
Current Analytical Strategies for mRNA-Based Therapeutics

 , , ,
, , ,
Abstract
:1. Introduction
2. Electrophoretic Approaches
3. Chromatographic Approaches
3.1. Anion-Exchange Chromatography
3.2. Size Exclusion Chromatography
3.3. Slalom Chromatography
3.4. Ion-Pair Reversed Phase Liquid Chromatography
4. Mass Spectrometric Approaches
4.1. Advancements in Oligonucleotide Mapping

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA/ Lenght (nt) | Analysis Level | Analytical Method Conditions | Target QA | General Comments | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| LC Mode | LC Column | Mobile Phase | MS/Software for Data Analysis | |||||
| SARS-CoV-2 mRNA vaccine | Oligo mapping | IP-RP | ACQUITY Premier Oligonucleotide C18 (130 Å, 1.7 µm, 2.1 × 150 mm) | MPA: 0.1% TEA, 1% HFIP, H2O MPB: 0.1% TEA, 1% HFIP, MeOH | Orbitrap Exploris 240 MS/Biopharma Finder | Sequence confirmation, mRNA chemistry modifications, 5′-capping efficiency, and 3′ poly(A)-tail length | 100% maximum sequence coverage, optimized MS/MS HCD fragmentation to sequence isomers. | [79] |
| - Epo (859 nts) - FLuc (~2000 nts) - α-catenin (900 nts) | Oligo mapping | IP-RP | ACQUITY UPLC Oligonucleotide BEH C18 (130 Å, 1.7 μm, 2.1 × 100 mm) | MPA: is 1% HFIP, 0.1% DIPEA, H2O MPB: 0.075% HFIP, 0.0375% DIPEA, 65% ACN, 35% H2O | 6550 Q-TOF MS/Agilent MassHunter data | Sequence confirmation and sequence impurities (SNPs) | Use of multiple endonucleases (RNase T1, colicin E5, and mazF), enabling complementary sequence coverage. | [80] |
| eGFP, SARS-CoV-2 spike protein mRNA, Fluc (5moU) mRNA | Oligo mapping | IP-RP | DNAPac RP (150 mm × 2.1 mm) | MPA: 0.2% TEA and 50 mM HFIP MPB: 0.2% TEA, 50 mM HFIP, and 20% v/v ACN | Orbitrap Exploris 240 MS/Biopharma Finder | Sequence confirmation and impurity analysis | Partial RNase digestions using RNase T1 immobilized on magnetic particles >80% sequence of coverage. | [81] |
| p- and s-mimBNT162b2 mRNAs | Oligo mapping | IP-RP | RP Develosil C30-UG column (3 μm particle size, 150 μm × 240 mm) | MPA: 10 mM TEAA, pH 7, in (90:10, v/v) H20, MeOH MPB: (60:40, v/v) 10 mM TEAA, ACN | Q Exactive Orbitrap MS | 5′ capping efficiency and 3′ poly(A)-tail length | Isotope-dilution LC–MS method to sequence 200–4300 nts mRNAs Direct Nanoflow LC-MS/MS. | [82] |
| eGFP, eGFP (5moU), Fluc, Nickase Cas9 (5moU), and Cas9 | Oligo mapping | HILIC | 1D: immobilized RNase T1, and RNase A cartridges (2.1 × 33 mm) 2D: Premier BEH amide column (130 Å, 1.7 μm, 2.1 × 50 mm) | MPA: 10 mM NH4OAc in H2O/acetonitrile (3:97, v/v) MPB: 25 mM NH4OAc in H2O/ACN (60:40, v/v) | Q-Exactive Orbitrap MS/Biopharma Finder version 5.0 software | Sequence confirmation, mRNA chemistry modifications | Online nucleotide mapping of mRNAs using 2D LC-MS system with an 1D immobilized RNase cartridge, followed by HILIC-MS analysis. | [83] |
| eGFP, eGFP (5moU), Epo (5moU), and Cas9 (5moU) | Oligo mapping | IP-RP | Oligonucleotide BEH C18 (130 Å, 1.7 μm, 2.1 mm × 150 mm) | MPA: 1% HFIP, 0.1% DIPEA in H2O MPB: 0.075% HFIP, 0.0375% DIPEA in 65% ACN, 35% H2O | Thermo Q-Exactive plus MS/Byonic software—Byologic “Digested Oligonucleotides” | Sequence confirmation, mRNA chemistry modifications | Flow through-based strategy to achieve the limited RNase T1 digestion, which boosted the overall sequence coverage (over 93%). Automated digestion workflow using the AssayMAP platform. | [84] |
| - EPO (859 nts) - Fluc (~2000 nts) | Poly(A) tail analysis | IP-RP | ACQUITY Premier Oligonucleotide C18 (130 Å, 1.7 μm, 50 × 2.1 mm) | MPA: 8 mM DIPEA, 40 mM HFIP in H2O, pH 8.8 MPB: 4 mM DIPEA, 4 mM HFIP in 75% EtOH | Waters BioAccord LC MS system, MassLynx 4.2 | Poly(A) tail length and heterogeneity | Evaluation of IP RP LC method for poly(A) tail length measurement, demonstrating robustness and suitability for routine mRNA quality control. | [10] |
| - Largest mRNA: eGFP (758 nts) | Mid-level oligonucleotide | No separation, nano ESI | In-house pulled quartz emitters | 150 mM NH4OAc | Bruker solariX XR FTICR MS equipped with a 7 T magnet, Bruker DataAnalysis, RiboDynamics, SeqRead | Mid-level mRNA sequencing with CID | Proof-of-concept study for mid-level mRNA sequencing using RNA-cleaving deoxyribozymes. Decreased complexity for sequencing larger fragments and increased assignment confidence. | [87] |
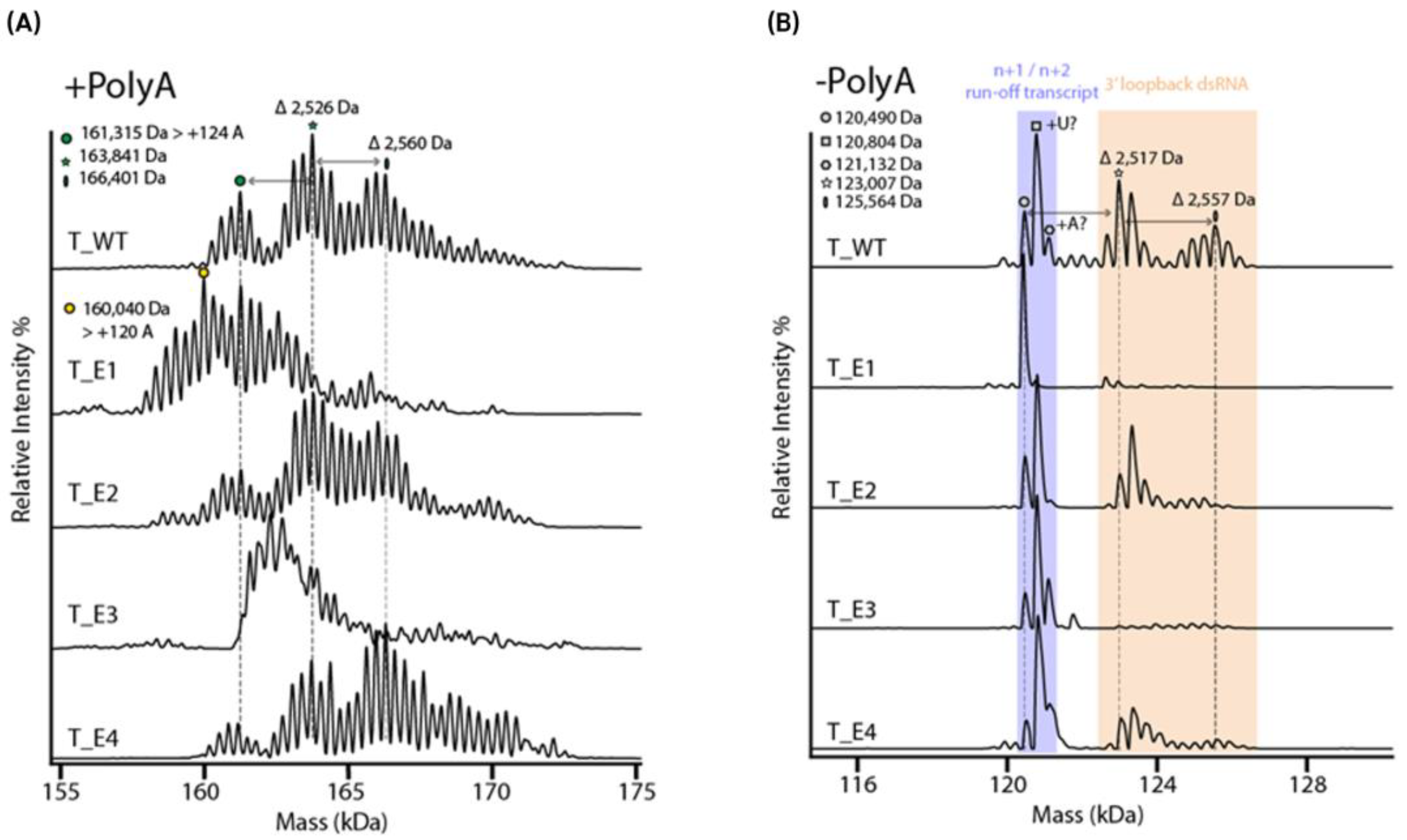
| - Poly(A) tail: T1 cleavage, 100 nts in-house produced -mRNA +/- Poly(A) tail (783/683 nts) - IP-RP fractionated mRNA (580 nts) | Intact mass analysis, Poly(A) tail analysis | No separation, static ESI | Borosilicate emitter | Buffer: 200 mM NH4OAc | Orbitrap Q Exactive UHMR MS, FreeStyle (v 1.8) for isotopically resolved Poly(A) tail, UniDec (v 6.0.3.) for deconvolution of intact mRNA data | Integrity, PolyA heterogeneity | Isotopic resolution of Poly(A) tails up to 100 nts under native MS conditions in positive mode. Intact mass analysis mRNA, revealed new insights on integrity from IP RP LC fractions. | [30] |
| - mRNAs with/without Poly(A) tail using 5 different T7 polymerases | Intact mass analysis, Poly(A) tail analysis | No separation, static ESI | Borosilicate emitter | Buffer: 200 mM NH4OAc + 100 mM TEAA for charge reduction | Orbitrap Q Exactive UHMR MS, UniDec (v 6.0.3.) for deconvolution of data | Integrity, poly(A) tail, 5′loopback ds dsRNA | Functionality testing correlation Native MS analysis can detect short 3′loopback dsRNA impurities. | [51] |
4.2. Advancements in Intact MS-Based Approaches
5. Advanced mRNA Sequencing Approaches
5.1. Sanger Sequencing
5.2. Illumina Sequencing for Short-Read Precision
5.3. PacBio Sequencing for Long-Read Accuracy
5.4. Oxford Nanopore Sequencing for Versatile Long-Read Solutions
6. Functionality Testing
7. Overview of Analytical Technologies for mRNA Characterization
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parhiz, H.; Atochina-Vasserman, E.N.; Weissman, D. mRNA-based therapeutics: Looking beyond COVID-19 vaccines. Lancet 2024, 403, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Chancellor, D.; Barrett, D.; Nguyen-Jatkoe, L.; Millington, S.; Eckhardt, F. The state of cell and gene therapy in 2023. Mol. Ther. 2023, 31, 3376–3388. [Google Scholar] [CrossRef] [PubMed]
- Boros, L.G.; Kyriakopoulos, A.M.; Brogna, C.; Piscopo, M.; McCullough, P.A.; Seneff, S. Long-lasting, biochemically modified mRNA, and its frameshifted recombinant spike proteins in human tissues and circulation after COVID-19 vaccination. Pharmacol. Res. Perspect. 2024, 12, e1218. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Wei, J.; Hui, A.-M. The Delivery of mRNA Vaccines for Therapeutics. Life 2022, 12, 1254. [Google Scholar] [CrossRef]
