
The Polymorphism of Orlyum White 520T, an Ultraviolet Luminescent Security Ink

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
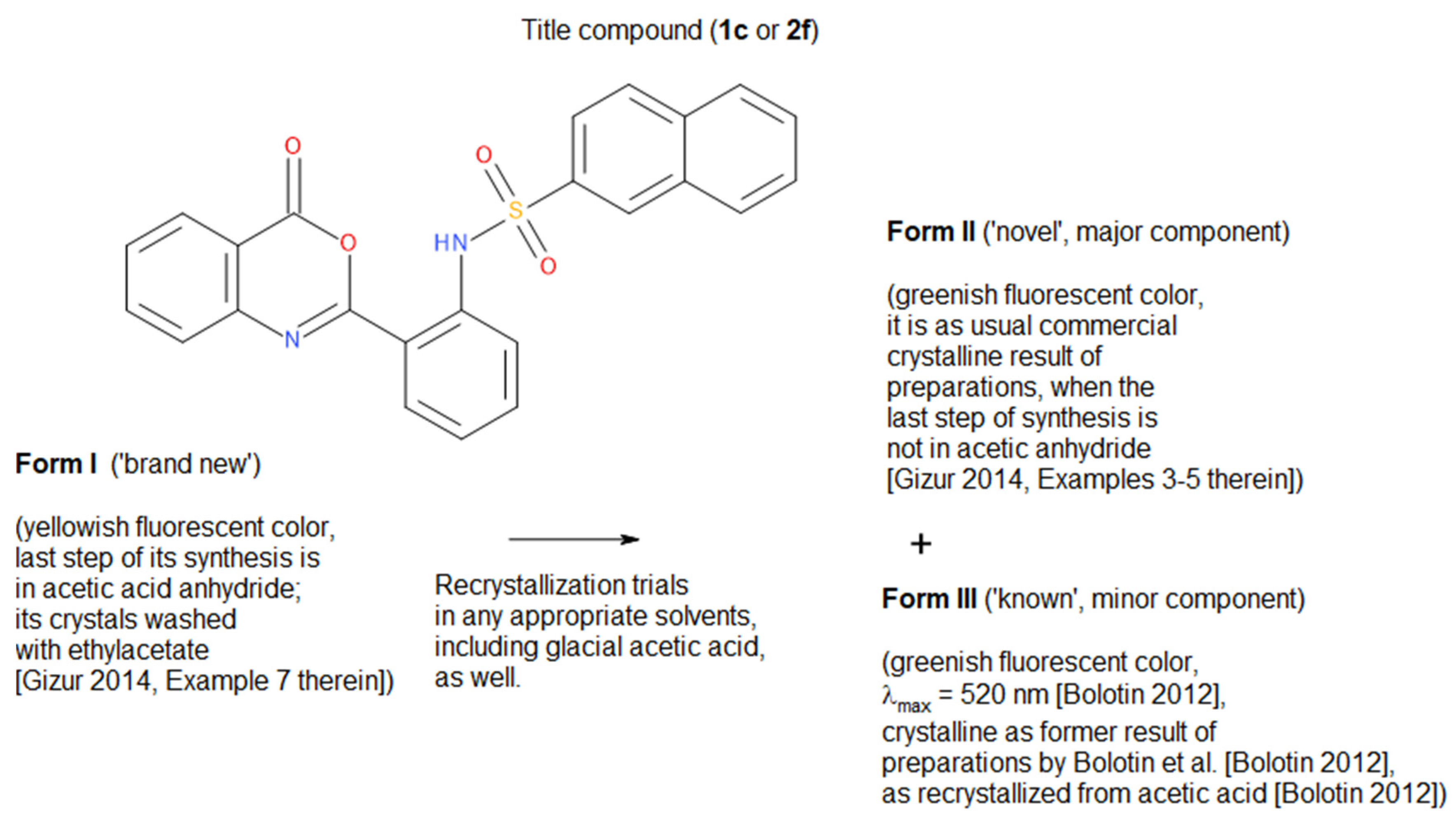
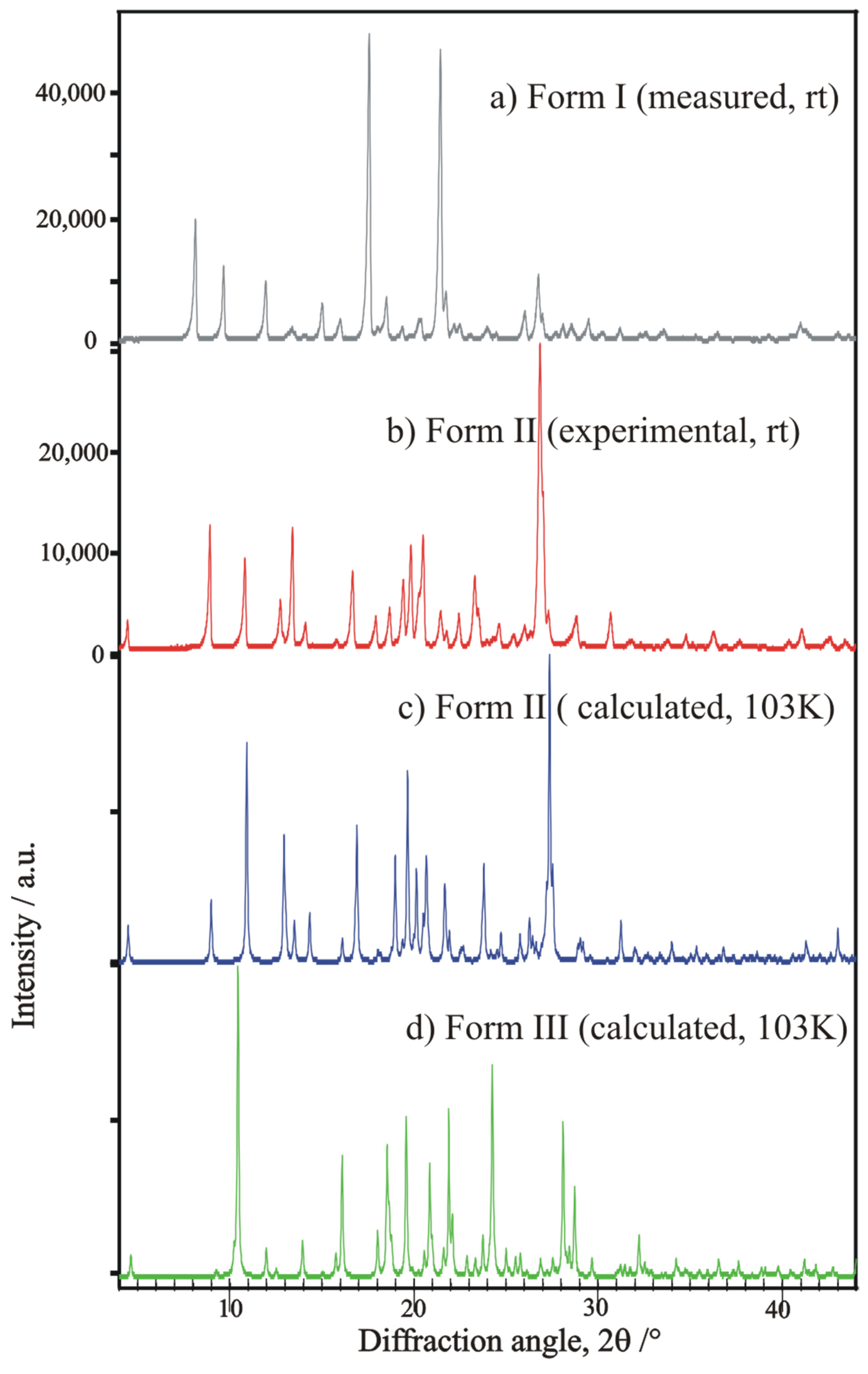
2.1. Relation Among Unreported New Forms I and II and the Old Form III
2.2. Identical Composition of Form I and II Proven by CHN(S) Elemental Analysis and NMR Spectroscopy
