

The Antiproliferative Activity and NO Inhibition of Neo-Clerodane Diterpenoids from Salvia guevarae in RAW 264.7 Macrophages
 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Characterization
2.2. Biological Activity
2.2.1. Antiproliferative Activity
2.2.2. Anti-Inflammatory Activity Due to the Inhibition of NO Production in RAW 264.7 Macrophage Cells
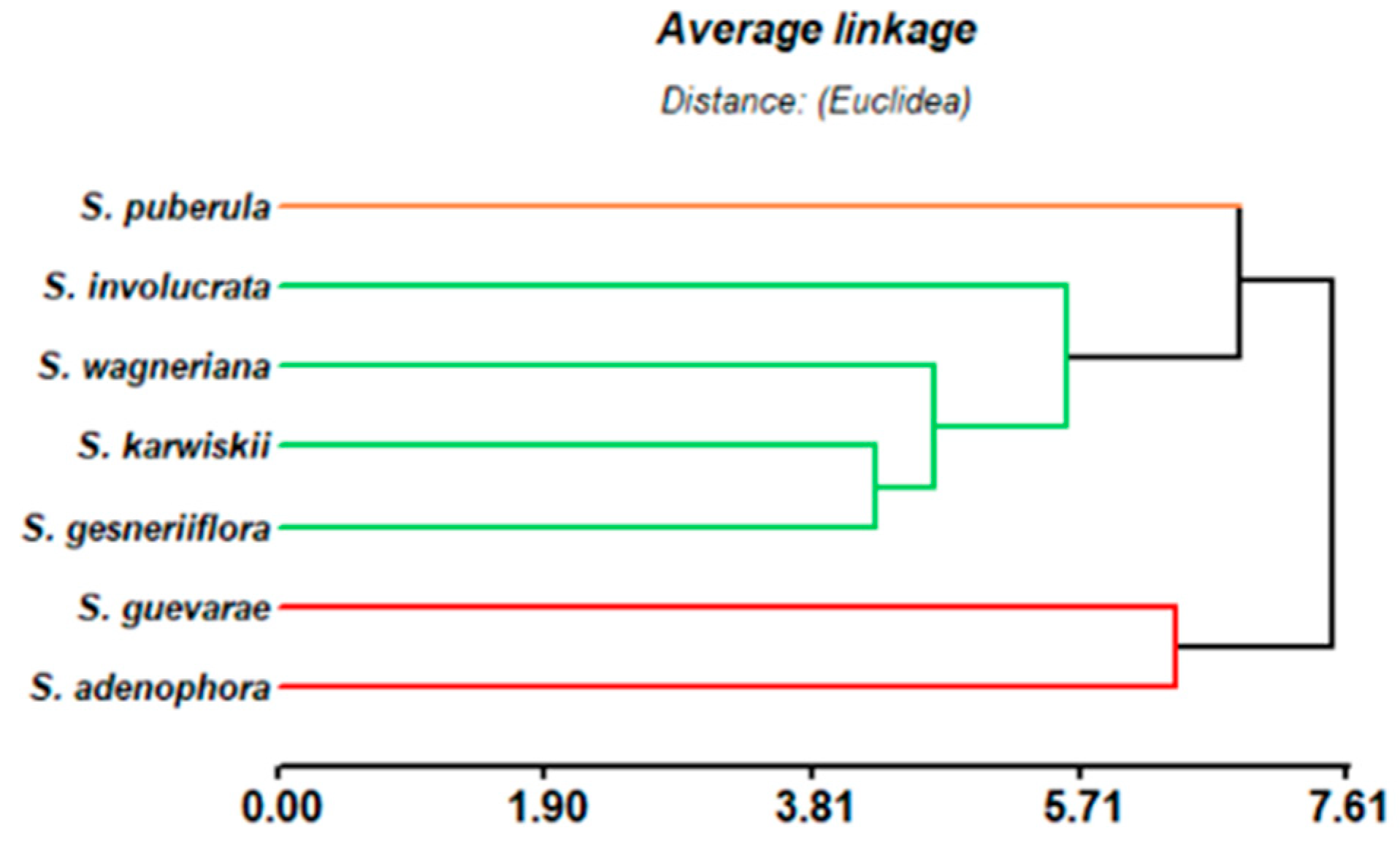
2.3. Taxonomic Considerations
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Materials
3.3. Extraction and Isolation
3.4. Electronic Circular Dichroism Calculations
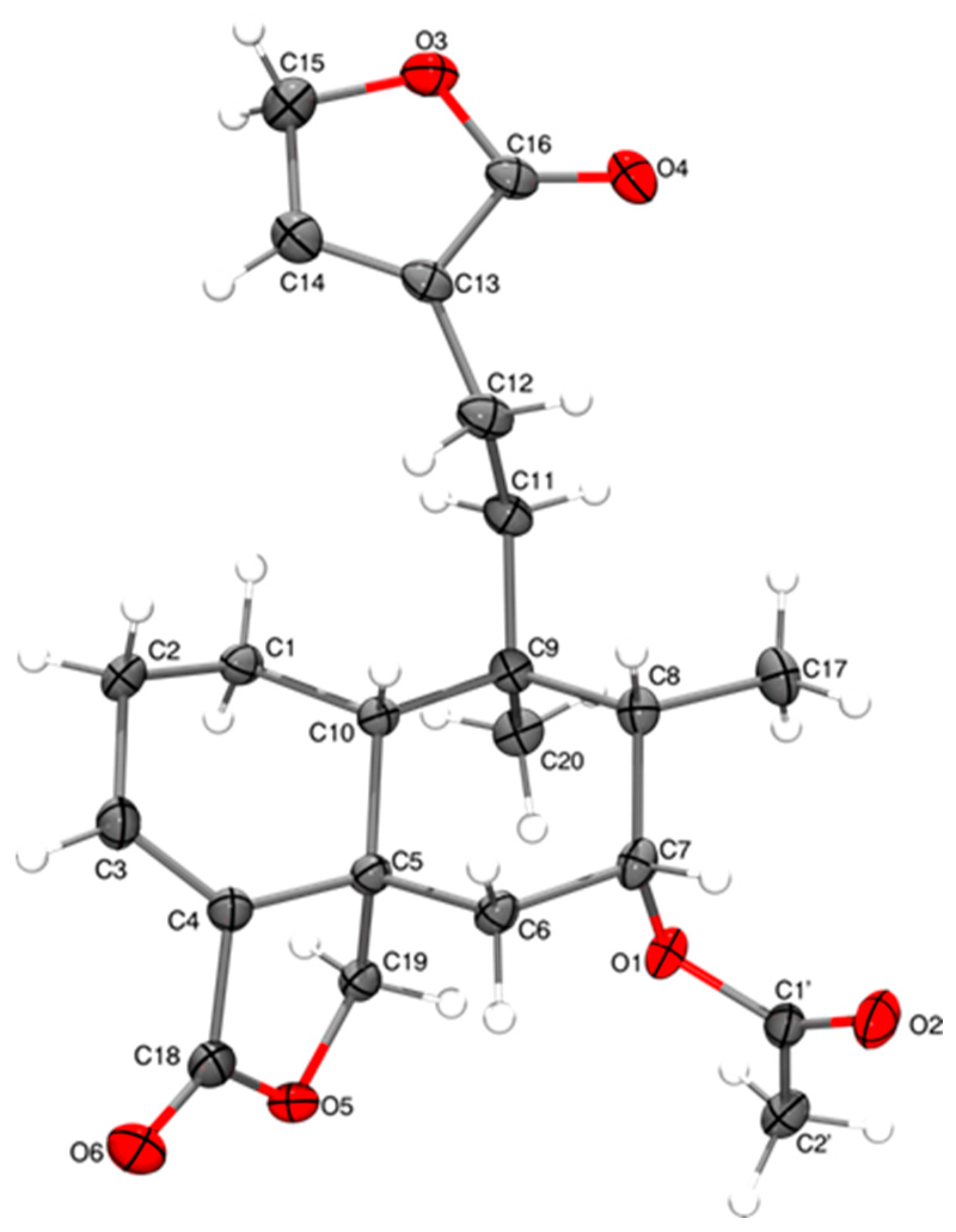
3.5. Single-Crystal X-Ray Diffraction Analysis of 1 and 10
3.6. Chemometric Analysis
3.7. Antiproliferative Activity
3.8. NO Inhibition in RAW 264.7 Macrophages
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lara-Cabrera, S.I.; de la Luz Perez-Garcia, M.; Maya-Lastra, C.A.; Montero-Castro, J.C.; Godden, G.T.; Cibrian-Jaramillo, A.; Fisher, A.E.; Porter, J.M. Phylogenomics of Salvia L. Subgenus Calosphace (Lamiaceae). Front. Plant Sci. 2021, 12, 725900. [Google Scholar] [CrossRef]
- Hu, G.X.; Takano, A.; Drew, B.T.; Liu, E.; De Soltis, D.E.; Soltis, P.S.; Peng, H.; Xiang, C.L. Phylogeny and Staminal Evolution of Salvia (Lamiaceae, Nepetoideae) in East Asia. Ann. Bot. 2018, 122, 649–668. [Google Scholar] [CrossRef] [PubMed]
- González-Gallegos, J.G.; Bedolla-García, B.Y.; Cornejo-Tenorio, G.; Fernández-Alonso, J.L.; Fragoso-Martínez, I.; García-Peña, M.D.R.; Harley, R.M.; Klitgaard, B.; Martínez-Gordillo, M.J.; Wood, J.R.I.; et al. Richness and Distribution of Salvia Subg. Calosphace (Lamiaceae). Int. J. Plant Sci. 2020, 181, 831–856. [Google Scholar] [CrossRef]
- Jenks, A.A.; Kim, S.C. Medicinal Plant Complexes of Salvia Subgenus Calosphace: An Ethnobotanical Study of New World Sages. J. Ethnopharmacol. 2013, 146, 214–224. [Google Scholar] [CrossRef]
- Li, R.; Morris-Natschke, S.L.; Lee, K.H. Clerodane Diterpenes: Sources, Structures, and Biological Activities. Nat. Prod. Rep. 2016, 33, 1166–1226. [Google Scholar] [CrossRef] [PubMed]
- Bustos-Brito, C.; Nieto-Camacho, A.; Hernandez-Ortega, S.; Rivera-Chávez, J.; Quijano, L.; Esquivel, B. Structural Elucidation of Malonylcommunol and 6β-Hydroxy-Trans-Communic Acid, Two Undescribed Diterpenes from Salvia cinnabarina. First Examples of Labdane Diterpenoids from a Mexican Salvia Species. Molecules 2020, 25, 1808. [Google Scholar] [CrossRef]