- Taguchi, Y.-H. RNA m6A Modification and microRNAs. In MicroRNA; Elsevier: Amsterdam, The Netherlands, 2022; pp. 169–180. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Tinari, S. The EMA COVID-19 data leak, and what it tells us about mRNA instability. BMJ 2021, 372, n627. [Google Scholar] [CrossRef]
- Morreel, K.; T’kindt, R.; Debyser, G.; Jonckheere, S.; Sandra, P. Diving into the Structural Details of In Vitro Transcribed mRNA Using Liquid Chromatography–Mass Spectrometry-Based Oligonucleotide Profiling. LCGC Eur. 2022, 35, 220–236. [Google Scholar] [CrossRef]
- Beverly, M.; Dell, A.; Parmar, P.; Houghton, L. Label-free analysis of mRNA capping efficiency using RNase H probes and LC-MS. Anal. Bioanal. Chem. 2016, 408, 5021–5030. [Google Scholar] [CrossRef]
- Gilar, M.; Doneanu, C.; Gaye, M.M. Liquid Chromatography Methods for Analysis of mRNA Poly(A) Tail Length and Heterogeneity. Anal. Chem. 2023, 95, 14308–14316. [Google Scholar] [CrossRef]
- Brouze, A.; Krawczyk, P.S.; Dziembowski, A.; Mroczek, S. Measuring the tail: Methods for poly(A) tail profiling. Wiley Interdiscip. Rev. RNA 2022, 14, e1737. [Google Scholar] [CrossRef]
- Siew, Y.Y.; Zhang, W. Removing immunogenic double-stranded RNA impurities post in vitro transcription synthesis for mRNA therapeutics production: A review of chromatography strategies. J. Chromatogr. A 2024, 1740, 465576. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Hur, S. Immunogenicity of In Vitro-Transcribed RNA. Accounts Chem. Res. 2021, 54, 4012–4023. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Dousis, A.; Ravichandran, K.; Hobert, E.M.; Moore, M.J.; Rabideau, A.E. An engineered T7 RNA polymerase that produces mRNA free of immunostimulatory byproducts. Nat. Biotechnol. 2022, 41, 560–568. [Google Scholar] [CrossRef]
- de Faria, I.J.; Imler, J.-L.; Marques, J.T. Protocol for the analysis of double-stranded RNAs in virus-infected insect cells using anti-dsRNA antibodies. STAR Protoc. 2023, 4, 102033. [Google Scholar] [CrossRef]
- Aramburu, J.; Navas-Castillo, J.; Moreno, P.; Cambra, M. Detection of double-stranded RNA by ELISA and dot immunobinding assay using an antiserum to synthetic polynucleotides. J. Virol. Methods 1991, 33, 1–11. [Google Scholar] [CrossRef]
- Whitley, J.; Zwolinski, C.; Denis, C.; Maughan, M.; Hayles, L.; Clarke, D.; Snare, M.; Liao, H.; Chiou, S.; Marmura, T.; et al. Development of mRNA manufacturing for vaccines and therapeutics: mRNA platform requirements and development of a scalable production process to support early phase clinical trials. Transl. Res. J. Lab. Clin. Med. 2021, 242, 38–55. [Google Scholar] [CrossRef]
- Cross, D.R.; Miller, B.J.; James, S.J. A simplified HPLC method for simultaneously quantifying ribonucleotides and deoxyribonucleotides in cell extracts or frozen tissues. Cell Prolif. 1993, 26, 327–336. [Google Scholar] [CrossRef]
- Blenke, E.O.; Örnskov, E.; Schöneich, C.; Nilsson, G.A.; Volkin, D.B.; Mastrobattista, E.; Almarsson, Ö.; Crommelin, D.J. The Storage and In-Use Stability of mRNA Vaccines and Therapeutics: Not A Cold Case. J. Pharm. Sci. 2022, 112, 386–403. [Google Scholar] [CrossRef]
- Mulroney, T.E.; Pöyry, T.; Yam-Puc, J.C.; Rust, M.; Harvey, R.F.; Kalmar, L.; Horner, E.; Booth, L.; Ferreira, A.P.; Stoneley, M.; et al. N1-methylpseudouridylation of mRNA causes +1 ribosomal frameshifting. Nature 2023, 625, 189–194. [Google Scholar] [CrossRef]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA) quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.B.; Barrett, D.M.; Karikó, K. The Emerging Role of In Vitro-Transcribed mRNA in Adoptive T Cell Immunotherapy. Mol. Ther. 2019, 27, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Zhang, K.; Utegg, R.; Stephens, E.; Salem, S.; Welch, H.; Grobe, S.; Schlereth, J.; Kuhn, A.N.; Ryczek, J.; et al. Characterization of BNT162b2 mRNA to Evaluate Risk of Off-Target Antigen Translation. J. Pharm. Sci. 2023, 112, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, D.E.; Swank, A.; Thomas, R.S.; Strynar, M.; Lee, M.-Y.; Carmichael, P.L.; Simmons, S.O. mRNA transfection retrofits cell-based assays with xenobiotic metabolism. J. Pharmacol. Toxicol. Methods 2018, 92, 77–94. [Google Scholar] [CrossRef]
- Bloom, K.; Ely, A.; Arbuthnot, P. A T7 Endonuclease I Assay to Detect Talen-Mediated Targeted Mutation of HBV cccDNA. In Hepatitis B Virus; Guo, H., Cuconati, A., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1540, pp. 85–95. [Google Scholar] [CrossRef]
- Lu, T.; Klein, L.J.; Ha, S.; Rustandi, R.R. High-Resolution capillary electrophoresis separation of large RNA under non-aqueous conditions. J. Chromatogr. A 2020, 1618, 460875. [Google Scholar] [CrossRef]
- Raffaele, J.; Loughney, J.W.; Rustandi, R.R. Development of a microchip capillary electrophoresis method for determination of the purity and integrity of mRNA in lipid nanoparticle vaccines. Electrophoresis 2021, 43, 1101–1106. [Google Scholar] [CrossRef]
- Di Grandi, D.; Dayeh, D.M.; Kaur, K.; Chen, Y.; Henderson, S.; Moon, Y.; Bhowmick, A.; Ihnat, P.M.; Fu, Y.; Muthusamy, K.; et al. A single-nucleotide resolution capillary gel electrophoresis workflow for poly(A) tail characterization in the development of mRNA therapeutics and vaccines. J. Pharm. Biomed. Anal. 2023, 236, 115692. [Google Scholar] [CrossRef]
- Camperi, J.; Lippold, S.; Ayalew, L.; Roper, B.; Shao, S.; Freund, E.; Nissenbaum, A.; Galan, C.; Cao, Q.; Yang, F.; et al. Comprehensive Impurity Profiling of mRNA: Evaluating Current Technologies and Advanced Analytical Techniques. Anal. Chem. 2024, 96, 3886–3897. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Li, Z.; Zhu, X.; Liu, C.; Zhang, D.; Dou, X. Polyethylene Oxide (PEO) and Polyethylene Glycol (PEG) Polymer Sieving Matrix for RNA Capillary Electrophoresis. PLoS ONE 2015, 10, e0123406. [Google Scholar] [CrossRef]
- De Scheerder, L.; Sparén, A.; Nilsson, G.A.; Norrby, P.-O.; Örnskov, E. Designing flexible low-viscous sieving media for capillary electrophoresis analysis of ribonucleic acids. J. Chromatogr. A 2018, 1562, 108–114. [Google Scholar] [CrossRef]