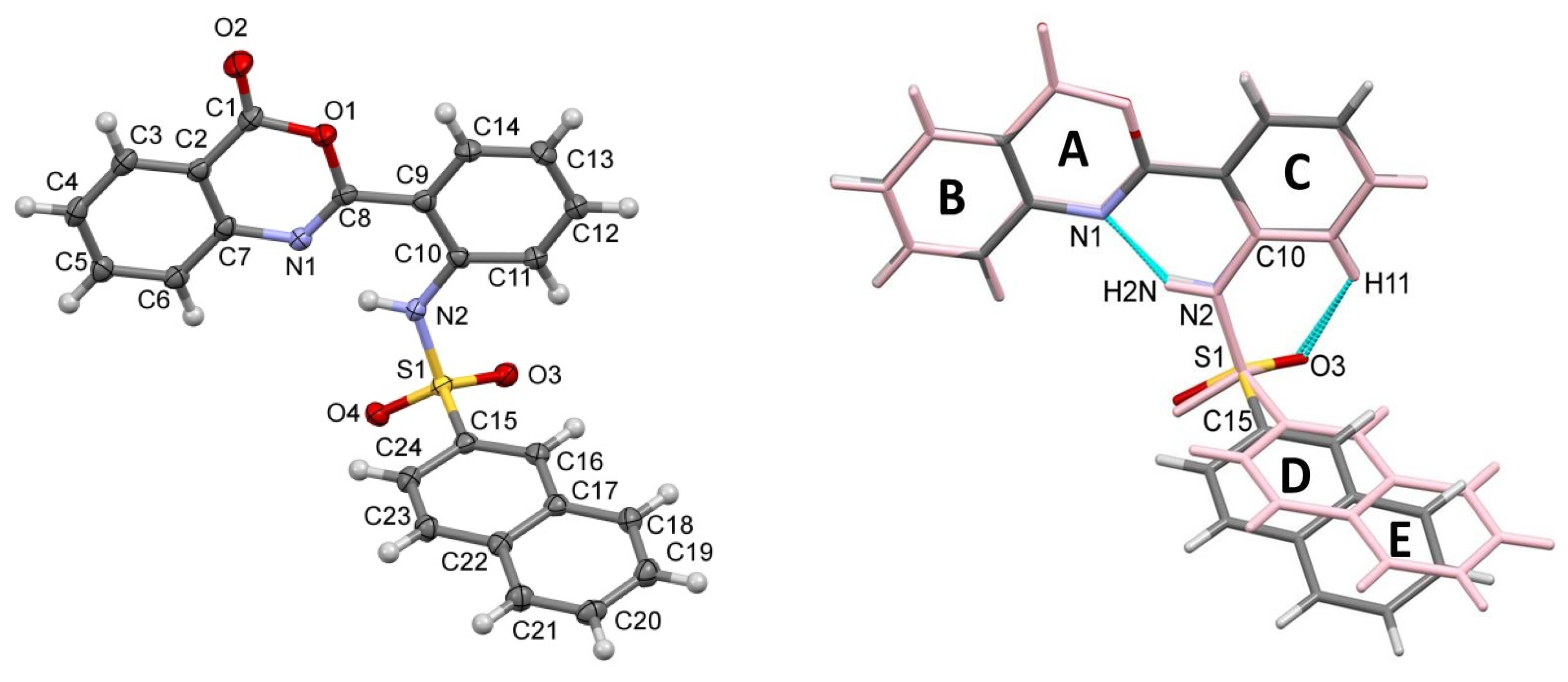
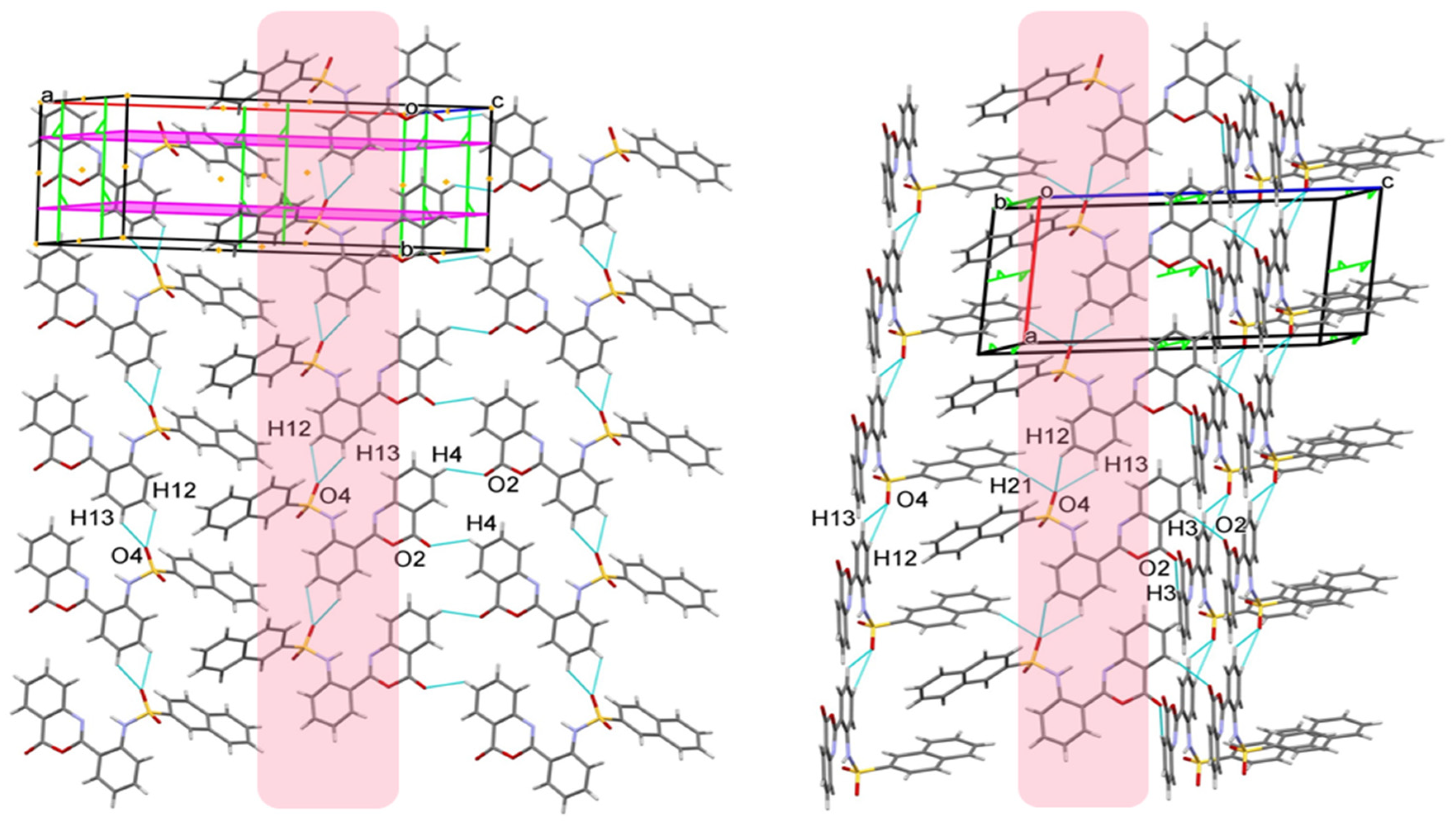
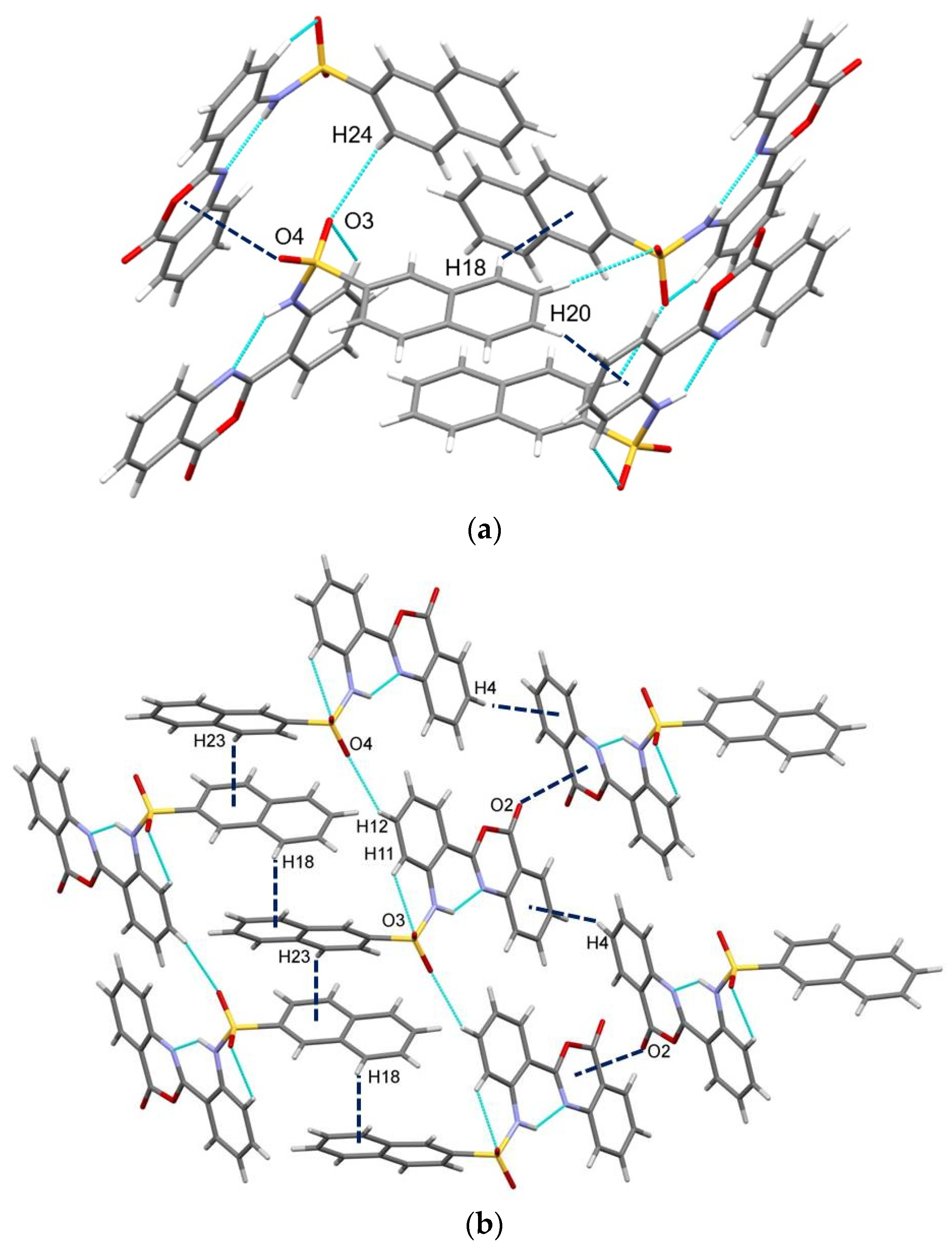
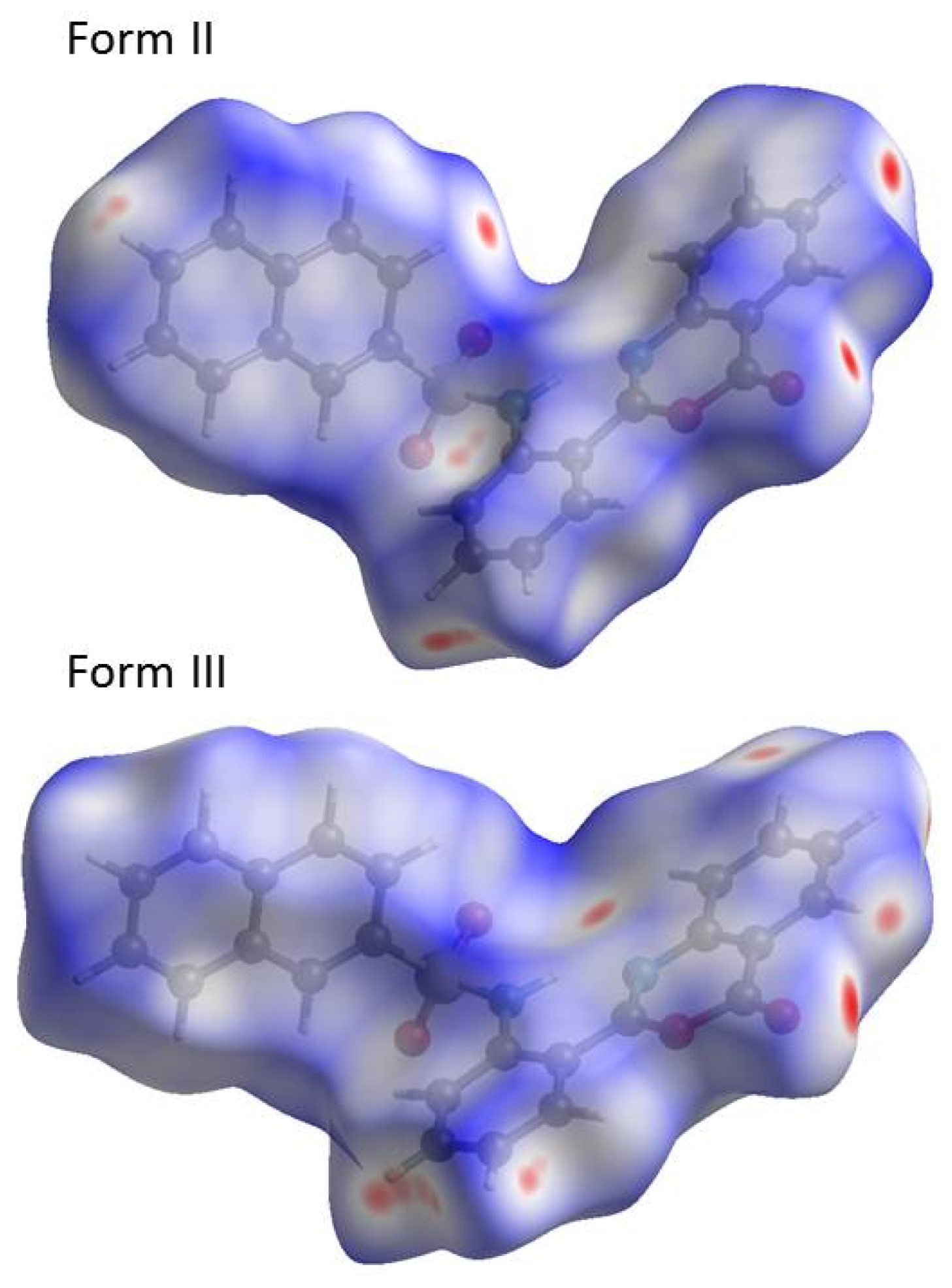
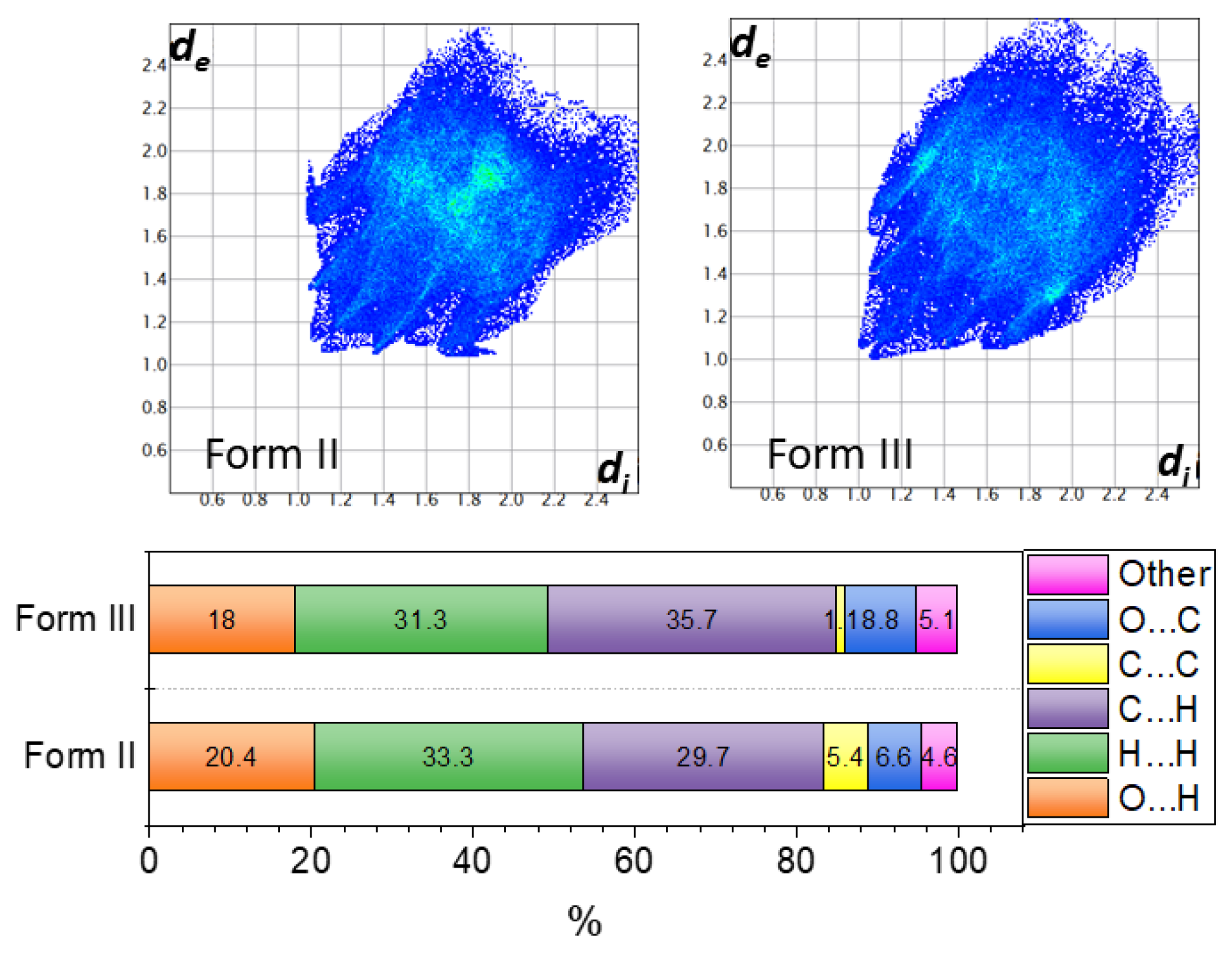
2.3. Similarities and Differences in Single-Crystal Structures of Form II and III
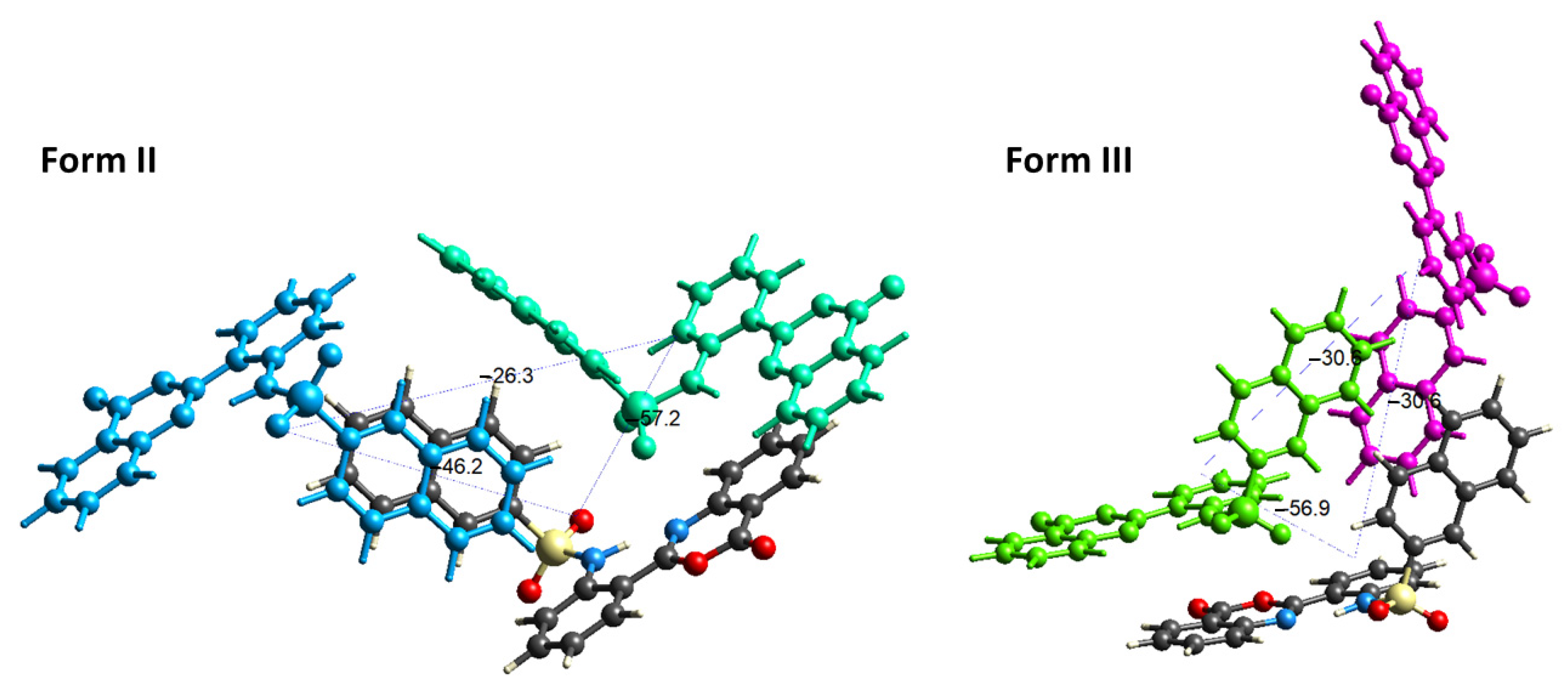
