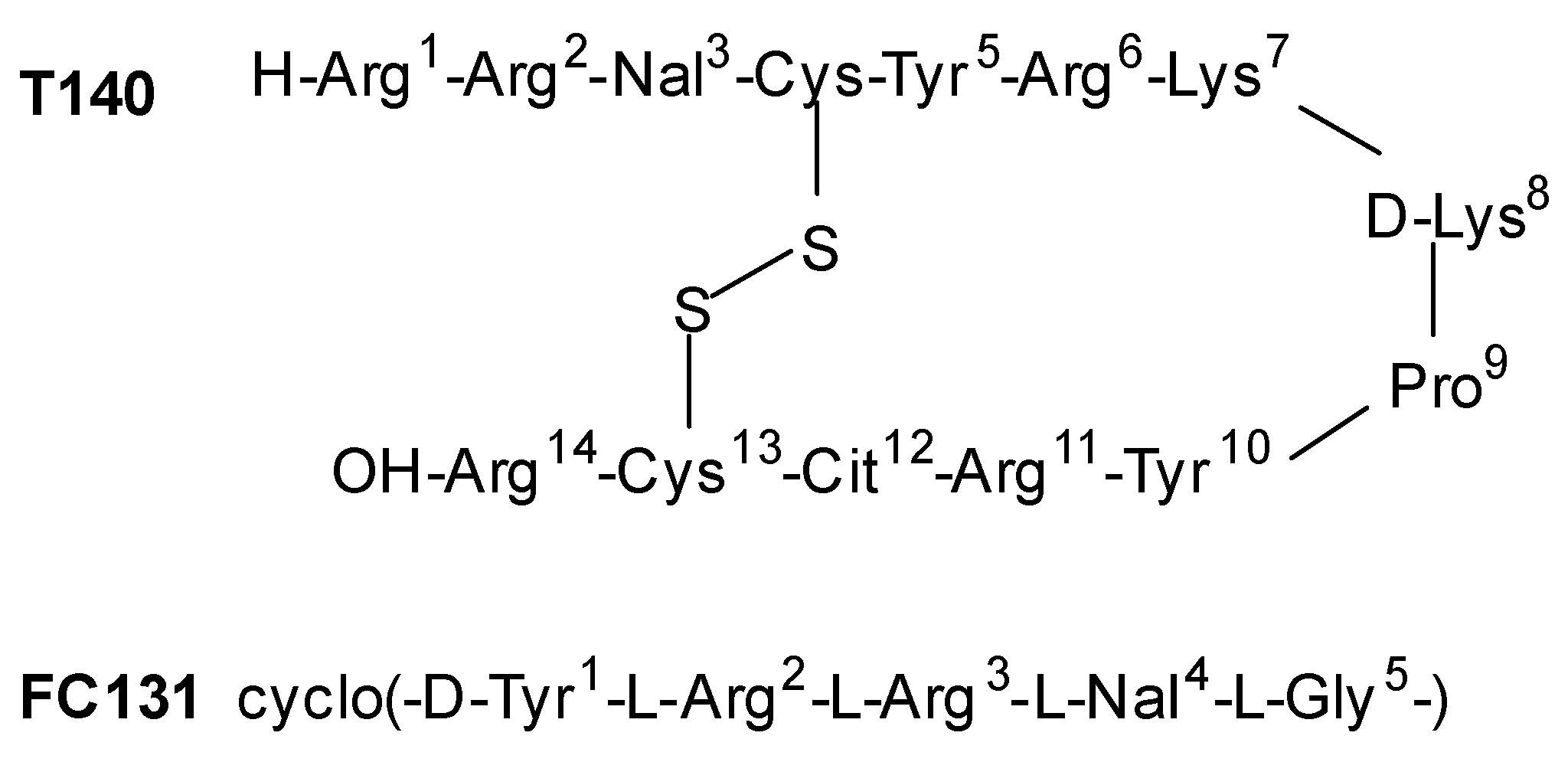




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Mechanism | Application |
|---|---|---|---|
| AMD3100 |  | Mobilization of hematopoietic stem cells. | Currently used to prepare patients for stem cell transplant. |
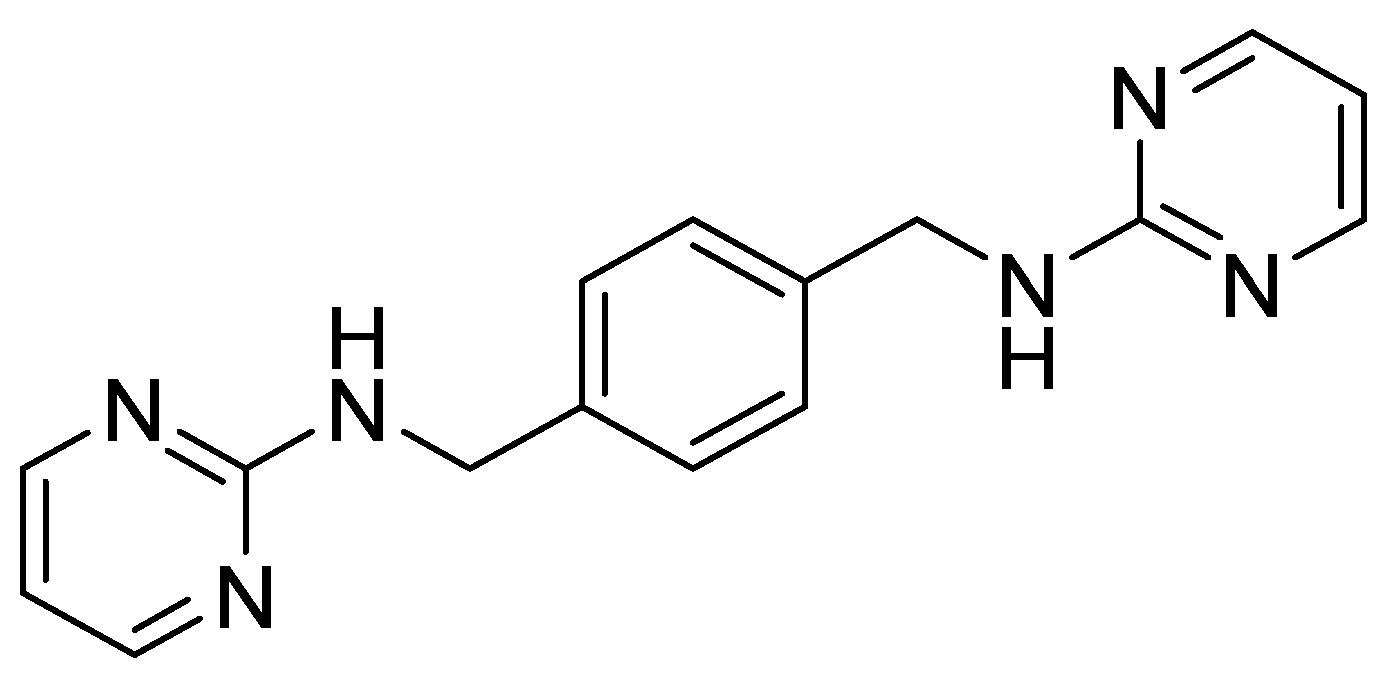
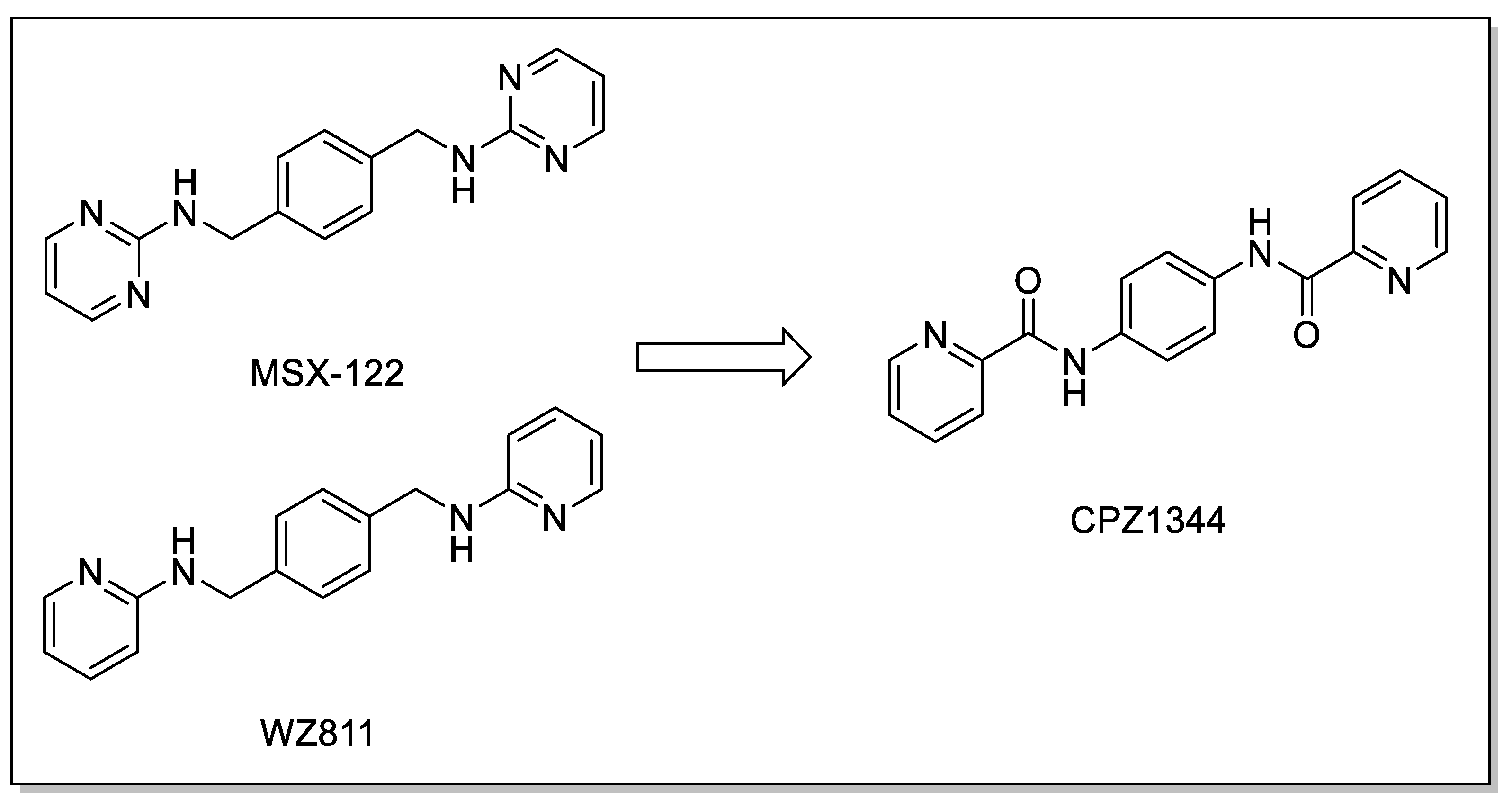
| AMD3100 derivative MSX-122 |  | Inhibition of metastasis and inflammation. | Withdrawn from clinical trials for its toxicity |
| Amide-based scaffold CPZ1344 | 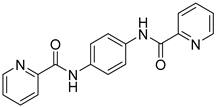 | Anticancer effects on glioma cell lines. | Under investigation. |
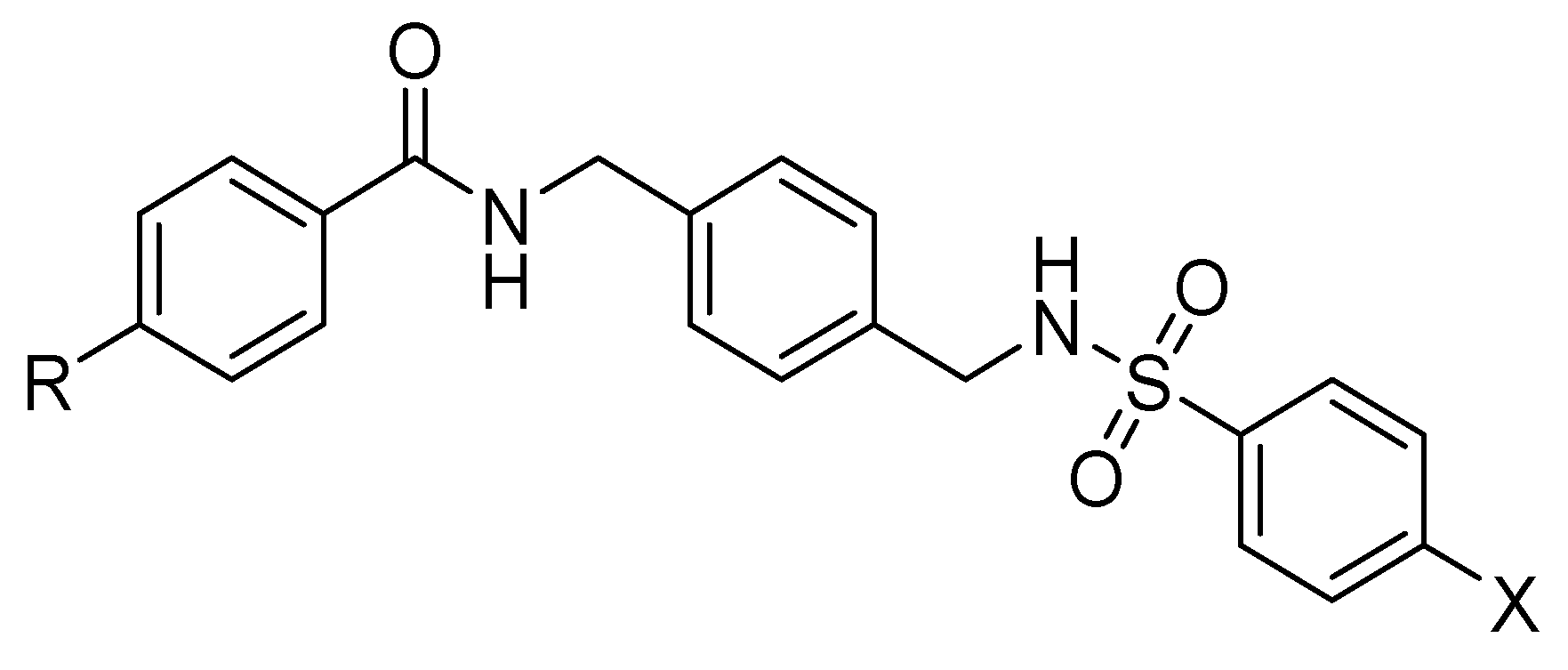