- Skeidsvoll, J.; Ueland, P.M. Analysis of RNA by capillary electrophoresis. Electrophoresis 1996, 17, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Todorov, T.I.; de Carmejane, O.; Walter, N.G.; Morris, M.D. Capillary electrophoresis of RNA in dilute and semidilute polymer solutions. Electrophoresis 2001, 22, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, C.; Zhang, D.; Luo, S.; Yamaguchi, Y. Capillary electrophoresis of RNA in hydroxyethylcellulose polymer with various molecular weights. J. Chromatogr. B 2016, 1011, 114–120. [Google Scholar] [CrossRef]
- Mantri, P.; Juneja, B.; Henderson, S.; Koufos, E.; Moon, Y.; Dayeh, D.M.; Di Grandi, D.; Fu, Y.; Muthusamy, K.; Ihnat, P.M.; et al. Comparison of capillary electrophoresis-based methods for the analytical characterization of purity and stability of in vitro transcribed mRNA. J. Pharm. Biomed. Anal. 2024, 249, 116352. [Google Scholar] [CrossRef]
- Rollo, D.; Kulkarni, A.; Yu, K.; Fabris, D. Investigating the Merits of Microfluidic Capillary Zone Electrophoresis–Mass Spectrometry (CZE-MS) in the Bottom-Up Characterization of Larger RNAs. J. Am. Soc. Mass Spectrom. 2024, 35, 561–574. [Google Scholar] [CrossRef]
- Abe, B.T.; Wesselhoeft, R.A.; Chen, R.; Anderson, D.G.; Chang, H.Y. Circular RNA migration in agarose gel electrophoresis. Mol. Cell 2022, 82, 1768–1777.e3. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Zhou, S.; Dain, L.; Mei, L.; Zhu, G. Circular RNA: An emerging frontier in RNA therapeutic targets, RNA therapeutics, and mRNA vaccines. J. Control. Release 2022, 348, 84–94. [Google Scholar] [CrossRef]
- Han, F.; Huynh, B.H.; Ma, Y.; Lin, B. High-Efficiency DNA Separation by Capillary Electrophoresis in a Polymer Solution with Ultralow Viscosity. Anal. Chem. 1999, 71, 2385–2389. [Google Scholar] [CrossRef]
- Webb, A.L.; Welbourne, E.N.; Evans, C.A.; Dickman, M.J. Characterisation and analysis of mRNA critical quality attributes using liquid chromatography based methods. J. Chromatogr. A 2025, 1745, 465724. [Google Scholar] [CrossRef]
- Fekete, S.; Doneanu, C.; Addepalli, B.; Gaye, M.; Nguyen, J.; Alden, B.; Birdsall, R.; Han, D.; Isaac, G.; Lauber, M. Challenges and emerging trends in liquid chromatography-based analyses of mRNA pharmaceuticals. J. Pharm. Biomed. Anal. 2022, 224, 115174. [Google Scholar] [CrossRef]
- Guimaraes, G.J.; Kim, J.; Bartlett, M.G. Characterization of mRNA therapeutics. Mass Spectrom. Rev. 2023, 43, 1066–1090. [Google Scholar] [CrossRef] [PubMed]
- Welbourne, E.N.; Loveday, K.A.; Nair, A.; Nourafkan, E.; Qu, J.; Cook, K.; Kis, Z.; Dickman, M.J. Anion exchange HPLC monitoring of mRNA in vitro transcription reactions to support mRNA manufacturing process development. Front. Mol. Biosci. 2024, 11, 1250833. [Google Scholar] [CrossRef] [PubMed]
- Kanavarioti, A. HPLC methods for purity evaluation of man-made single-stranded RNAs. Sci. Rep. 2019, 9, 1019. [Google Scholar] [CrossRef]
- Fekete, S.; Yang, H.; Wyndham, K.; Lauber, M. Salt gradient and ion-pair mediated anion exchange of intact messenger ribonucleic acids. J. Chromatogr. Open 2022, 2, 100031. [Google Scholar] [CrossRef]
- Goyon, A.; Tang, S.; Fekete, S.; Nguyen, D.; Hofmann, K.; Wang, S.; Shatz-Binder, W.; Fernandez, K.I.; Hecht, E.S.; Lauber, M.; et al. Separation of Plasmid DNA Topological Forms, Messenger RNA, and Lipid Nanoparticle Aggregates Using an Ultrawide Pore Size Exclusion Chromatography Column. Anal. Chem. 2023, 95, 15017–15024. [Google Scholar] [CrossRef]
- D’atri, V.; Lardeux, H.; Goyon, A.; Imiołek, M.; Fekete, S.; Lauber, M.; Zhang, K.; Guillarme, D. Optimizing Messenger RNA Analysis Using Ultra-Wide Pore Size Exclusion Chromatography Columns. Int. J. Mol. Sci. 2024, 25, 6254. [Google Scholar] [CrossRef]
- De Vos, J.; Morreel, K.; Alvarez, P.; Vanluchene, H.; Vankeirsbilck, R.; Sandra, P.; Sandra, K. Evaluation of size-exclusion chromatography, multi-angle light scattering detection and mass photometry for the characterization of mRNA. J. Chromatogr. A 2024, 1719, 464756. [Google Scholar] [CrossRef]
- Currie, J.; Dahlberg, J.R.; Eriksson, J.; Schweikart, F.; Nilsson Gunilla, A.; Örnskov, E. Stability Indicating Ion-Pair Reversed-Phase Liquid Chromatography Method for Modified mRNA. ChemRxiv 2024. [Google Scholar] [CrossRef]
- Camperi, J.; Roper, B.; Freund, E.; Leylek, R.; Nissenbaum, A.; Galan, C.; Caothien, R.; Hu, Z.; Ko, P.; Lee, A.; et al. Exploring the Impact of In Vitro-Transcribed mRNA Impurities on Cellular Responses. Anal. Chem. 2024, 96, 17789–17799. [Google Scholar] [CrossRef]
- Cheng, F.; Li, J.; Hu, C.; Bai, Y.; Liu, J.; Liu, D.; He, Q.; Jin, Q.; Mao, Q.; Liang, Z.; et al. Study on the Characterization and Degradation Pattern of Circular RNA Vaccines Using an HPLC Method. Chemosensors 2024, 12, 120. [Google Scholar] [CrossRef]
- Gagnon, P. Purification of Nucleic Acids: A Handbook for Purification of Plasmid DNA and mRNA for Gene Therapy and Vaccines; BIA Separations: Ajdovščina, Slovenia, 2020. [Google Scholar]
- Fekete, S.; Yang, H.; Koza, S.M.; Wyndham, K.; Lauber, M.A. Methods for the Anion Exchange Chromatographic Analysis of mRNAs; Waters Application Note; Waters Corporation: Milford, MA, USA, 2021. [Google Scholar]
- Barth, H.G.; Jackson, C.; Boyes, B.E. Size Exclusion Chromatography. Anal. Chem. 1994, 66, 595–620. [Google Scholar] [CrossRef]
- Camperi, J.; Moshref, M.; Dai, L.; Lee, H.Y. Physicochemical and Functional Characterization of Differential CRISPR-Cas9 Ribonucleoprotein Complexes. Anal. Chem. 2021, 94, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Brophy, P.; Shion, H.; Doneanu, C.H.; Yang, B.D.; Botamanenko, D.; Abbatiello, S.; Jarrell, A.; Koza, S.; Yu, Y.; Giles, K.; et al. Characterization of Intact mRNA Using IP-RP-TOF-MS, SEC MALS and CDMS. ©2022 Water Corporation. Available online: https://lcms.cz/paper/17854 (accessed on 9 February 2025).
- Feng, X.; Su, Z.; Cheng, Y.; Ma, G.; Zhang, S. Messenger RNA chromatographic purification: Advances and challenges. J. Chromatogr. A 2023, 1707, 464321. [Google Scholar] [CrossRef] [PubMed]
- Kloczewiak, M.; Banks, J.M.; Jin, L.; Brader, M.L. A Biopharmaceutical Perspective on Higher-Order Structure and Thermal Stability of mRNA Vaccines. Mol. Pharm. 2022, 19, 2022–2031. [Google Scholar] [CrossRef]
- Foley, E.D.B.; Kushwah, M.S.; Young, G.; Kukura, P. Mass photometry enables label-free tracking and mass measurement of single proteins on lipid bilayers. Nat. Methods 2021, 18, 1247–1252. [Google Scholar] [CrossRef]
- Deslignière, E.; Yin, V.C.; Ebberink, E.H.T.M.; Rolland, A.D.; Barendregt, A.; Wörner, T.P.; Nagornov, K.O.; Kozhinov, A.N.; Fort, K.L.; Tsybin, Y.O.; et al. Ultralong transients enhance sensitivity and resolution in Orbitrap-based single-ion mass spectrometry. Nat. Methods 2024, 21, 619–622. [Google Scholar] [CrossRef]