2.4. Interaction Energy and Energy Frameworks
2.5. Characterization of Polymorphic Modifications by Powder XRD, DSC, and FTIR Spectroscopy
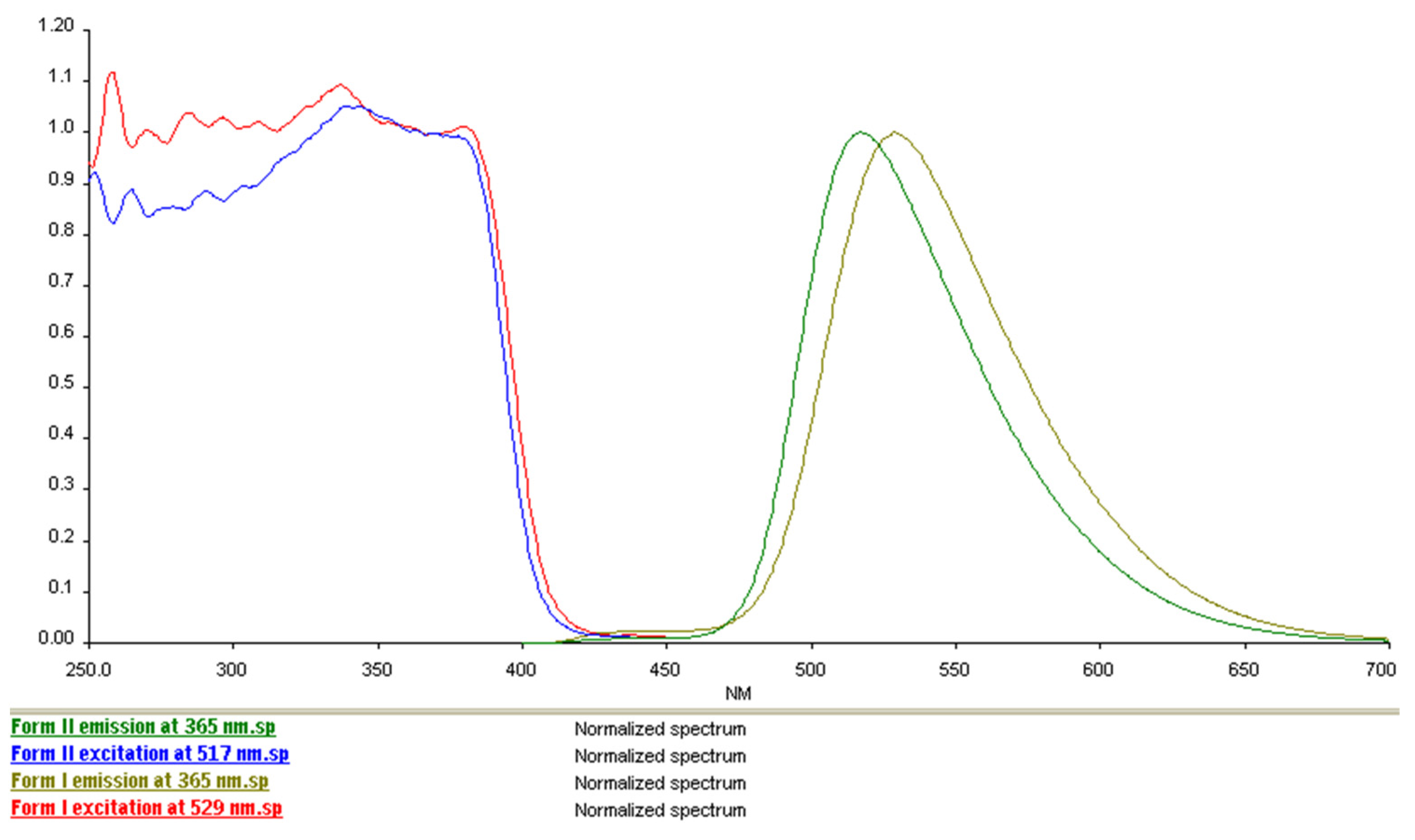
2.6. Comparison of Solid-State Fluorescence of the Polymorphic Forms of Sample 1 (Form I, “UV-Yellowish”) and Sample 2 (Form II, “UV-Greenish”)