| Amide–sulfonamide derivatives | 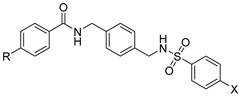 | Block of CXCR4+ cancer cell invasion. | Under investigation. |
| Tetrahydroquinoline–benzimidazole-based scaffold AMD11070 | 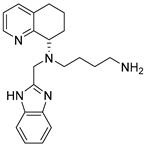 | Inhibition migration of the melanoma cell line A375. | Approved by FDA in WHIM syndrome. |
| Indole derivative 10g | 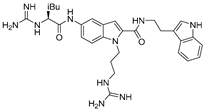 | CXCR4 antagonist. | Under investigation. |
| IT1t |  | Inhibition of CXCR4/CXCL12 interaction. | Under investigation on triple-negative breast cancer. |
| Guanidine-based derivative PHIA | 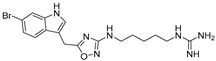 | Inhibition of proliferation and CXCL12-dependent migration in GH4C1 cells of the rat pituitary adenoma cell line. | Under investigation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smaldone, G.; Di Matteo, F.; Castelluccio, R.; Napolitano, V.; Miranda, M.R.; Manfra, M.; Campiglia, P.; Vestuto, V. Targeting the CXCR4/CXCL12 Axis in Cancer Therapy: Analysis of Recent Advances in the Development of Potential Anticancer Agents. Molecules 2025, 30, 1380. https://doi.org/10.3390/molecules30061380
Smaldone G, Di Matteo F, Castelluccio R, Napolitano V, Miranda MR, Manfra M, Campiglia P, Vestuto V. Targeting the CXCR4/CXCL12 Axis in Cancer Therapy: Analysis of Recent Advances in the Development of Potential Anticancer Agents. Molecules. 2025; 30(6):1380. https://doi.org/10.3390/molecules30061380
Chicago/Turabian StyleSmaldone, Gerardina, Francesca Di Matteo, Roberta Castelluccio, Valeria Napolitano, Maria Rosaria Miranda, Michele Manfra, Pietro Campiglia, and Vincenzo Vestuto. 2025. "Targeting the CXCR4/CXCL12 Axis in Cancer Therapy: Analysis of Recent Advances in the Development of Potential Anticancer Agents" Molecules 30, no. 6: 1380. https://doi.org/10.3390/molecules30061380
APA StyleSmaldone, G., Di Matteo, F., Castelluccio, R., Napolitano, V., Miranda, M. R., Manfra, M., Campiglia, P., & Vestuto, V. (2025). Targeting the CXCR4/CXCL12 Axis in Cancer Therapy: Analysis of Recent Advances in the Development of Potential Anticancer Agents. Molecules, 30(6), 1380. https://doi.org/10.3390/molecules30061380