- Gritti, F.; Wyndham, K. Retention mechanism in combined hydrodynamic and slalom chromatography for analyzing large nucleic acid biopolymers relevant to cell and gene therapies. J. Chromatogr. A 2024, 1730, 465075. [Google Scholar] [CrossRef]
- Gritti, F. Ultra-high pressure slalom chromatography: Application to the characterization of large DNA and RNA samples relevant in cell and gene therapy. J. Chromatogr. A 2024, 1738, 465487. [Google Scholar] [CrossRef]
- Wei, B.; Wang, J.; Cadang, L.; Goyon, A.; Chen, B.; Yang, F.; Zhang, K. Development of an ion pairing reversed-phase liquid chromatography-mass spectrometry method for characterization of clustered regularly interspaced short palindromic repeats guide ribonucleic acid. J. Chromatogr. A 2022, 1665, 462839. [Google Scholar] [CrossRef]
- Donegan, M.; Nguyen, J.M.; Gilar, M. Effect of ion-pairing reagent hydrophobicity on liquid chromatography and mass spectrometry analysis of oligonucleotides. J. Chromatogr. A 2022, 1666, 462860. [Google Scholar] [CrossRef]
- Azarani, A. RNA analysis by ion-pair reversed-phase high performance liquid chromatography. Nucleic Acids Res. 2001, 29, e7. [Google Scholar] [CrossRef] [PubMed]
- Lokras, A.; Chakravarty, A.; Rades, T.; Christensen, D.; Franzyk, H.; Thakur, A.; Foged, C. Simultaneous quantification of multiple RNA cargos co-loaded into nanoparticle-based delivery systems. Int. J. Pharm. 2022, 626, 122171. [Google Scholar] [CrossRef] [PubMed]
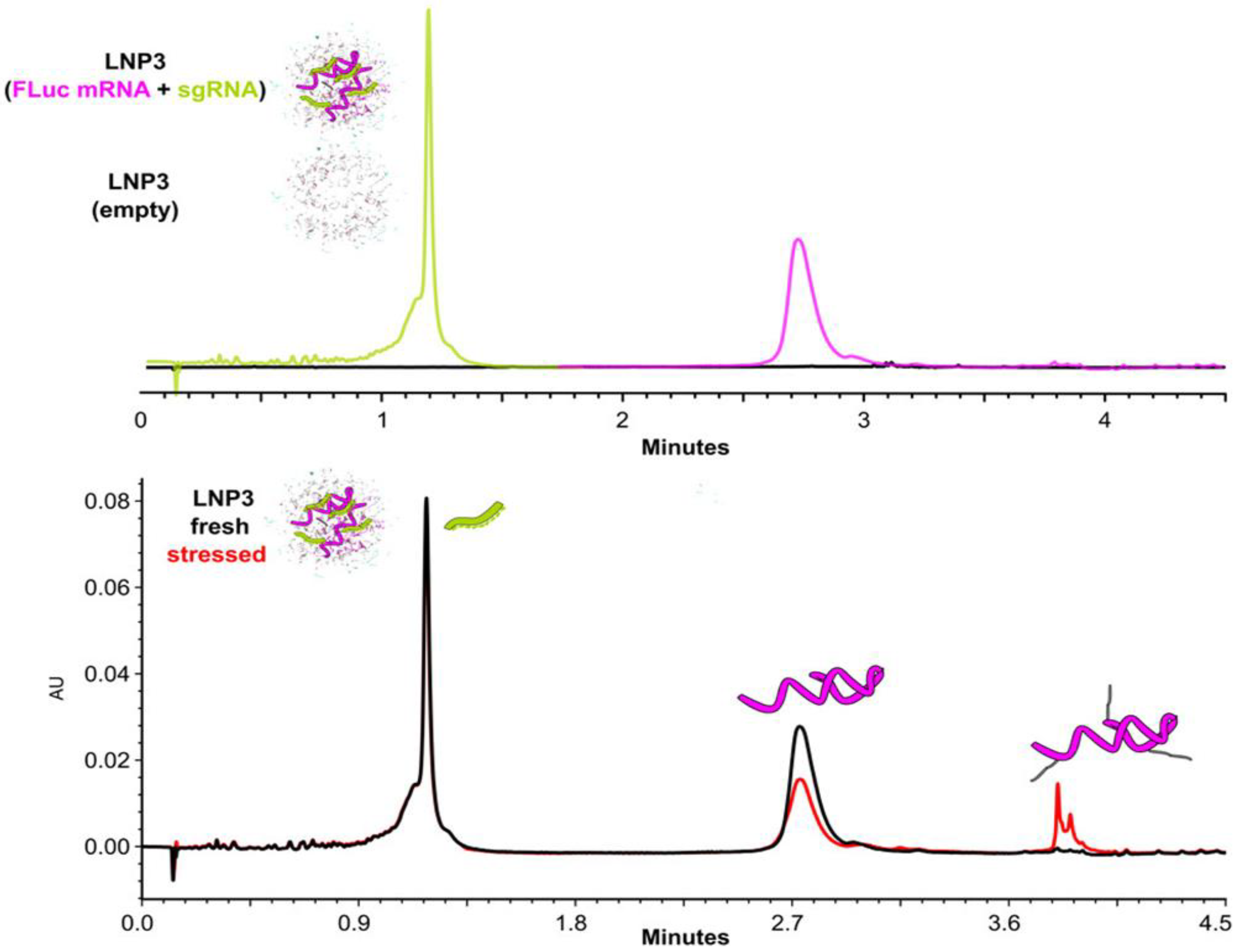
- Imiołek, M.; Cojocaru, R.; Fekete, S.; Le Huray, J.; Lauber, M. High-Throughput Quantification and Characterization of Dual Payload mRNA/LNP Cargo via Deformulating Size Exclusion and Ion Pairing Reversed Phase Assays. Anal. Chem. 2025, 97, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Blenke, E.O.; Evers, M.J.; Baumann, V.; Winkler, J.; Storm, G.; Mastrobattista, E. Critical evaluation of quantification methods for oligonucleotides formulated in lipid nanoparticles. Int. J. Pharm. 2018, 548, 793–802. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Yus, B.I.; Aldon, Y.; Shattock, R.J. Inside out: Optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 2019, 26, 363–372. [Google Scholar] [CrossRef]
- Beverly, M.; Hagen, C.; Slack, O. Poly A tail length analysis of in vitro transcribed mRNA by LC-MS. Anal. Bioanal. Chem. 2018, 410, 1667–1677. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Kong, J.; Yerabolu, R.; Hullen, K.; Zhao, K.; Wen, E.; Gunsch, M.J.; Foley, D.; He, Y. DNAzyme approach for simultaneous mRNA cap and poly(A) tail length analysis: A one-step method to multiple quality attributes. J. Pharm. Biomed. Anal. 2025, 257, 116695. [Google Scholar] [CrossRef]
- Campuzano, I.D.G.; Sandoval, W. Denaturing and Native Mass Spectrometric Analytics for Biotherapeutic Drug Discovery Research: Historical, Current, and Future Personal Perspectives. J. Am. Soc. Mass Spectrom. 2021, 32, 1861–1885. [Google Scholar] [CrossRef]
- Pourshahian, S. Therapeutic oligonucleotides, impurities, degradants, and their characterization by mass spectrometry. Mass Spectrom. Rev. 2019, 40, 75–109. [Google Scholar] [CrossRef]
- Parikh, R.A.; Miller, L.M.; Draper, B.E.; Kizekai, L.; Addepalli, B.; Chen, M.; Lauber, M.A.; Jarrold, M.F. Coupling of Size Exclusion Chromatography to High Throughput Charge Detection Mass Spectrometry for the Analysis of Large Proteins and Virus-like Particles. Anal. Chem. 2025, 97, 3036–3044. [Google Scholar] [CrossRef]
- Jarrold, M.F. Single-Ion Mass Spectrometry for Heterogeneous and High Molecular Weight Samples. J. Am. Chem. Soc. 2024, 146, 5749–5758. [Google Scholar] [CrossRef] [PubMed]
- Wörner, T.P.; Snijder, J.; Friese, O.; Powers, T.; Heck, A.J. Assessment of genome packaging in AAVs using Orbitrap-based charge-detection mass spectrometry. Mol. Ther.-Methods Clin. Dev. 2021, 24, 40–47. [Google Scholar] [CrossRef] [PubMed]
- van Schaick, G.; Haselberg, R.; Somsen, G.W.; Wuhrer, M.; Domínguez-Vega, E. Studying protein structure and function by native separation–mass spectrometry. Nat. Rev. Chem. 2022, 6, 215–231. [Google Scholar] [CrossRef] [PubMed]
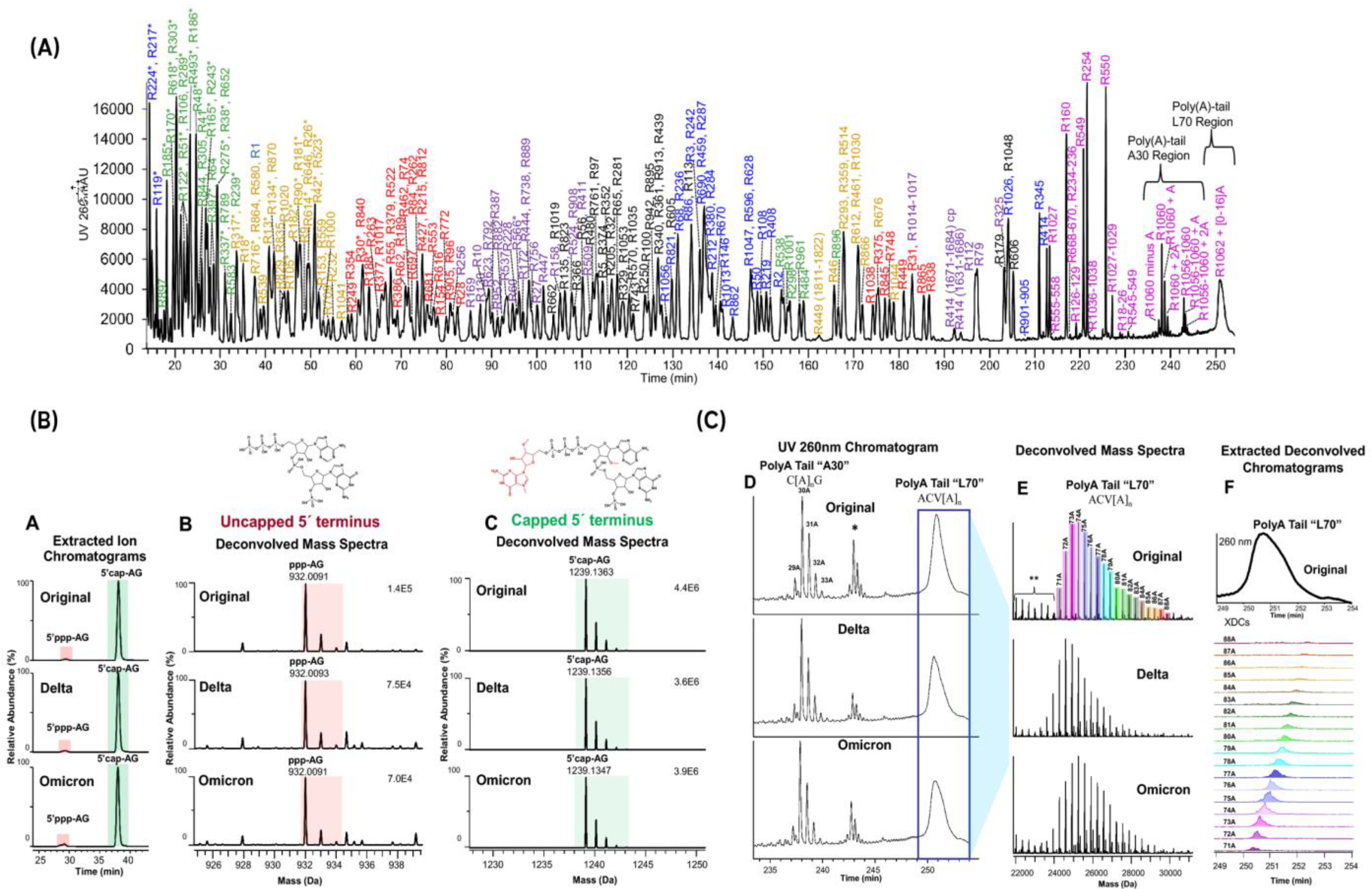
- Gau, B.C.; Dawdy, A.W.; Wang, H.L.; Bare, B.; Castaneda, C.H.; Friese, O.V.; Thompson, M.S.; Lerch, T.F.; Cirelli, D.J.; Rouse, J.C. Oligonucleotide mapping via mass spectrometry to enable comprehensive primary structure characterization of an mRNA vaccine against SARS-CoV-2. Sci. Rep. 2023, 13, 9038. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, N.; Kim, J.; Murgo, J.-R.; Kissai, M.; Ravichandran, K.R.; Miracco, E.J.; Presnyak, V.; Hua, S. Oligonucleotide Sequence Mapping of Large Therapeutic mRNAs via Parallel Ribonuclease Digestions and LC-MS/MS. Anal. Chem. 2019, 91, 8500–8506. [Google Scholar] [CrossRef]
- Vanhinsbergh, C.J.; Criscuolo, A.; Sutton, J.N.; Murphy, K.; Williamson, A.J.K.; Cook, K.; Dickman, M.J. Characterization and Sequence Mapping of Large RNA and mRNA Therapeutics Using Mass Spectrometry. Anal. Chem. 2022, 94, 7339–7349. [Google Scholar] [CrossRef]
- Nakayama, H.; Nobe, Y.; Koike, M.; Taoka, M. Liquid Chromatography–Mass Spectrometry-Based Qualitative Profiling of mRNA Therapeutic Reagents Using Stable Isotope-Labeled Standards Followed by the Automatic Quantitation Software Ariadne. Anal. Chem. 2022, 95, 1366–1375. [Google Scholar] [CrossRef]
- Goyon, A.; Scott, B.; Yehl, P.; Zhang, K. Online Nucleotide Mapping of mRNAs. Anal. Chem. 2024, 96, 8674–8681. [Google Scholar] [CrossRef]
- Tang, S.; Liu, G.-Y.; Yan, Y.; Wang, S.; Li, N. Development of a Flow Through-Based Limited Digestion Approach for High-Throughput and High-Sequence Coverage Mapping of Therapeutic mRNAs. Anal. Chem. 2024, 96, 16994–17003. [Google Scholar] [CrossRef]
- Lobue, P.A.; Jora, M.; Addepalli, B.; Limbach, P.A. Oligonucleotide analysis by hydrophilic interaction liquid chromatography-mass spectrometry in the absence of ion-pair reagents. J. Chromatogr. A 2019, 1595, 39–48. [Google Scholar] [CrossRef]
- Hayashi, Y.; Sun, Y. Overcoming Challenges in Oligonucleotide Therapeutics Analysis: A Novel Nonion Pair Approach. J. Am. Soc. Mass Spectrom. 2024, 35, 2034–2037. [Google Scholar] [CrossRef] [PubMed]