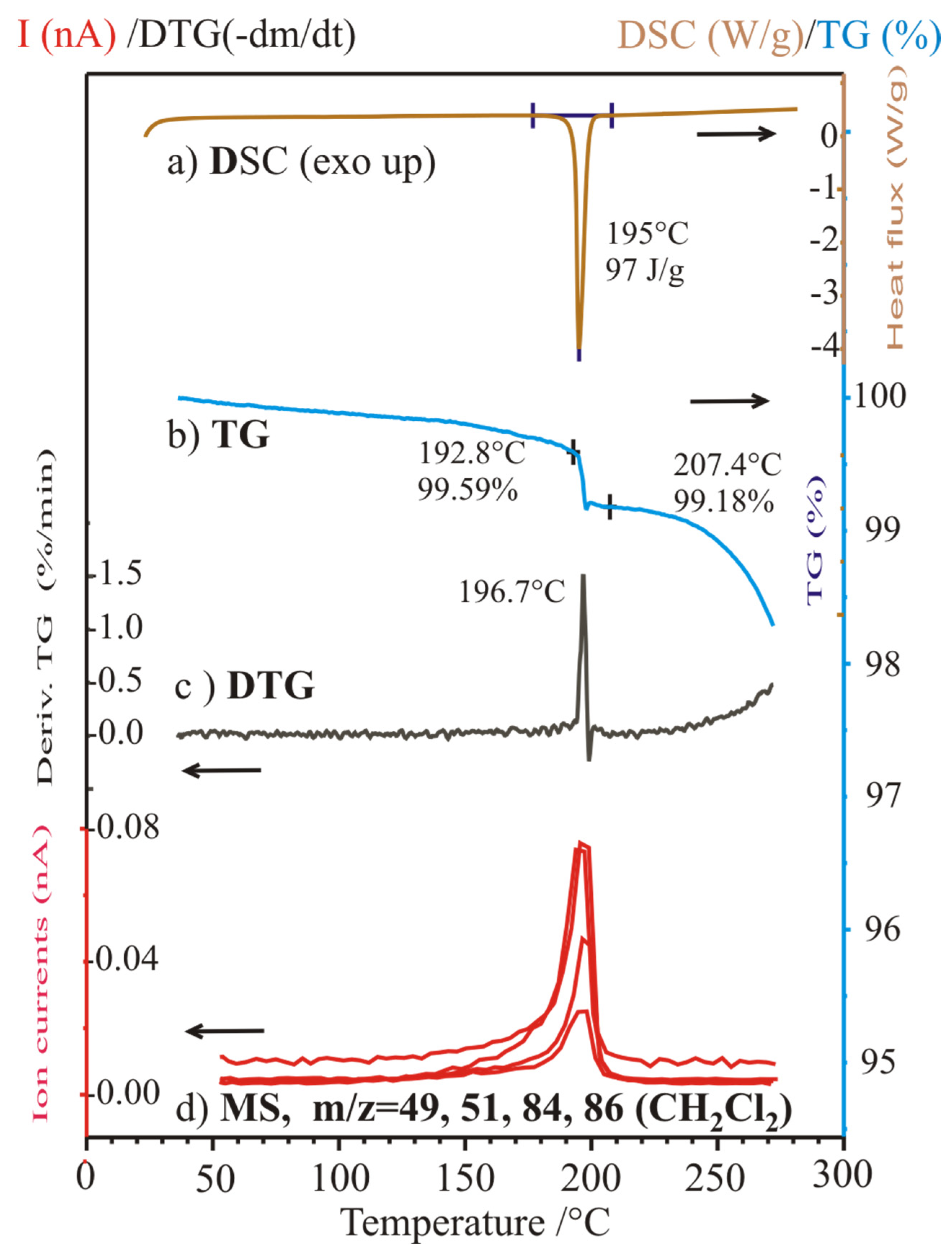
2.7. Simultaneous Thermogravimetric and Differential Thermal Analysis (TG/DTA) Coupled with Quadrupole Mass Spectroscopy for Evolved Gas Analysis of the Polymorphic Forms Sample 2 (Form II, “UV-Greenish”) Prepared in Dichloromethane
3. Materials and Methods
3.1. X-Ray Data Collection, Structure Solution and Refinement
3.2. Hirshfeld Surface Analysis and Energy Calculations
3.3. Further Analytical Methods (CHN, FTIR, NMR, DSC, TG/DTA-EGA-MS, XRD, UV)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CAS SciFinder(n). Literature and Chemical Compound Database. Available online: https://scifinder-n.cas.org/search/substance/66bb509d5d94650a0109de8f/1 (accessed on 15 August 2024).