- Bustos-Brito, C.; Torres-Medicis, J.P.; Bedolla-García, B.Y.; Zamudio, S.; Ramírez-Apan, T.O.; Macías-Rubalcava, M.L.; Quijano, L.; Esquivel, B. Structure, Absolute Configuration, Antiproliferative and Phytotoxic Activities of Icetexane and Abietane Diterpenoids from Salvia carranzae and Chemotaxonomic Implications. Molecules 2024, 29, 1226. [Google Scholar] [CrossRef]
- Bedolla-García, B.Y.; Zamudio, S. Nueva Especie de Salvia (Lamiaceae) del Centro de México. Phytoneuron 2017, 66, 1–12. [Google Scholar]
- Ramamoorthy, T.P. Typifications in Salvia (Lamiaceae). Taxon 1984, 33, 322–324. [Google Scholar]
- Adou, E.; Williams, R.B.; Schilling, J.K.; Malone, S.; Meyer, J.; Wisse, J.H.; Frederik, D.; Koese, D.; Werkhoven, M.C.M.; Snipes, C.E.; et al. Cytotoxic Diterpenoids from Two Lianas from the Suriname Rainforest. Bioorganic Med. Chem. 2005, 13, 6009–6014. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Esquivel, B.; Hernandez, M.; Cardenas, J.; Ramamoorthy, T.P.; Rodríguez-Hahn, L. Further ent-Clerodane Diterpenoids from Salvia melissodora. Phytochemistry 1989, 28, 566. [Google Scholar] [CrossRef]
- Omosa, L.K.; Midiwo, J.O.; Derese, S.; Yenesew, A.; Peter, M.G.; Heydenreich, M. Neo-Clerodane Diterpenoids from the Leaf Exudate of Dodonaea angustifolia. Phytochem. Lett. 2010, 3, 217–220. [Google Scholar] [CrossRef]
- Yürüker, A.; Orjala, J.; Sticher, O.; Rali, T. Triterpenes from Rhus taitensis. Phytochemistry 1988, 48, 863–866. [Google Scholar]
- Ikuta, A.; Itokawa, H. The Triterpenes from Stauntonia hexaphylla Callus Tissues and Their Biosynthetic Significance. J. Nat. Prod. 1989, 52, 37. [Google Scholar] [CrossRef]
- Kolak, U.; Topçu, G.; Birteksöz, S.; Ötük, G.; Ulubelen, A. Terpenoids and Steroids from the Roots of Salvia blepharochlaena. Turk. J. Chem. 2005, 29, 177–186. [Google Scholar]
- Migas, P.; Cisowski, W.; Dembinska-Migas, W. Isoprene Derivatives from the Leaves and Callus Cultures of Vaccinium corymbosum Var. Bluecrop. Acta Pol. Pharmacéutica-Drug Res. 2025, 62, 45–51. [Google Scholar]