- Mutchek, S.; Kenderdine, T.; Turner, K.; Ring, J.; German, M.; Fabris, D. Strand-Cleaving Deoxyribozymes Enable the Mid-Down Sequencing of mRNA by Mass Spectrometry with No Front-End Separations. Anal. Chem. 2025, 97, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Lippens, J.L.; Timmons, H.C.; Welch, C.; Kulkarni, A.; Flick, T.G. Rapid Intact Mass Analysis and Evaluation of the Separation Potential of Microfluidic Capillary Electrophoresis Mass Spectrometry for Oligonucleotides. J. Am. Soc. Mass Spectrom. 2023, 34, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Strezsak, S.R.; Pimentel, A.J.; Hill, I.T.; Beuning, P.J.; Skizim, N.J. Novel Mobile Phase to Control Charge States and Metal Adducts in the LC/MS for mRNA Characterization Assays. ACS Omega 2022, 7, 22181–22191. [Google Scholar] [CrossRef]
- Tamara, S.; Boer, M.A.D.; Heck, A.J.R. High-Resolution Native Mass Spectrometry. Chem. Rev. 2021, 122, 7269–7326. [Google Scholar] [CrossRef]
- Jain, M.; Abu-Shumays, R.; Olsen, H.E.; Akeson, M. Advances in nanopore direct RNA sequencing. Nat. Methods 2022, 19, 1160–1164. [Google Scholar] [CrossRef]
- Subtelny, A.O.; Eichhorn, S.W.; Chen, G.R.; Sive, H.; Bartel, D.P. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 2014, 508, 66–71. [Google Scholar] [CrossRef]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide Determination of Poly(A) Tail Length and 3′ End Modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef]
- Lim, J.; Lee, M.; Son, A.; Chang, H.; Kim, V.N. mTAIL-seq reveals dynamic poly(A) tail regulation in oocyte-to-embryo development. Genes Dev. 2016, 30, 1671–1682. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, Y.; Cheng, C.; Wang, W.; Zhou, Z.; Rang, W.; Yu, H.; Wei, Y.; Wu, Q.; Zhang, Y. Poly(A)-seq: A method for direct sequencing and analysis of the transcriptomic poly(A)-tails. PLoS ONE 2020, 15, e0234696. [Google Scholar] [CrossRef]
- Harrison, P.F.; Powell, D.R.; Clancy, J.L.; Preiss, T.; Boag, P.R.; Traven, A.; Seemann, T.; Beilharz, T.H. PAT-seq: A method to study the integration of 3′-UTR dynamics with gene expression in the eukaryotic transcriptome. RNA 2015, 21, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.D.; Slevin, M.K.; Tatomer, D.C.; Duronio, R.J.; Prins, J.F.; Marzluff, W.F. EnD-Seq and AppEnD: Sequencing 3′ ends to identify nontemplated tails and degradation intermediates. RNA 2015, 21, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Scheer, H.; De Almeida, C.; Sikorska, N.; Koechler, S.; Gagliardi, D.; Zuber, H. High-Resolution Mapping of 3’ Extremities of RNA Exosome Substrates by 3’ RACE-Seq. In The Eukaryotic RNA Exosome; LaCava, J., Vaňáčová, Š., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2062, pp. 147–167. [Google Scholar] [CrossRef]
- Woo, Y.M.; Kwak, Y.; Namkoong, S.; Kristjánsdóttir, K.; Lee, S.H.; Lee, J.H.; Kwak, H. TED-Seq Identifies the Dynamics of Poly(A) Length during ER Stress. Cell Rep. 2018, 24, 3630–3641.e7. [Google Scholar] [CrossRef] [PubMed]
- Brar, G.A.; Weissman, J.S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 651–664. [Google Scholar] [CrossRef]
- Ingolia, N.T. Ribosome Footprint Profiling of Translation throughout the Genome. Cell 2016, 165, 22–33. [Google Scholar] [CrossRef]
- Gunter, H.M.; Idrisoglu, S.; Singh, S.; Han, D.J.; Ariens, E.; Peters, J.R.; Wong, T.; Cheetham, S.W.; Xu, J.; Rai, S.K.; et al. mRNA vaccine quality analysis using RNA sequencing. Nat. Commun. 2023, 14, 5663. [Google Scholar] [CrossRef]
- Smola, M.J.; Rice, G.M.; Busan, S.; Siegfried, N.A.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 2015, 10, 1643–1669. [Google Scholar] [CrossRef]
- Begik, O.; Lucas, M.C.; Pryszcz, L.P.; Ramirez, J.M.; Medina, R.; Milenkovic, I.; Cruciani, S.; Liu, H.; Vieira, H.G.S.; Sas-Chen, A.; et al. Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat. Biotechnol. 2021, 39, 1278–1291. [Google Scholar] [CrossRef]
- Long, Y.; Jia, J.; Mo, W.; Jin, X.; Zhai, J. FLEP-seq: Simultaneous detection of RNA polymerase II position, splicing status, polyadenylation site and poly(A) tail length at genome-wide scale by single-molecule nascent RNA sequencing. Nat. Protoc. 2021, 16, 4355–4381. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Legnini, I.; Alles, J.; Karaiskos, N.; Ayoub, S.; Rajewsky, N. FLAM-seq: Full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods 2019, 16, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nie, H.; Liu, H.; Lu, F. Poly(A) inclusive RNA isoform sequencing (PAIso−seq) reveals wide-spread non-adenosine residues within RNA poly(A) tails. Nat. Commun. 2019, 10, 5292. [Google Scholar] [CrossRef] [PubMed]
- Mattijssen, S.; Iben, J.R.; Li, T.; Coon, S.L.; Maraia, R.J. Single molecule poly(A) tail-seq shows LARP4 opposes deadenylation throughout mRNA lifespan with most impact on short tails. eLife 2020, 9, e59186. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Gazestani, V.H.; Hampton, M.; Abrahante, J.E.; Salavati, R.; Zimmer, S.L. circTAIL-seq, a targeted method for deep analysis of RNA 3′ tails, reveals transcript-specific differences by multiple metrics. RNA 2016, 22, 477–486. [Google Scholar] [CrossRef]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef]
- Bicknell, A.A.; Reid, D.W.; Licata, M.C.; Jones, A.K.; Cheng, Y.M.; Li, M.; Hsiao, C.J.; Pepin, C.S.; Metkar, M.; Levdansky, Y.; et al. Attenuating ribosome load improves protein output from mRNA by limiting translation-dependent mRNA decay. Cell Rep. 2024, 43, 114098. [Google Scholar] [CrossRef]
- Zheng, D.; Persyn, L.; Wang, J.; Liu, Y.; Montoya, F.U.; Cenik, C.; Agarwal, V. Predicting the Translation Efficiency of Messenger RNA in Mammalian Cells. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, H.; Zhou, J.; He, X.; Jiang, T.; Zeng, J. Analysis of Ribosome Stalling and Translation Elongation Dynamics by Deep Learning. Cell Syst. 2017, 5, 212–220.e6. [Google Scholar] [CrossRef]
- Begik, O.; Diensthuber, G.; Liu, H.; Delgado-Tejedor, A.; Kontur, C.; Niazi, A.M.; Valen, E.; Giraldez, A.J.; Beaudoin, J.-D.; Mattick, J.S.; et al. Nano3P-seq: Transcriptome-wide analysis of gene expression and tail dynamics using end-capture nanopore cDNA sequencing. Nat. Methods 2022, 20, 75–85. [Google Scholar] [CrossRef]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef] [PubMed]
- Leppek, K.; Byeon, G.W.; Kladwang, W.; Wayment-Steele, H.K.; Kerr, C.H.; Xu, A.F.; Kim, D.S.; Topkar, V.V.; Choe, C.; Rothschild, D.; et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 2022, 13, 1–22. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, H.; Wei, Z.; Hong, H.; Huang, D.; Liu, G.; Qin, Q.; Rong, R.; Gao, P.; Meng, J.; et al. NanoMUD: Profiling of pseudouridine and N1-methylpseudouridine using Oxford Nanopore direct RNA sequencing. Int. J. Biol. Macromol. 2024, 270, 132433. [Google Scholar] [CrossRef]