- Bolotin, B.M.; Ryabokobylko, Y.S.; Drapkina, D.A.; Brudz, V.G. Synthesis and optical properties of 2-phenyl-4H-3,1-benzoxazin-4-one and its derivatives. Tr. Vses. Nauchn.-Issled. Inst. Khim. Reakt. I Osob. Chist. Khim. Veshchestv 1965, 27, 289–301. (In Russian) [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Struct. Sci. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre (CCDC). Crystal Structure Database (CSD), Release v.5.45, + 2 Updates. Available online: https://www.ccdc.cam.ac.uk/solutions/csd/ (accessed on 15 August 2024).
- Filipenko, O.S.; Ponomarev, V.I.; Bolotin, B.M.; Atovmyan, L.O. Crystal and molecular structure of a new organic luminophor 2-(2-tosylaminophenyl)-4H-3,1-benzoxazin-4-one. Crystallogr. Rep. 1979, 24, 957–963. [Google Scholar]
- Bolotin, B.M.; Mikhlina, Y.A.; Arkhipova, S.A.; Kuz’mina, L.G. X-ray diffraction study of crystals of the organic luminophore Orlyum White 520T. Crystallogr. Rep. 2012, 57, 246–251. [Google Scholar]
- Bolotin, B.M.; Drapkina, D.A.; Brudz, V.G.; Sudiyarova, R.U. 2-Aryl-4H-3,1-benzoxazin-4-ones and their analogs. II. Some reactions of o-acylaminobenzamides In Zhurnal Vsesoyuznogo Khimicheskogo Obshchestva im. D. I. Mendeleeva; Izdatel’stvo Khimiya: Moscow, Russia, 1968; Volume 13, pp. 475–476. [Google Scholar]
- Wakankar, D.M.; Hosangadi, B.D. Studies in large ring compounds: Part IV. Reactions of N-substituted anthranilic acids with polyphosphate ester and dicyclohexylcarbodiimide. Indian J. Chem. Sect. B Org. Chem. Incl. Med. Chem. 1978, 16B, 393–395. [Google Scholar] [CrossRef]
- Davis, M.; Hook, R.J.; Wu, W.Y. 2,1-Benzisothiazoles. XIII. Intermediates and products in the sulfur extrusion reaction of 2,1-benzisothiazolin-3(1H)-one. A new synthesis of 11H-pyrido[2,1-b]quinazolin-11-one (“pyracridone”). J. Heterocycl. Chem. 1984, 21, 369–373. [Google Scholar] [CrossRef]
- Khimich, M.N.; Birgen, E.A.; Bolotin, B.M.; Uzhinov, B.M. Intramolecular photoinduced proton transfer in 2-(2-aminophenyl)-4H-3,1-benzoxazin-4-ones with different electron-withdrawing N-substituents. High Energy Chem. 2009, 43, 123–128. [Google Scholar] [CrossRef]
- All-Union Scientific-Research Institute of Chemical Reagents and Pure Chemical Substances. 2-(2-Arylsulfonamidophenyl)-4H-3,1-Benzoxazin-4-Ones. French Patent application FR1392448, 12 March 1965.
- Filipenko, O.S.; Atovmyan, L.O.; Ponomarev, V.I.; Bolotin, B.M. Molecular and crystal structure of the organic luminophor 2-(2-tosylaminophenyl)-6-bromo-4H-3,1-benzoxazin-4-one. Bull. Acad. Sci. USSR Div. Chem. Sci. 1981, 30, 592–595. [Google Scholar] [CrossRef]
- Filipenko, O.S.; Ponomarev, V.I.; Atovmyan, L.O.; Bolotin, B.M. Molecular and crystal structure of organic phosphors 2-(2′-tosylaminophenyl)-6-bromo-4H-3,1-benzoxazin-4-one at 300 K and 2-(2′-tosylamino-4-methoxyphenyl)-4H-3,1-benzoxazin-4-one at 300 and 100 K. Crystallogr. Rep. 1987, 32, 102–112. [Google Scholar]
- Filipenko, O.S.; Bolotin, B.M.; Atovmyan, L.O.; Ponomarev, V.I. Molecular and crystal structure of the organic luminophor 2-(2-tosylamino-5-nitrophenyl)-4H-3,1-benzoxazin-4-one. Sov. Phys. Dokl. 1980, 25, 679. [Google Scholar]
- Utenyshev, A.N.; Filipenko, O.S.; Bolotin, B.M.; Ponomarev, V.I. Low-temperature study of the molecular and crystal structures of 2-(2′-tosylamino-5′-nitrophenyl)-4H-3,1-benzoxazin-4-one, an organic luminophor. Crystallogr. Rep. 2001, 46, 265–270. [Google Scholar] [CrossRef]
- Filipenko, O.S.; Atovmyan, L.O.; Bolotin, B.M.; Ponomarev, V.I. Crystal and molecular structure of the organic luminophor 2-(2-tosylamino-4-nitrophenyl)-4H-3,1-benzoxazin-4-one. Crystallogr. Rep. 1981, 26, 711–716. [Google Scholar]
- Bolotin, B.M.; Kurnosova, L.S.; Koroljkova, O.N.; Drapkina, D.A.; Terskoi, J.A.; Sinjaver, L.G.; Brudz, V.G.; Jarovaja, G.D. Process of Manufacturing Substitued Benzoxazin-4-Ones. Patent Specification GB1070326A, 1 June 1967. Assignee: Vsesojuzny Nauchno-Issledovateljsky Institute, Example No. 2. [Google Scholar]
- Bolotin, B.M.; Kurnosova, L.S.; Koroljkova, O.N.; Drapkina, D.A.; Terskoi, J.A.; Sinjaver, L.G.; Brudz, V.G.; Jarovaja, G.D. Process of Manufacturing Substitutied 2(21-Arylsulphonyl Aminophenyl)-4H,3,1-Benzoxazin-4-Ones. Patent US3335137A2, 8 August 1967. Assignee: Vsesojuzny Nauchno-Issledovateljsky Institute, Example No. 2. [Google Scholar]
- Cevasco, A.A. Substituierte Sulfonamido-Aund Carboxamidophenylchazolone. German (BRD) Patent Application Offenlegegungsschrift. DE2209872A1, 7 September 1972. Assignee: American Cyanamide Co. Beispiel 9. [Google Scholar]
- Cevasco, A.A. Certain 2-(o-Sulfonamidophenyl)-4(3H)-Quinazolineones. U.S. Patent Application USPO 3,740,402, 19 June 1973. Assignee: American Cyanamide Co. Example 9. [Google Scholar]
- Bratscley, R.; Cotterill, P.J.; Haslop, J.M.; Smith, P.N.; Tyson, R.G. Fluorescent Compounds and Their Use. European Patent Application EP0314350A1, 14 October 1988. Assignee Thomas De La Rue & Company Limited, Example 11. [Google Scholar]
- Gizur, T.; Nagy, A.; Derzsi, B. New Industrial Procedure for Production of a Sulfonamide Derivative. Hungarian Patent Application HU230 888 B1, 18 September 2014. Assignee: Első Vegyi Industria ZRt, Budapest, Hungary. [Google Scholar]
- Boultif, A.; Louer, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Cryst. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Higashi, T. NUMABS rev. 2002; Rigaku/MSC Inc.: The Woodlands, TX, USA, 1998. [Google Scholar]
- Rigaku/MSC Inc. CrystalClear SM 1.4.0; Rigaku/MSC Inc.: The Woodlands, TX, USA, 2008. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- SHELXL, version 2013; Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 2013.