- Sun, I.-C.; Wang, H.-K.; Kashiwada, Y.; Shen, J.-K.; Cosentino, L.M.; Chen, C.-H.; Yang, L.-M.; Lee, K.-H. Anti-AIDS Agents. 34. Synthesis and Structure−Activity Relationships of Betulin Derivatives as Anti-HIV Agents. J. Med. Chem. 1998, 41, 4648–4657. [Google Scholar] [CrossRef]
- Symon, A.V.; Veselova, N.N.; Kaplun, A.P.; Vlasenkova, N.K.; Fedorova, G.A.; Lyutik, A.I.; Gerasimova, G.K.; Shvets, V.I. Synthesis and Antitumor Activity of Cyclopropane Derivatives of Betulinic and Betulonic Acids. Russ. J. Bioorganic Chem. 2005, 31, 286–291. [Google Scholar] [CrossRef]
- Melchior Larsen, L.; Kvist Nielsen, J. Identification of 3-O-[2-O-(β-D-Xylopyranosyl)-β-D-Glactopyranosil] Flavonoids in Horseradish Leaves Acting as Feeding Stimulants for a Flea Beetle. Phytochemistry 1982, 21, 1029–1033. [Google Scholar]
- Hofmann, R.W.; Swinny, E.E.; Bloor, S.J.; Markham, K.R.; Ryan, K.G.; Campbell, B.D.; Jordan, B.R.; Fountain, D.W. Responses of Nine Trifolium repens L. Populations to Ultraviolet-B Radiation: Differential Flavonol Glycoside Accumulation and Biomass Production. Ann. Bot. 2000, 86, 527–537. [Google Scholar] [CrossRef]
- Sikorska, M.; Matławska, I. Quercetin and Its Glycosides in the Flowers of Asclepias syriaca L. Acta Pol. Pharm. 2000, 57, 321–324. [Google Scholar]
- Olszewska, M.A.; Kwapisz, A. Metabolite Profiling and Antioxidant Activity of Prunus padus L. Flowers and Leaves. Nat. Prod. Res. 2011, 25, 1115–1131. [Google Scholar] [CrossRef]
- Iannuzzi, A.M.; Giacomelli, C.; De Leo, M.; Pietrobono, D.; Camangi, F.; De Tommasi, N.; Martini, C.; Trincavelli, M.L.; Braca, A. Antioxidant Activity of Compounds Isolated from Elaeagnus umbellata Promotes Human Gingival Fibroblast Well-Being. J. Nat. Prod. 2020, 83, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Markham, K.R.; Webby, R.F.; Vilain, C. 7-O-Methyl-(2R: 3R)-Dihidroquercetin 5-O-B-D-Glucoside and Other Flavonooids from Podocarpus nivalis. Phytochemistry 1984, 23, 2049–2052. [Google Scholar] [CrossRef]
- Yeap Foo, L.; Karchesy, J.J. Polyphenolic Glycosides from Douglas Fir Inner Bark. Phytochemistry 1989, 28, 1237–1240. [Google Scholar] [CrossRef]
- Slimestad, R.; Andersen, Ø.M.; Francis, G.W. Ampelopsin 7-Glucoside and Other Dihydroflavonol 7-Glucosides from Needles of Picea abies. Phytochemistry 1994, 35, 550–552. [Google Scholar] [CrossRef]