- Tavakoli, S.; Nabizadeh, M.; Makhamreh, A.; Gamper, H.; McCormick, C.A.; Rezapour, N.K.; Hou, Y.-M.; Wanunu, M.; Rouhanifard, S.H. Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing. Nat. Commun. 2023, 14, 1–12. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Liu-Wei, W.; van der Toorn, W.; Bohn, P.; Hölzer, M.; Smyth, R.P.; von Kleist, M. Sequencing accuracy and systematic errors of nanopore direct RNA sequencing. BMC Genom. 2024, 25, 528. [Google Scholar] [CrossRef]
- Dhungel, P.; Cantu, F.; Hernandez, C.; Yang, Z. In Vitro Transcribed RNA-based Luciferase Reporter Assay to Study Translation Regulation in Poxvirus-infected Cells. J. Vis. Exp. 2019, 147, 59626. [Google Scholar] [CrossRef]
- Patel, N.; Davis, Z.; Hofmann, C.; Vlasak, J.; Loughney, J.W.; DePhillips, P.; Mukherjee, M. Development and Characterization of an In Vitro Cell-Based Assay to Predict Potency of mRNA–LNP-Based Vaccines. Vaccines 2023, 11, 1224. [Google Scholar] [CrossRef]
- Stiving, A.Q.; Roose, B.W.; Tubbs, C.; Haverick, M.; Gruber, A.; Rustandi, R.R.; Kuiper, J.; Schombs, M.; Schuessler, H.; Li, X. Enabling Functionality and Translation Fidelity Characterization of mRNA-Based Vaccines with a Platform-Based, Antibody-Free Mass Spectrometry Detection Approach. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.S.; Sattar, M.N.; Iqbal, Z.; Raza, A.; Al-Sadi, A.M. Next-Generation Sequencing and the CRISPR-Cas Nexus: A Molecular Plant Virology Perspective. Front. Microbiol. 2021, 11, 609376. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-H.; Völse, K.; Senft, D.; Jeremias, I. A reporter system for enriching CRISPR/Cas9 knockout cells in technically challenging settings like patient models. Sci. Rep. 2021, 11, 12649. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Hur, S. Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 2021, 23, 286–301. [Google Scholar] [CrossRef]





| RNA Samples | Analytical Method Conditions | Target QA | General Comments | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| CE Mode | CE Capillary/Gel Type | Background Electrolyte (BGE) | Detection Mode | Separation Time | ||||
| - mRNA-LNP (~2000 nts) | mCE | RNA labchips | RNA reagent kits (Catalog# CLS960010) | LIF | 70 s | Purity and integrity | Development and optimization of a purity and integrity assay for mRNA-based vaccines encapsulated in LNPs. | [28] |
| - eGFP (996 nts) - FLuc (1909 nts) - β-Gal (3420 nts) | mCE | RNA labchips | RNA reagent kits (Catalog# CLS960010) | LIF | 70 s | Purity and aggregate contents | mCE used in non-denaturing conditions (heated at 70 °C for 10 min) to determine the percentage of covalent and non-covalent aggregates. | [30] |
| - eGFP (996 nts) - FLuc (1909 nts) - β-Gal (3420 nts) | CGE | Fused-silica capillary length of 60 cm (50 μm i.d.) | SCIEX RNA 9000 Purity and Integrity kit | LIF | 100 min | Purity and aggregate contents | CGE used in denaturing conditions to monitor covalent aggregates. | [30] |
| - eGFP (996 nts) - Luciferase (2000 nts) - Cas9 (4500 nts) | CGE | Fused-silica capillary length of 30 cm (100 μm i.d.) | Tris-Borate with urea and methylcellulose | UV | 40 min | Poly(A) tail length | CGE is used for the determination of the poly(A) tail length, with a resolution comparable with the IP-RP LC method. | [29] |
| RNA marker (100 to 10,000 nts) | CGE | Fused-silica capillary length of 15 cm (75 μm i.d.) | PEG polymer, TBE, 4 M urea | LIF | 10 min | N/A | Investigation of the separation of RNA fragments in PEG and PEO solutions. | [31] |
| - EPO (859 nts) | CGE | Fused-silica capillary length of 72 cm (50 μm i.d.) | Tris-Borate-EDTA HEPES buffer (pH 7.5) with polymer solution (PVP and glycerol) | UV | 130 min | Purity | Design a flexible, multi-objective CGE method for analysis of modified mRNA by focusing on the components of the low-viscous polymer matrix. | [32] |
| - Single-stranded RNA size marker (size ranging from 281 to 6583 nts) | CGE | Fused-silica capillary length of 21.5 cm (50 μm i.d.) | Tris-Borate-EDTA buffer with urea and HEC as the polymer solution | UV | 10 min or 20 min | N/A | Investigation of electrophoretic separation of large RNA (over 6000 nts) in dilute and semidilute polymer matrices. | [33,34] |
| BNT162b2 mRNA (active substance) | CGE | Fused-silica capillary (50 μm i.d.) | Agilent RNA Analysis Kit | LIF | 60 min | Purity | Analysis of short impurity species to be 5’-end BNT162b2 fragments generated from premature transcription stop during the IVT reaction. | [24] |
| RNA marker (100 to 10,000 nts) | CGE | Fused-silica capillary length of 9 cm (75 μm i.d.) | 10X TBE, HEC | LIF | 8 min | N/A | Investigation of the effect of MW of HEC on the separation performance of long RNA. HEC favors the separation of short RNA fragments (<1000 nts). | [35] |
| - RNA size marker (size ranging from 200 to 6000 nts) | CGE | Fused-silica and PVA-coated capillary length of 56 cm (50 μm i.d.) | A stock formamide buffer (pH 6.0) prepared in formamide or water and containing MES and EDTA with or without urea | UV | 20 and 50 min | N/A | CGE method using high MW polymers and formamide, enhancing the resolution for mRNAs by approximately six-fold compared with standard aqueous CGE methods. | [27] |
| - eGFP (996 nts) - Luciferase (2000 nts) - Cas9 (4500 nts) | CGE | Fused-silica capillary length of 30 cm (100 μm i.d.) | ssDNA 100-R Gel reconstituted in Tris-Borate-7 M urea buffer | UV | 28, 32, and 40 min for eGFP, Luciferase, and Cas9, respectively | Poly(A) tail lengh | CGE method having the same resolution as an LC-MS method for the characterization of the poly(A) tail length. | [29] |
| - eGFP (996 nts) - Ovalbumine (1437 nts) - Luciferase (1929 nts) - Cas9 (4521 nts) | CGE |
|
|
|
| Integrity, purity | Sciex RNA 9000 Purity and Integrity Kit provides the highest selectivity and resolving power for characterization of mRNA. Agilent 600 Nano Kit, Revvity RNA Reagent Kit, and Agilent HS RNA Kit offer faster analysis times, making them more suitable for high-throughput and screening applications. | [36] |
| tRNAPhe (75 nt) and HIV-1 5′-UTR (364 nt) | CZE | High-resolution bare glass chips (HRB) | Mix of ammonium acetate with H2O+ 25% IPA | MS | 5 min | Sequence confirmation and impurities | CZE-MSl for the bottom-up characterization of nucleic acids, using microfluidic devices that combine both capillary and transmitter in the same chip. | [37] |