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Westrip, S.P. publCIF: Software for editing, validating and formatting crystallographic information files. J. Appl. Crystallogr. 2010, 43, 920–925. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Cryst. B 2004, 60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- Shöniger, W. Analytical procedures for the flask combustion method. In Proceedings of the International Symposium on Microchemistry, Birmingham, UK, 20–27 August 1958; pp. 93–95. [Google Scholar]
- Available online: https://github.com/ccdc-opensource/dash/releases (accessed on 1 January 2023).
- Markvardsen, A.J.; David, W.I.F.; Johnson, J.C.; Shankland, K. A probabilistic approach to space-group determination from powder diffraction data. Acta Crystallogr. Sect. A 2001, 57, 47–54. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; International Union of Crystallography Monographs on Crystallography; Oxford University Press: Oxford, UK, 2002; Chapter 4; pp. 94–150. [Google Scholar]
- Castle, R.N.; Witt, N.F. The Polymorphism of Sulfapyridine. J. Am. Chem. Soc. 1946, 68, 64–66. [Google Scholar] [CrossRef]
- Bar, J.; Bernstein, J. Conformational Polymorphism VI: The Crystal and Molecular Structures of Form II, Form 111, and Form V of 4-Amino-N-2-pyridinylbenzenesulfonamide (Sulfapyridine). J. Pharm. Sci. 1985, 74, 255–263. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorph IV of 4-amino-N-2-pyridinylbenzenesulfonamide (sulfapyridine). Acta Cryst. 1988, C44, 900–902. [Google Scholar] [CrossRef] [PubMed]
- Gelbrich, T.; Threlfall, T.L.; Bingham, A.L.; Hursthouse, M.B. Hursthouse, Polymorph VI of sulfapyridine: Interpenetrating two- and three-dimensional hydrogen-bonded nets formed from two tautomeric forms. Acta Cryst. 2007, C63, o323–o326. [Google Scholar] [CrossRef]
- Pratt, J.; Hutchinson, J.; Stevens, C.L.K. Sulfapyridine (polymorph III), sulfapyridine dioxane solvate, sulfapyridine tetrahydrofuran solvate and sulfapyridine piperidine solvate, all at 173 K. Acta Cryst. 2011, C67, o487–o491. [Google Scholar] [CrossRef]
- Bora, P.; Saikia, B.; Sarma, B. Oriented Crystallization on Organic Monolayers to Control Concomitant Polymorphism. Chem. Eur. J. 2020, 26, 699–710. [Google Scholar] [CrossRef]
- Sovago, I.; Gutmann, M.J.; Grant Hill, J.; Senn, H.M.; Thomas, L.H.; Wilson, C.C.; Farrugia, L.J. Experimental Electron Density and Neutron Diffraction Studies on the Polymorphs of Sulfathiazole. Cryst. Growth Des. 2014, 14, 1227–1239. [Google Scholar] [CrossRef]
- Hughes, D.S.; Hursthouse, M.B.; Threlfall, T.; Tavener, S. A new polymorph of sulfathiazole. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1999, 55, 1831. [Google Scholar] [CrossRef]
- Gelbrich, T.; Hughes, D.S.; Hursthouse, M.B.; Threlfall, T.L. Packing similarity in polymorphs of sulfathiazole. CrystEngComm 2008, 10, 1328–1334. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds, CAS Reg. No., mp(s), [Ref(s)] | Compounds, CSD Code(s), [Ref(s)], SG |
|---|---|
 |  |
| 1a, Ar = Ph, R1 = H (CAS 10128-51-5, 214–215 °C [2], 218–219 °C [7], 219–220 °C [8]) | 2a, R1 = R2 = R3 = H, Ar = 4-MePh (TAPBZO) [5], P-1 |
| 1b, Ar = 4-MePh, R1 = H (CAS 3808-20-6, 220–220.8 °C [2], 219 °C [9], 221 °C [10], 221 °C [11]) | 2b, R1 = Br, R2 = R3 = H, Ar = 4-MePh (BEHHOL [12], BEHHOL10 [13]), P21/a |
| 1c, Ar = β-naphthyl, R1 = H (CAS 10128-55-9, 184.8–185.5 °C [2]) | 2c, R1 = R3 = H, R2 = OCH3, Ar = 4-MePh (FEYJEY and FEYJEY01 [13]), P21/b |
| 1c, Ar = α(β?)-naphthyl, R1 = H (CAS 3779-52-0, 184.8–185.5 °C [11]) | 2d, R1 = R2 = H, R3 = NO2, Ar = 4-MePh (TSBZON [14], TSBZON01 [15]), Pbn21 |
| 1e, Ar = 2,4,6-Me3Ph, R1 = H (CAS 3780-70-9, 223–223.5 °C [2] 224 °C [11]) | 2e, R1 = R3 = H, R2 = NO2, Ar = 4-MePh (TSBZOX [16]), P-1 |
| 1f, Ar = 2-MePh, R1 = H (CAS 10128-56-0, 222–223 °C [2]) | 2f, R1 = R2 = R3 = H, Ar = β-naphthyl (YOCTAO [6]), P21 |
| 1g, Ar = 4-iPrPh, R1 = H (CAS 3808-65-9, 185 °C [11]) | 2g, R1 = R2 = R3 = H, Ar = β-naphthyl, 0.5 p-xylene solvate (YOCSUH [6]), P-1 |
| 1h, Ar = 4-NO2Ph, R1 = H (CAS 10128-57-1, 225.5–226.5 °C [2]) | |
| 1i, Ar = 4-MePh, R1 = CH3 (CAS 10129-14-3, 176–177 °C [2]) |
 | 13C Chemical Shifts Peak No. | Sample 1 δ (ppm) | Sample 2 δ (ppm) |
| 1 | 157.7 | 157.7 | |
| 2 | 157.0 | 157.0 | |
| 3 | 144.8 | 144.7 | |
| 4 | 139.5 | 139.5 | |
| 5 | 137.0 | 137.0 | |
| 6 | 136.2 | 136.2 | |
| 7 | 134.8 | 134.8 | |
| 8 | 134.0 | 133.9 | |
| 9 | 131.8 | 131.8 | |
| 10 | 129.6 | 129.6 | |
| 11 | 129.4 | 129.4 | |
| 12–13, not resolved | 129.1 | 129.1 | |
| 14 | 128.9 | 128.9 | |
| 15 | 128.8 | 128.7 | |
| 16 | 128.7 | 128.7 | |
| 17 | 127.8 | 127.8 | |
| 18 | 127.5 | 127.5 | |
| 19 | 126.5 | 126.5 | |
| 20 | 123.3 | 123.3 | |
| 21 | 121.9 | 121.9 | |
| 22 | 119.4 | 119.4 | |
| 23 | 116.5 | 116.5 | |
| 24 | 115.4 | 115.4 |
| Form II | Form III | |
|---|---|---|
| Empirical formula | C24H16N2O4S | C24H16N2O4S |
| Formula weight (g/mol) | 428.45 | 428.45 |
| Crystal color and shape | Colorless, Platelet | Colorless, Block |
| Temperature (K) | 103(2) | 103(2) |