- Álvarez-Fernández, M.A.; Hornedo-Ortega, R.; Cerezo, A.B.; Troncoso, A.M.; García-Parrilla, M.C. Determination of Nonanthocyanin Phenolic Compounds Using High-Resolution Mass Spectrometry (UHPLC-Orbitrap-MS/MS) and Impact of Storage Conditions in a Beverage Made from Strawberry by Fermentation. J. Agric. Food Chem. 2016, 64, 1367–1376. [Google Scholar] [CrossRef]
- Xu, J.; Yu, Y.; Shi, R.; Xie, G.; Zhu, Y.; Wu, G.; Qin, M. Organ-Specific Metabolic Shifts of Flavonoids in Scutellaria baicalensis at Different Growth and Development Stages. Molecules 2018, 23, 428. [Google Scholar] [CrossRef]
- Rayyan, S.; Fossen, T.; Solheim Nateland, H.; Andersen, M. Isolation and Identification of Flavonoids, Including Flavone Rotamers, from the Herbal Drug “Crateagi Folium Cum Flore” (Hawthorn). Phytochem. Anal. 2005, 16, 334–341. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, S.; Yan, L.; Tai, J.; Xiao, Q.; Zou, K.; Zhou, Y.; Wu, J. Separation of Flavanone Enantiomers and Flavanone Glucoside Diastereomers from Balanophora involucrata Hook. f. by Capillary Electrophoresis and Reversed-Phase High-Performance Liquid Chromatography on a C18 Column. J. Chromatogr. A 2008, 1185, 117–129. [Google Scholar] [CrossRef]
- da Rosa, E.; do Amaral, Q.D.F.; Duarte, J.A.; Limberger, J.T.; Chaves, P.E.E.; Zuravski, L.; de Oliveira, L.F.S.; Machado, M.M. Antigenotoxic, Antimutagenic and Cytoprotective Potential of Salvia hispanica L. Seed Extract on Human Leukocytes Exposed to Oxidative Damage. J. Funct. Foods 2017, 38, 505–509. [Google Scholar] [CrossRef]
- Kim, M.H.; Jung, K.; Nam, K.H.; Jang, H.J.; Lee, S.W.; Kim, Y.; Park, C.S.; Lee, T.H.; Park, J.H.; Choi, J.H.; et al. Salvia Plebeia R.Br. Inhibits Signal Transduction of IL-6 and Prevents Ovariectomy-Induced Bone Loss by Suppressing Osteoclastogenesis. Arch. Pharm. Res. 2016, 39, 1671–1681. [Google Scholar] [CrossRef]
- Bahadori, M.B.; Asghari, B.; Dinparast, L.; Zengin, G.; Sarikurkcu, C.; Abbas-Mohammadi, M.; Bahadori, S. Salvia nemorosa L.: A Novel Source of Bioactive Agents with Functional Connections. LWT-Food Sci. Technol. 2017, 75, 42–50. [Google Scholar] [CrossRef]
- Petersen, M.; Simmonds, M.S.J. Molecules of Interest Rosmarinic Acid. Phytochemistry 2003, 62, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Sharma, Y.; Velamuri, R.; Fagan, J.; Schaefer, J. Full-Spectrum Analysis of Bioactive Compounds in Rosemary (Rosmarinus officinalis L.) as Influenced by Different Extraction Methods. Molecules 2020, 25, 4599. [Google Scholar] [CrossRef] [PubMed]