| - Modified linear GLuc mRNA - Modified hEpo mRNA - GLuc APIE CVB3 pAC (circRNA) | AGE | Agarose E- EX gel (2%) | Tris-acetate-EDTA buffer with formamide | Bands are visualized using blue light transillumination | 80 min | Purity | AGE for the separation of circular splicing products (i.e., linear precursor molecules, nicked circles, splicing intermediates, and excised introns). | [38,39] |
| RNA Samples | Analytical Method Conditions | Target QA | General Comments | Ref. | |||
|---|---|---|---|---|---|---|---|
| LC Mode | LC Column | Mobile Phase, Column Temperature | Detector | ||||
| mRNA samples produced from eGFP, C-Spike and NLuc plasmid DNA templates | AEX | DNAPac PA200 (50 mm × 2.1 mm) | MPA: 10 mM NaOH MPB: 2 M NaCl, 25 °C | UV | Method to separate IVT components (e.g., NTPs, Cap analogue, plasmid DNA, and mRNA) | In process IVT mRNA impurities. | [44] |
| Short oligonucleotides and EGFP (996 nts) mRNA | AEX | DNAPac PA200 (50 mm × 2.1 mm) | Neutral pH—sodium chloride or sodium perchlorate salt gradient—60 °C, or high pH (pH = 12)—salt gradient separation—10 °C | UV | Purity—degradation RNA products | AEX using denaturing conditions. | [45] |
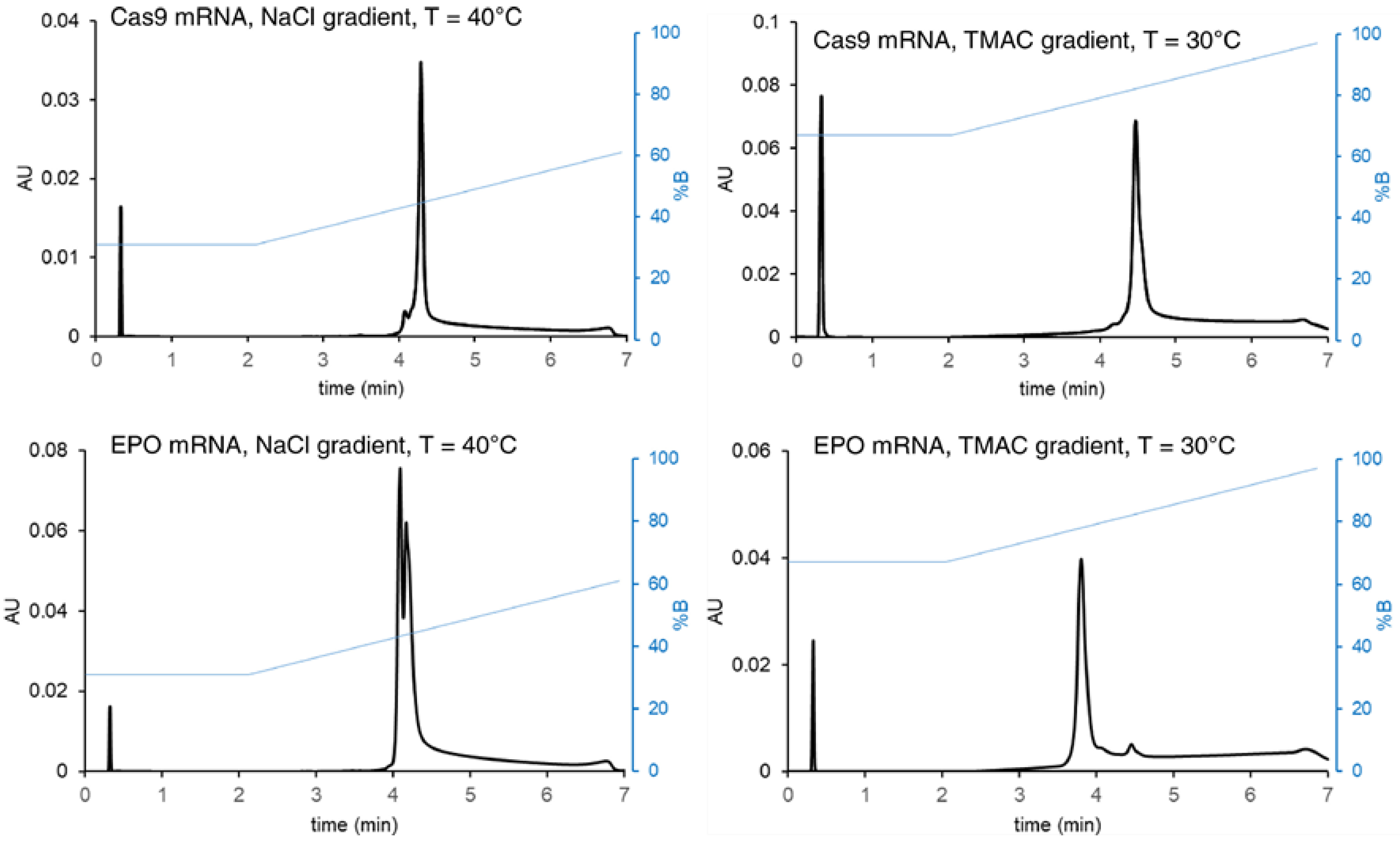
| - Cas9 (4500 nts) - EPO (859 nts) | AEX (IPAX) | Protein-Pak Hi Res Q strong anion exchange (50 × 2.1 mm, 5 µm) | 25 mM HEPES and TRIS buffers (pH 7.5–8) and 1–3 M TMAC gradients, up to 60 °C | UV | Purity—degradation RNA products | Using a gradient of weak ion-pairing cations (e.g., TMAC) to provide different recovery and selectivity effects. | [46] |
| - EPO (859 nts) - β-FLuc (~2000 nts) | SEC | ACQUITY Premier protein SEC (250 Å, 1.7 μm, 4.6 × 150 mm) | 0.1 M phosphate buffer, pH 8, 25 °C | UV | Poly(A) tail length | Average poly(A) tail length. | [10] |
| - EGFP (996 nts) - Cre (1350 nts) | SEC | Ultrawide pore prototype SEC (4.6 mm I.D. x 300, 3.0 μm, 1275 Å) column—silica-based packing material modified with an OH-PEG bonding | 50 mM Tris and 200 mM potassium chloride (pH adjusted to 7.5, 25 °C) | UV, RI, and MALS | Purity—short fragments and aggregates | Use of prototype, low adsorption ultrawide pore SEC columns. | [47] |
| Various mRNA samples (996–4521 nt), including modifications (5moU), stressed samples, and from different suppliers | SEC | GTxResolve Premier BEH™ SEC (450 Å, 2.5 µm, 150 × 4.6 mm) GTxResolve Premier SEC 1000 (1000 Å, 3 µm, 150 × 4.6 mm and 300 × 4.6 mm) Ultrawide pore prototype SEC (2500 Å, 5 µm, 150 and 300 × 4.6 mm) | 50 mM Tris buffer, pH 7.5, 200 mM KCl and 10 mM MgCl2. 25 °C | UV | Purity—short fragments and aggregates | Use of ultrawide pore columns Addition of 10 mM MgCl2 to improve chromatographic resolution and/or preserve mRNA from confounding effects (in some cases). | [48] |
| FLuc (~2000 nts) | SEC | Agilent Bio SEC-5 (4.6 × 300 mm, 5 μm) with varying pore size (300 Å, 1000 Å and 2000 Å) | 100 mM phosphate, pH 7.0, 25 °C | UV, RI, and MALS | Purity—MW, short fragments, and aggregates | Optimization of LC conditions on the separation performance and structural integrity of mRNAs. | [49] |
| - eGFP (996 nts) - FLuc (1909 nts) - β-Gal (3420 nts) | SEC | SRT(R) SEC-1000 gel (4.6 mm I.D. × 300 mm, 5 μm particle size, 1000 Å, stainless steel) | 100 mM Tris–HCl and 300 mM NaCl at pH 7.5, 25 °C | UV | Purity—MW, short fragments, and aggregates | Level of mRNA aggregates can be significantly reduced after a heating step. Limitation of SEC mode for mRNA aggregates. | [30] |
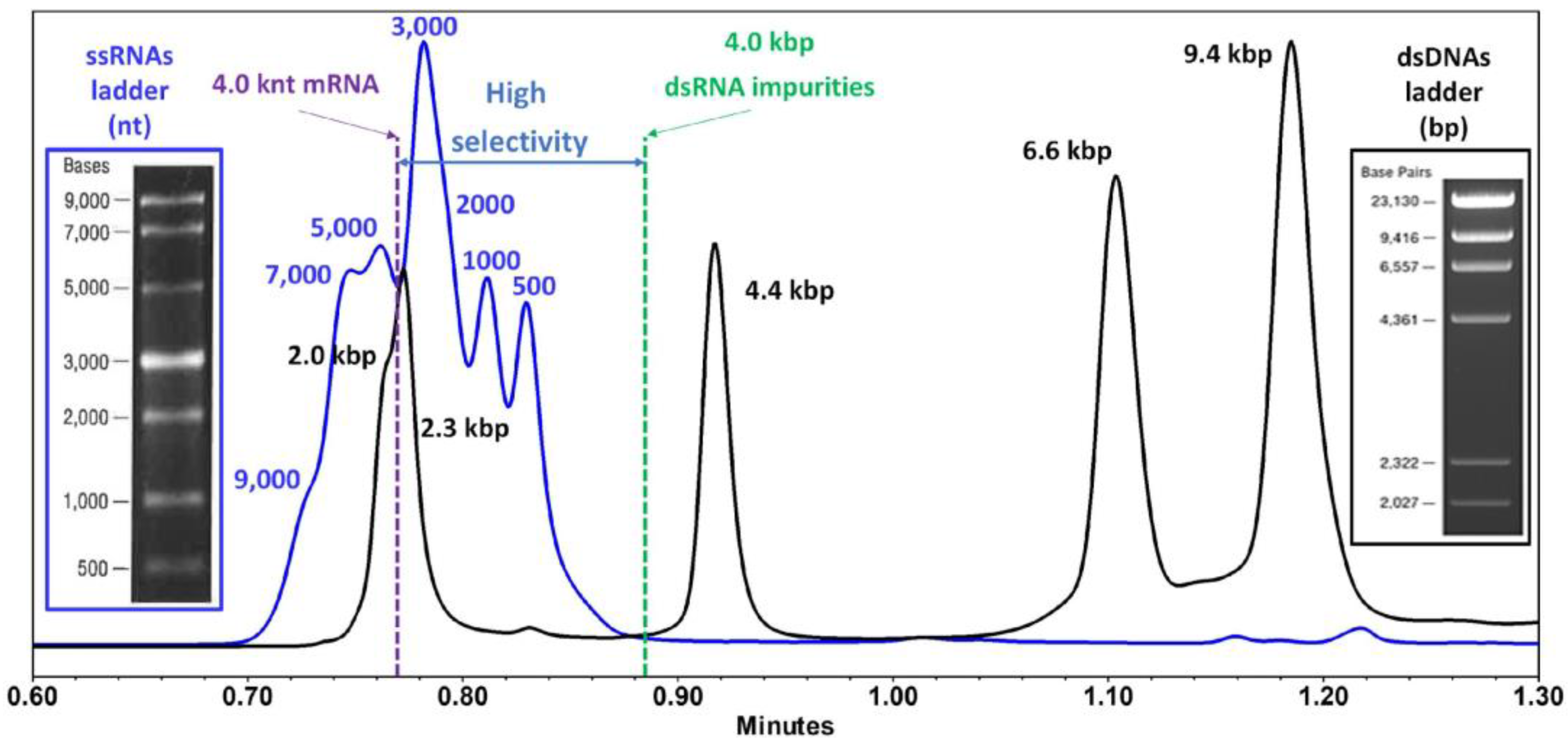
| - Large RNA, up to 1000 nts - eGFP (996 nts) | IP-RP | DNAPac RP (150 mm × 2.1 mm i.d.) | MPA: 100 mM TEA in H2O MPB: 100 mM TEA in 40% ACN, 80 °C | UV | Purity—degradation RNA products, short fragments and aggregates | Stability indicating method (direct exposure to heat, hydrolytic conditions, and treatment with ribonucleases). | [50] |
| - EPO (859 nts) - β-FLuc (~2000 nts) | IP-RP | ACQUITY premier oligonucleotide BEH C18 (300 Å, 1.7 μm, 2.1 × 150 m) | MPA: 0.1 M OAA, 1% HFIP in 40% ACN MPB: 0.1 M OAA, 1% HFIP in 90% ACN | UV | Poly(A) tail length | DIPEA/HFIP is a suitable ion-pairing system for sensitive LC MS analysis. | [10] |
| - eGFP (996 nts) - FLuc (1909 nts) - β-Gal (3420 nts) | IP-RP | DNAPac RP (150 mm × 2.1 mm i.d.) | MPA: 50 mM TEA, 50 mM HFIP in H2O MPB: 25 mM TEA, 25 mM HFIP in 90% MeOH. 80 °C | UV | Purity—short fragments and aggregates (5′loopback dsRNA) | Aggregates observed in IP-RP are associated with 5′loopback dsRNA impurities. | [30] |
| - zsGreen (~1000 nts) - mScarlet (~1000 nts) | IP-RP | DNAPac RP (150 mm × 2.1 mm i.d.) | MPA: 50 mM TEA, 50 mM HFIP in H2O Mobile phase B: 25 mM TEA, 25 mM HFIP in 90% MeOH. 80 °C | UV | Purity—short fragments | The use of different T7 polymerase directly impacted the level of short mRNA fragments. | [51] |
| Circular eGFP RNA | IP-RP | AcclaimTM 300 C18 (3 μm, 150 × 4.6 mm) | MPA: 100 mM TEAA in H2O MPB: 5% 100 mM TEAA in 95% ACN | UV | Purity, impurity analysis | Identification of circRNAs and nicked RNAs, and elucidated the degradation pattern of the circRNA substance. | [52] |
| Platform | Method/Application | Method Overview | Advantages | Limitations | Ref |
|---|---|---|---|---|---|
| Illumina | Sequence identity | Library preparation involves cDNA synthesis, fragmentation, adapter ligation, and PCR amplification | High-throughput, high-quality data | Length (standard < 300 bp; 600 bp max on MiSeq and NextSeq1000/2000 instruments) | [91] |
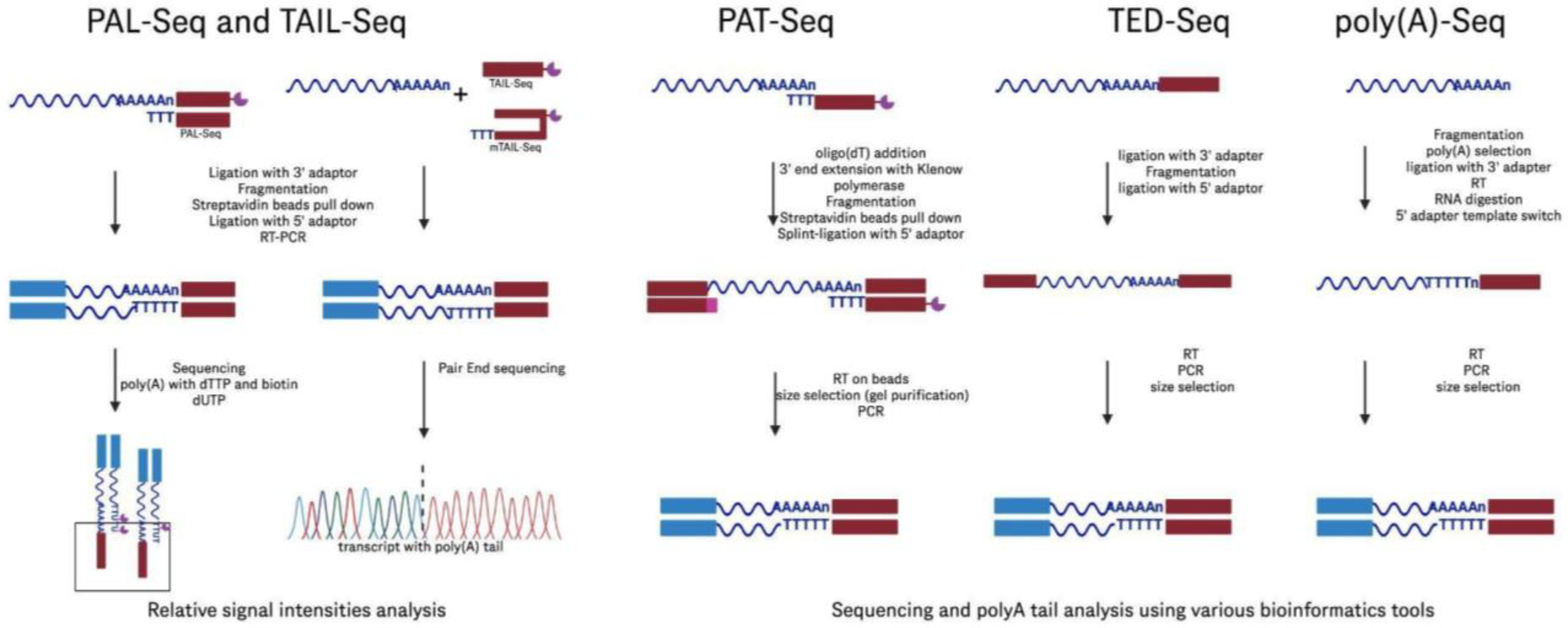
| PAL-Seq: Poly(A) tail profiling | Labels poly(A) tails with biotinylated dUTP for fluorochrome detection | High throughput; effective poly(A) tail measurement | Requires outdated technology; limited by reliance on older sequencers | [92] | |
| TAIL-Seq: Poly(A) tail profiling | Uses fluorescence quantification to estimate poly(A) tail length | Compatible with modern sequencers; measures both the length of the poly(A) tails and their modifications, such as uridylation | Complex data analysis: requires tailored control software; intermediate fluorescence data storage; needs a substantial amount of RNA | [93] | |
| mTAIL-Seq: Poly(A) tail profiling | Modified TAIL-Seq with hairpin adapters for improved efficiency | Enhances the efficiency of ligation, reducing the amount of starting material needed, providing similar capabilities to those of TAIL-Seq | Shares the technical complexity and data analysis challenges of TAIL-Seq | [94] | |