| Radiation and wavelength (Å) | Mo-Kα, 0.71073 | Mo-Kα, 0.71073 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/c | P21 |
| Unit cell dimensions a | ||
| a (Å) | 20.2155(10) | 8.7804(7) |
| b (Å) | 8.8822(4) | 5.7461(6) |
| c (Å) | 11.0016(4) | 19.3481(19) |
| β (°) | 103.6160(10) | 100.425(2) |
| Volume (Å3) | 1919.91(15) | 960.06(16) |
| Z/Z’ | 4/1 | 2/1 |
| Density (calc.) (mg m−3) | 1.482 | 1.482 |
| Absorption coefficient, μ (mm−1) | 0.206 | 0.206 |
| F(000) | 888 | 444 |
| Crystal size (mm) | 0.50 × 0.30 × 0.10 | 0.50 × 0.12 × 0.12 |
| Absorption correction | Numerical | Numerical |
| Max. and min. transmission | 0.9297, 0.9774 | 0.9543, 0.9781 |
| θ?range for data collection (°) | 3.006 ≤ θ ≤ 27.473 | 3.212 ≤ θ ≤ 25.350 |
| Index ranges | −26 ≤ h ≤ 26; −11 ≤ k ≤ 11; −14 ≤ l ≤ 14 | −10 ≤ h ≤ 10; −6 ≤ k ≤ 6; −23 ≤ l ≤ 23 |
| Reflections collected | 42568 | 12343 |
| Completeness to 2θ | 0.999 | 0.997 |
| Independent reflections (Rint) | 4392, 0.0645 | 3510, 0.0924 |
| Reflections I > 2σ(I) | 3339 | 2514 |
| Refinement method | full-matrix least-squares on F2 | full-matrix least-squares on F2 |
| Data/restraints/parameters | 4392/0/280 | 3510/0/280 |
| Goodness-of-fit b, on F2 | 1.095 | 1.048 |
| Final R indices c,d [I > 2σ(I)] (R1, wR2) | 0.0425, 0.1016 | 0.0551, 0.1069 |
| R indices c,d (all data) (R1, wR2) | 0.0589, 0.1098 | 0.0881, 0.1249 |
| Max. and mean shift/esd | 0.000; 0.000 | 0.000; 0.000 |
| Largest diff. peak and hole (e.Å−3) | 0.370; −0.304 | 0.340; −0.347 |
| D—X…A | D—X (Å) | X…A (Å) | D…A (Å) | D—X…A (°) | Symmetry Code |
|---|---|---|---|---|---|
| Form II | |||||
| N2-H2N…N1 | 0.88 | 1.94 | 2.654(2) | 137 | intra |
| C11-H11…O3 | 0.95 | 2.31 | 2.988(2) | 128 | intra |
| C14-H14…O1 | 0.95 | 2.66 | 2.295(2) | 102 | intra |
| C16-H16…O3 | 0.95 | 2.91 | 2.554(2) | 103 | intra |
| C4-H4…O2 | 0.95 | 2.51 | 3.270(2) | 138 | −x, −1/2 + y, 3/2 − z |
| C12-H12…O4 | 0.95 | 2.51 | 3.130(2) | 123 | x, 1 + y, z |
| C13-H13…O4 | 0.95 | 2.61 | 3.168(2) | 118 | x, 1 + y, z |
| C24-H24…O3 | 0.95 | 2.54 | 3.249(2) | 132 | x, 3/2 − y, 1/2 + z |
| C18-H18…Cg(D) a | 0.95 | 2.84 | 3.6457(19) | 143 | 1 − x, 1/2 + y, 1/2 − z |
| C20-H20…Cg(C) a | 0.95 | 2.58 | 3.512(2) | 167 | 1 − x, 2 − y, 1 − z |
| S1-O4…Cg(A) a | 1.4317(12) | 3.0870(14) | 3.8940(8) | 113.92(6) | x, 3/2 − y, −1/2 + z |
| Form III | |||||
| N2-H2N…N1 | 0.88 | 1.95 | 2.665(7) | 137 | intra |
| C11-H11…O3 | 0.95 | 2.45 | 3.092(7) | 125 | intra |
| C14-H14…O1 | 0.95 | 2.67 | 2.309(7) | 102 | intra |
| C16-H16…O3 | 0.95 | 2.84 | 2.438(7) | 105 | intra |
| C3-H3…O2 | 0.95 | 2.62 | 3.564(7) | 171 | 1 − x, −1/2 + y,1 − z |
| C12-H12…O4 | 0.95 | 2.52 | 3.105(6) | 120 | 1 + x, y, z |
| C13-H13…O4 | 0.95 | 2.56 | 3.133(7) | 119 | 1 + x, y, z |
| C21-H21…O4 | 0.95 | 2.64 | 3.485(7) | 149 | −x, −1/2 + y, − z |
| C4-H4…Cg(B) a | 0.95 | 2.97 | 3.819(6) | 149 | −x, −1/2 + y, 1 − z |
| C18-H18…Cg(E) a | 0.95 | 2.77 | 3.514(6) | 135 | 1−x, 1/2 + y, − z |
| C23-H23…Cg(D) a | 0.95 | 2.83 | 3.551(6) | 134 | −x, −1/2 + y, − z |
| C1-O2…Cg(A) a | 1.196(7) | 3.050(4) | 4.063(6) | 142.3(4) | 1−x, −1/2 + y, 1 − z |
| Unit Cell Parameters | Form I, Powder Pattern Indexing at RT, by DASH [This Work] | Form II, Powder Pattern Indexing at RT, by DASH [This Work] | Form II Single-Crystal Structure Determination at T = 103 K [This Work] | Form III Single-Crystal Structure Determination at T = 103 K [This Work] | Form III Single-Crystal Structure Determination at T = 173 K [6] |
|---|---|---|---|---|---|
| space group | P 21/a No. 14 | P 21/c No. 14 | P 21/c No. 14 | P 21 No. 4 | P 21 No. 4 |
| a (Å) | 21.945 | 20.269 | 20.2155(10) | 8.7804(7) | 8.778(4) |
| b (Å) | 10.128 | 8.9526 | 8.8822(4) | 5.7461(6) | 5.771(3) |
| c (Å) | 9.0333 | 11.205 | 11.0016(4) | 19.3481(19) | 19.308(9) |
| β (°) | 95.461 | 102.84 | 103.6160(10) | 100.425(2) | 100.446(7) |
| V (Å3) | 1998.7 | 1982.5 | 1919.91(15) | 960.06(16) | 962.0(8) |
| Z/Z′ | 4/1 | 4/1 | 4/1 | 2/1 | 2/1 |
| zero shift (°) | 0.0798 | 0.0145 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madarász, J.; May, N.V.; Bombicz, P.; Pokol, G.; Kocsis, R.; Hegymegi-Barakonyi, B.; Gizur, T. The Polymorphism of Orlyum White 520T, an Ultraviolet Luminescent Security Ink. Molecules 2025, 30, 1671. https://doi.org/10.3390/molecules30081671
Madarász J, May NV, Bombicz P, Pokol G, Kocsis R, Hegymegi-Barakonyi B, Gizur T. The Polymorphism of Orlyum White 520T, an Ultraviolet Luminescent Security Ink. Molecules. 2025; 30(8):1671. https://doi.org/10.3390/molecules30081671
Chicago/Turabian StyleMadarász, János, Nóra V. May, Petra Bombicz, György Pokol, Richárd Kocsis, Bálint Hegymegi-Barakonyi, and Tibor Gizur. 2025. "The Polymorphism of Orlyum White 520T, an Ultraviolet Luminescent Security Ink" Molecules 30, no. 8: 1671. https://doi.org/10.3390/molecules30081671
APA StyleMadarász, J., May, N. V., Bombicz, P., Pokol, G., Kocsis, R., Hegymegi-Barakonyi, B., & Gizur, T. (2025). The Polymorphism of Orlyum White 520T, an Ultraviolet Luminescent Security Ink. Molecules, 30(8), 1671. https://doi.org/10.3390/molecules30081671