- Forzato, C.; Nitti, P. New Diterpenes with Potential Antitumoral Activity Isolated from Plants in the Years 2017–2022. Plants 2022, 11, 2240. [Google Scholar] [CrossRef]
- Prakash, V. Terpenoids as Source of Anti-Inflamatory Compounds. Asian J. Pharm. Clin. Res. 2017, 10, 68. [Google Scholar] [CrossRef]
- Santos, E.P.; Harley, R.M. Notes on Salvia Section Nobiles (Lamiaceae) and Two New Species from Brazil. Kew Bull. 2004, 59, 103–109. [Google Scholar]
- Bisio, A.; Schito, A.M.; Ebrahimi, S.N.; Hamburger, M.; Mele, G.; Piatti, G.; Romussi, G.; Dal Piaz, F.; De Tommasi, N. Antibacterial Compounds from Salvia adenophora Fernald (Lamiaceae). Phytochemistry 2015, 110, 120–132. [Google Scholar] [CrossRef]
- Jimenez, M.; Moreno, E.D.; Díaz, E. Diterpenos de La Salvia gensneraefolia I. Estructuras de La Gensnerofolinas A y B. Rev. Latinoam. Quim. 1978, 10, 166–171. [Google Scholar]
- Esquivel, B.; Flores, E.A. A New neo-Clerodane Diterpenoid from Salvia gensneraeflora (Labiatae). Heterocycles 2001, 55, 505–509. [Google Scholar]
- Bustos-Brito, C.; Pérez-Juanchi, D.; Rivera-Chávez, J.A.; Hernández-Herrera, A.D.; Bedolla-García, B.Y.; Zamudio, S.; Ramírez-Apan, T.O.; Quijano, L.; Esquivel, B. Clerodane and 5,10-seco-Clerodane-Type Diterpenoids from Salvia involucrata. J. Mol. Struct. 2021, 1237, 130367. [Google Scholar] [CrossRef]
- Rodriguez-Hahn, L.; Esquivel, B.; Sánchez, A.-A.; Cárdenas, J.; Tovar, O.G.; Soriano-Garcia, M.; Toscano, A. Puberulin and Isopuberulin, Benzonorcaradiene and Benzocycloheptatriene Diterpenoids of Clerodanic Origin from Salvia puberula. J. Org. Chem. 1988, 53, 3933–3936. [Google Scholar]
- Esquivel, B.; Calderon, J.S.; Sánchez, L. Neo-Clerodane and Languidulane Diterpenoids from Salvia sousae and S. Karwinskii. Phytochemistry 1997, 45, 781–783. [Google Scholar] [CrossRef]
- Bisio, A.; De Tommasi, N.; Romussi, G. Diterpenoids from Salvia wagneriana. Planta Med. 2004, 70, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Del-Ángel, M.; Nieto, A.; Ramírez-Apan, T.; Delgado, G. Anti-Inflammatory Effect of Natural and Semi-Synthetic Phthalides. Eur. J. Pharmacol. 2015, 752, 40–48. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Lmmunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 a | 2 b | |||||