| Poly(A)-Seq: Poly(A) tail profiling | Analyzes the lengths and internal compositions of poly(A) tails, uses oligo(dT) selection and 3′ adapter ligation | Can detect detailed compositions, including guanosine insertions within tails | May bias toward longer tails due to the selection method used, loss of information on tails shorter than ten nucleotides due to data filtering | [95] | |
| PAT-Seq: Poly(A) tail profiling | Extends RNA’s 3′ end with Klenow DNA polymerase | Can measure poly(A) tails and analyze changes in gene expression | Requires precise size selection, which can impact result accuracy; constrained by the length of sequencing reads, making it difficult to detect very long tails | [96] | |
| EnD-Seq: Non-Poly(A) tail analysis | Targets short poly(A) tails and non-polyadenylated RNAs and specializes in analyzing non-poly(A) modifications at RNA 3′ ends | Highly sensitive to short and non-poly(A) tails, useful for examining RNA decay products | Not suitable for assessing long poly(A) tails, specific improvements in sensitivity may affect the results | [97] | |
| 3′RACE-Seq: Non-Poly(A) tail analysis | Analyzes specific transcripts’ 3′ ends | Enables detailed sequencing of particular RNA 3′ ends, can identify modifications at the 3′ end | Limited to specific RNA targets, nested PCR steps might introduce bias | [98] | |
| TED-Seq: 3′ end mapping | Maps 3′ cleavage and polyadenylation sites | Cost-effective; achieves 10 bp resolution; straightforward protocol | Lacks quantitative robustness; variable recovery rates; may require precise size selection | [99] | |
| Ribo-Seq: ribosome profiling, translation efficiency | Captures the positions of ribosomes on mRNA transcripts by sequencing ribosome-protected mRNA fragments | Can reveal translation efficiency and identify active reading frames providing insights into translation dynamics and protein synthesis | Complex sample preparation and data analysis, necessitating specialized expertise and resources, which might limit accessibility | [100,101] | |
| Oxford Nanopore Technologies | VAX-seq | Used for plasmid sequencing as well as IVT-produced RNA with both direct RNA sequencing and cDNA sequencing | Comprehensive workflow developed to assess mRNA vaccine quality by analyzing sequence identity, integrity, and poly(A) tail length | Expertise in long-read sequencing and complex data analysis required | [102] |
| nanoSHAPE: RNA structure | Chemical probing method to analyze RNA structure | Structural insights can inform the design and optimization of mRNA vaccines, potentially improving their stability and translation efficiency | Efficiency and specificity of the chemical modification can vary, potentially leading to incomplete or biased structural data if not carefully controlled | [103] | |
| Integrity analysis with Nano3P-seq | Captures both polyadenylated and non-polyadenylated RNA; uses a template-switching reverse transcriptase (TGIRT) to capture the full-length RNA | Long reads that can cover entire RNA molecules, including poly(A) tails to enable broad RNA analysis with no PCR bias | Higher error rate compared with other sequencing technologies, requires more complex data analysis | [104] | |
| FLEP-seq | Captures both polyadenylated and non-polyadenylated RNA; adds DNA adapter at 3′ end and RT | Long reads that can cover entire RNA molecules, RNA polymerase II positioning, splicing status, polyadenylation sites, and poly(A) tail lengths and translation efficiency | Higher error rate compared with other sequencing technologies | [105] | |
| RNA modification detection | Direct RNA sequencing | Direct sequencing of RNA uses RNA adapters, no need for reverse transcription or amplification and multiplexing capabilities | Higher error rates compared with other sequencing technologies | [91,102] | |
| PacBio | Sequence identity analysis, full-length mRNA sequencing | Library prep involves cDNA synthesis, adapter ligation, and circular consensus sequencing (CCS) | Long, high-fidelity/quality reads | High cost, PCR amplification bias, instrument generally less available than other platforms | [106] |
| FLAM-Seq: Poly(A) tail analysis | Poly(A) selection followed by enzymatically added G/I tail serving as anchor for oligo prior to RT | Low error rate, long reads, to analyze full-length mRNA and their poly(A) tails—can detect internal variations within the tails | May introduce biases from PCR amplification steps, requires a significant amount of RNA material | [107] | |
| PAIso-Seq: Poly(A) tail analysis | 3′-end extension of template oligo containing T stretch | Low error rates, long reads, to measure poly(A) tails without needing to pre-select polyadenylated RNA, can be performed with 100 ng of RNA or less | Potential issues with artifacts from the extension process | [108] | |
| SM-PAT Poly(A) tail analysis | Poly(A) selection followed by the addition of a 3′ adapter through splint ligation | Low error rates, long reads; very similar to FLAM-Seq and PAIso-Seq | May introduce biases through PCR amplification steps | [109] |
| Analytical Technology | Advantages | Limitations |
|---|---|---|
| Capillary gel electrophoresis |
|
|
| Anion-exchange chromatography |
|
|
| Size exclusion chromatography |
|
|
| Ion-pair reversed phase liquid chromatography |
|
|
| Mass photometry |
|
|
| Native mass spectrometry |
|
|
| Sanger sequencing |
|
|
| Illumina sequencing |
|
|
| PacBio sequencing |
|
|
| Oxford nanopore |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camperi, J.; Chatla, K.; Freund, E.; Galan, C.; Lippold, S.; Guilbaud, A. Current Analytical Strategies for mRNA-Based Therapeutics. Molecules 2025, 30, 1629. https://doi.org/10.3390/molecules30071629
Camperi J, Chatla K, Freund E, Galan C, Lippold S, Guilbaud A. Current Analytical Strategies for mRNA-Based Therapeutics. Molecules. 2025; 30(7):1629. https://doi.org/10.3390/molecules30071629
Chicago/Turabian StyleCamperi, Julien, Kamalakar Chatla, Emily Freund, Carolina Galan, Steffen Lippold, and Axel Guilbaud. 2025. "Current Analytical Strategies for mRNA-Based Therapeutics" Molecules 30, no. 7: 1629. https://doi.org/10.3390/molecules30071629
APA StyleCamperi, J., Chatla, K., Freund, E., Galan, C., Lippold, S., & Guilbaud, A. (2025). Current Analytical Strategies for mRNA-Based Therapeutics. Molecules, 30(7), 1629. https://doi.org/10.3390/molecules30071629