|---|---|---|---|---|---|---|
| Position | δC | Type | δH, Multiplicity (J in Hz) | δC | Type | δH, Multiplicity (J in Hz) |
| 1 | 29.2 | CH2 | 1.94, ddd (12.6, 7.0, 1.4) | 34.4 | CH2 | 2.48, dd (17.8, 13.9) |
| 1.49, td (12.6, 10.1) | 2.32, dd (17.8, 3.6) | |||||
| 2 | 69.1 | CH | 4.13, m | 199.7 | C | |
| 3 | 127.9 | CH | 5.19, brs | 126.5 | CH | 5.72, brs |
| 4 | 146.7 | C | 172.8 | C | ||
| 5 | 45.2 | C | 45.5 | C | ||
| 6 | 75.3 | CH | 3.48, dt (9.5, 5.6) | 73.5 | CH | 3.70, dd (11.1, 4.8) |
| 7 | 38.7 | CH2 | 1.57, m | 37.9 | CH2 | 1.69 ddd(13.0, 4.7, 3.7) |
| 1.62, ddd (13.0, 12.4, 11.1) | ||||||
| 8 | 35.2 | CH | 1.70, m | 34.4 | CH | 1.73, m |
| 9 | 38.8 | C | 38.7 | C | ||
| 10 | 45.4 | CH | 1.35, dd (12.6, 1.4) | 44.9 | CH | 1.88, dd (13.9, 3.6) |
| 11 | 36.8 | CH2 | 1.62, ddd (14.6, 12.7, 5.1) | 35.0 | CH2 | 1.58, m |
| 1.55, m | 1.47, ddd (14.6, 13.0, 4.9) | |||||
| 12 | 19.4 | CH2 | 2.14, m | 18.7 | CH2 | 2.16, m |
| 2.05, m | 2.00, m | |||||
| 13 | 134.3 | C | 134.3 | C | ||
| 14 | 146.2 | CH | 7.41, p (1.7) * | 144.0 | CH | 7.09, p (1.7) * |
| 15 | 71.0 | CH2 | 4.79, q (1.8) | 70.4 | CH2 | 4.76, p (1.7) |
| 16 | 174.7 | C | 174.3 | C | ||
| 17 | 16.0 | CH3 | 0.84, d (6.8) | 15.5 | CH3 | 0.89, d (6.8) |
| 18 | 22.6 | CH3 | 1.84, dd (1.6, 1.7) | 23.0 | CH3 | 2.14, d (1.3) |
| 19 | 15.6 | CH3 | 1.06, s | 13.6 | CH3 | 1.12, s |
| 20 | 18.3 | CH3 | 0.75, s | 17.4 | CH3 | 0.84, s |
| 2 OH | 3.6, d (6.1) | |||||
| 6 OH | 3.39, d (5.6) | |||||
| 1a a | 3 b | |||||
|---|---|---|---|---|---|---|
| Position | δC | Type | δH, Multiplicity (J in Hz) | δC | Type | δH, Multiplicity (J in Hz) |
| 1 | 34.2 | CH2 | 2.53, dd (17.0, 14.3) | 28.9 | CH2 | 2.02, ddt (12.5, 6.8, 1.3) |
| 2.39, dd (17.0, 3.1) | 1.57, td (12.3, 10.3) | |||||
| 2 | 197.8 | C | 69.5 | CH | 4.23, m | |
| 3 | 127.9 | CH | 5.82, brs | 129.6 | CH | 5.56, brs |
| 4 | 167.2 | C | 149.4 | C | ||
| 5 | 55.4 | C | 45.6 | C | ||
| 6 | 210.7 | C | 75.9 | CH | 3.58, m | |
| 7 | 43.7 | CH2 | 2.85, dd (12.7, 12.6) | 37.20 | CH2 | 1.64, m |
| 2.19, dd (12.7, 4.0) | ||||||
| 8 | 40.5 | CH | 2.13, m | 35.9 | CH | 1.76, m |
| 9 | 39.3 | C | 39.2 | C | ||
| 10 | 48.2 | CH | 2.25, dd (14.2, 3.2) | 45.6 | CH | 1.40, brd (12.3) |
| 11 | 35.4 | CH2 | 1.68, ddd (14.2, 12.7, 4.2) | 37.2 | CH2 | 1.68, ddd (13.7, 12.3, 4.9) |
| 1.53, ddd (14.6, 12.7, 4.9) | 1.61, m | |||||
| 12 | 19.1 | CH2 | 2.14, m | 19.7 | CH2 | 2.21, m |
| 2.00, m | 2.18, m | |||||
| 13 | 133.8 | C | 134.7 | C | ||
| 14 | 144.5 | CH | 7.10, p (1.6) * | 147.4 | CH | 7.36, p (1.5) * |
| 15 | 70.3 | CH2 | 4.76, q (1.8) | 72.1 | CH2 | 4.82, q (1.7) |
| 16 | 174.1 | C | 176.8 | C | ||
| 17 | 16.2 | CH3 | 1.01, d (6.6) | 16.0 | CH3 | 0.89, d (6.8) |
| 18 | 21.4 | CH3 | 2.17, d (1.3) | 65.5 | CH2 | 4.08, d (13.5) |
| 4.23, brd (13.5) | ||||||
| 19 | 18.4 | CH3 | 1.51, s | 16.5 | CH3 | 1.13, s |
| 20 | 17.9 | CH3 | 1.06, s | 18.6 | CH3 | 0.79, s |
| 4 | 5 | |||||
|---|---|---|---|---|---|---|
| Position | δC | Type | δH, Multiplicity (J in Hz) | δC | Type | δH, Multiplicity (J in Hz) |
| 1 | 35.3 | CH2 | 2.44, dd (17.8, 13.5) | 34.7 | CH2 | 2.54, dd (17.8, 13.9) |
| 2.37, dd (17.8, 4.3) | 2.37, dd (17.8, 3.4) | |||||
| 2 | 200.1 | C | 200.2 | C | ||
| 3 | 121.4 | CH | 6.10, br s | 125.4 | CH | 5.97, br s |
| 4 | 173.6 | C | 170.6 | C | ||
| 5 | 39.2, | C | 35.0 | C | ||
| 6 | 34.6 | CH2 | 1.57, m | 73.8 | CH | 3.83, dd (8.7, 6.9) |
| 1.45, m | ||||||
| 7 | 26.7 | CH2 | 1.53, m | 37.0 | CH2 | 1.63, m |
| 8 | 36.2 | CH | 1.58, m | 1.55, m | ||
| 9 | 38.9 | C | 34.5 | CH | 1.70, m | |
| 10 | 45.8 | CH | 1.98, dd (13.5, 4.3) | 45.1 | CH | 1.91, dd (13.9, 3.6) |
| 11 | 35.1 | CH2 | 1.54, m | 35.0 | CH2 | 2.51, ddd (13.7, 12.3, 4.9) |
| 1.49, m | ||||||
| 12 | 18.9 | CH2 | 2.21, m | 18.8 | CH2 | 2.13, m |
| 2.04, m | 1.96, m | |||||
| 13 | 134.4 | C | 134.2 | C | ||
| 14 | 143.9 | CH | 7.09, q (1.7) | 144.2 | CH | 7.10, p (1.4) |
| 15 | 70.4 | CH2 | 4.77, m | 70.4 | CH2 | 477, q (1.7) |
| 16 | 173.6 | C | 170.6 | C | ||
| 17 | 15.8 | CH3 | 0.87, d (6.4) | 14.8 | CH3 | 0.91, d (6.4) |
| 18 | 60.6 | CH2 | 4.40, dd (17.6, 1.8) | 65.1 | CH2 | 4.51, d (15.0) |
| 4.35, dd (17.6, 1.8) | 4.37, br d (15.0) | |||||
| 19 | 19.6 | CH3 | 1.20, s | 17.6 | CH3 | 1.22, s |
| 20 | 17.9 | CH3 | 0.85, s | 15.5 | CH3 | 0.85, s |
| 6 | 6 | ||||||
|---|---|---|---|---|---|---|---|
| Position | δC | Type | δH (J in Hz) | Position | δC | Type | δH (J in Hz) |
| 1 | 35.3 | CH2 | 2.65 dd (18.1, 14.1) | 11 | 35 | CH2 | 1.62, m |
| 2.56, dd (18.1, 3.6) | 1.51, ddd (14.7, 12.8, 4.8) | ||||||
| 2 | 199.9 | C | 12 | 18.7 | CH2 | 2.18, tdd (12.9, 4.3, 2.0) | |
| 3 | 141.1 | CH | 6.43, br s | 1.97, m | |||
| 4 | 163.8 | C | 13 | 134.1 | C | ||
| 5 | 45.8 | C | 14 | 144.1 | CH | 7.10, br s | |
| 6 | 72.1 | CH | 3.75, dd (11.1, 4.7) | 15 | 70.4 | CH2 | 4.77, q (2.1) |
| 7 | 35.5 | CH2 | 177, ddd (13.3, 4.8, 3.0) | 16 | 174.2 | C | |
| 1.65, m | 17 | 15.6 | CH3 | 0.84, d (6.6) | |||
| 8 | 34.2 | CH | 1.71, m | 18 | 197.5 | CH | 9.75, s |
| 9 | 38.6 | C | 19 | 15.2 | CH3 | 1.13, s | |
| 10 | 44.8 | CH | 1.95, dd (14.1, 3.5) | 20 | 17.1 | CH3 | 0.78, s |
| 6-OH | 4.63, s | ||||||
| Compounds | IC50 |
|---|---|
| 2 | 33.1 ± 1.3 |
| 7 | 39.8 ± 1.5 |
| Adriamycin | 0.2 ± 0.0 |
| Compound | IC50 (µM) |
|---|---|
| 6 | 26.4 ± 0.4 |
| 7 | 17.3 ± 0.5 |
| 10 | 13.7 ± 2.0 |
| Aminoguanidine | 26.2 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Médicis, J.P.; Bustos-Brito, C.; Quijano, L.; Bedolla-García, B.Y.; Zamudio, S.; Ramírez-Apan, T.; Martínez-Otero, D.; Esquivel, B. The Antiproliferative Activity and NO Inhibition of Neo-Clerodane Diterpenoids from Salvia guevarae in RAW 264.7 Macrophages. Molecules 2025, 30, 1628. https://doi.org/10.3390/molecules30071628
Torres-Médicis JP, Bustos-Brito C, Quijano L, Bedolla-García BY, Zamudio S, Ramírez-Apan T, Martínez-Otero D, Esquivel B. The Antiproliferative Activity and NO Inhibition of Neo-Clerodane Diterpenoids from Salvia guevarae in RAW 264.7 Macrophages. Molecules. 2025; 30(7):1628. https://doi.org/10.3390/molecules30071628
Chicago/Turabian StyleTorres-Médicis, Juan Pablo, Celia Bustos-Brito, Leovigildo Quijano, Brenda Y. Bedolla-García, Sergio Zamudio, Teresa Ramírez-Apan, Diego Martínez-Otero, and Baldomero Esquivel. 2025. "The Antiproliferative Activity and NO Inhibition of Neo-Clerodane Diterpenoids from Salvia guevarae in RAW 264.7 Macrophages" Molecules 30, no. 7: 1628. https://doi.org/10.3390/molecules30071628
APA StyleTorres-Médicis, J. P., Bustos-Brito, C., Quijano, L., Bedolla-García, B. Y., Zamudio, S., Ramírez-Apan, T., Martínez-Otero, D., & Esquivel, B. (2025). The Antiproliferative Activity and NO Inhibition of Neo-Clerodane Diterpenoids from Salvia guevarae in RAW 264.7 Macrophages. Molecules, 30(7), 1628. https://doi.org/10.3390/molecules30071628