Benzothiazolines Acting as Carbanion and Radical Transfer Reagents in Carbon–Carbon Bond Construction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
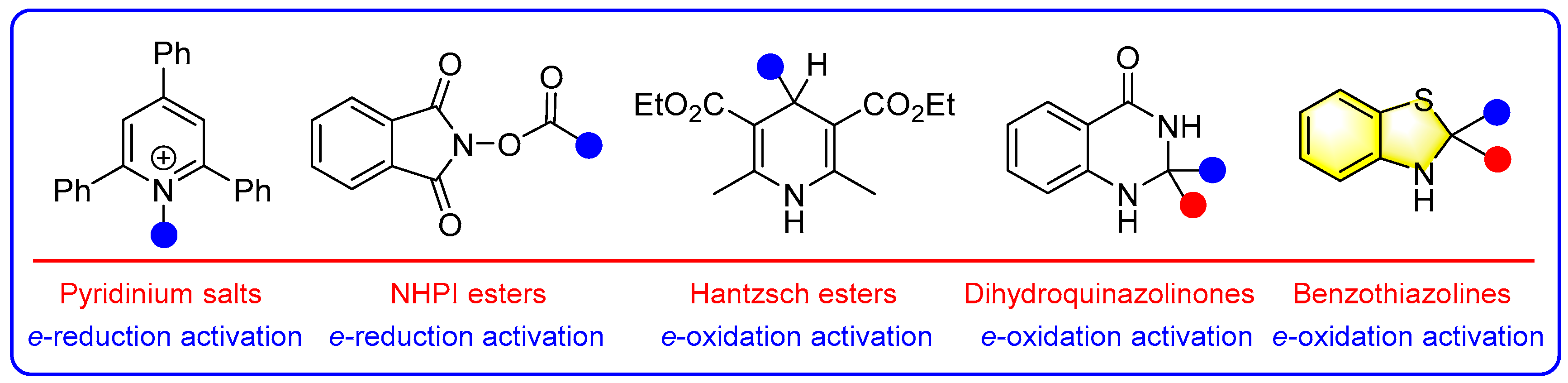
1. Introduction
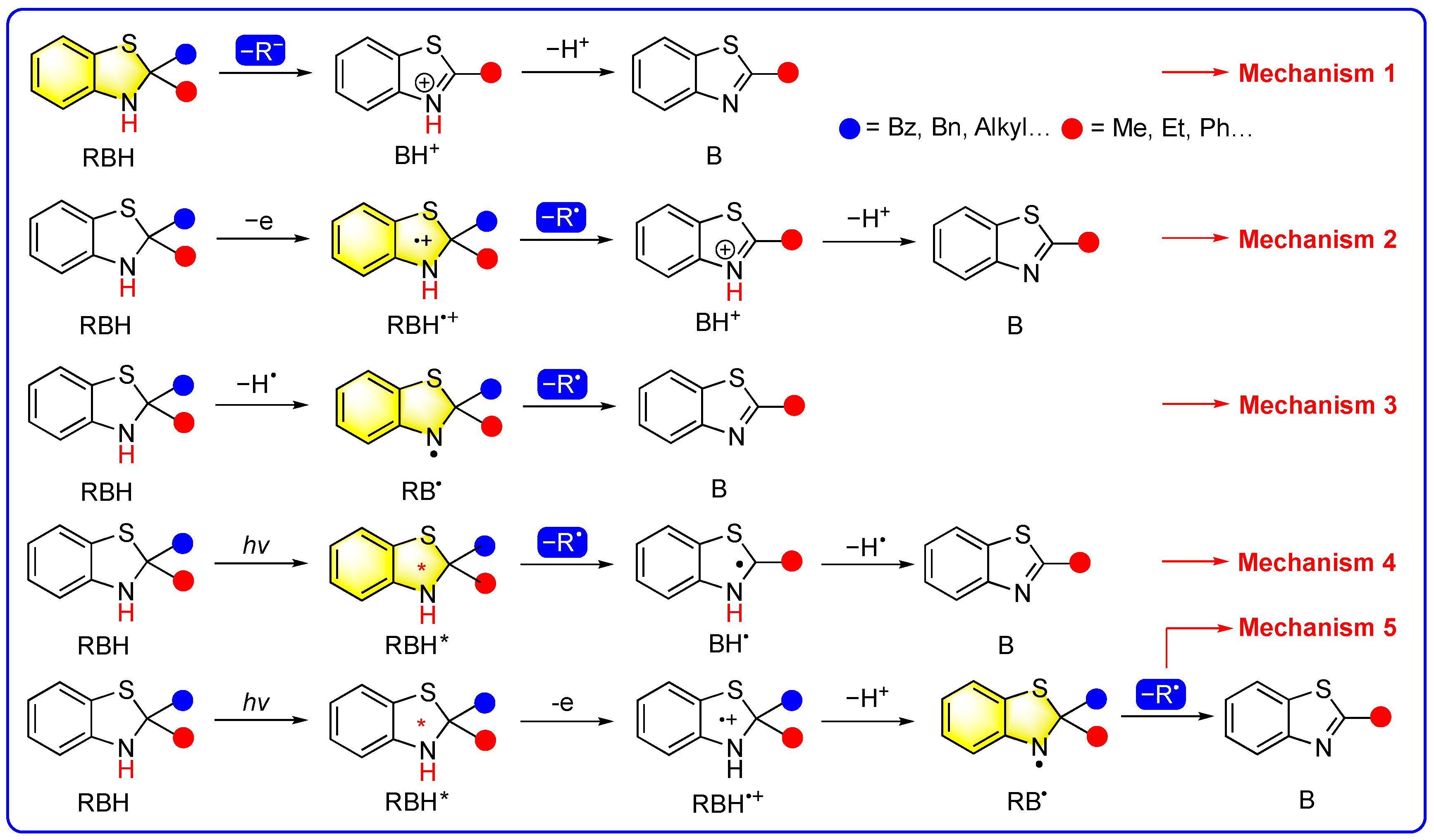
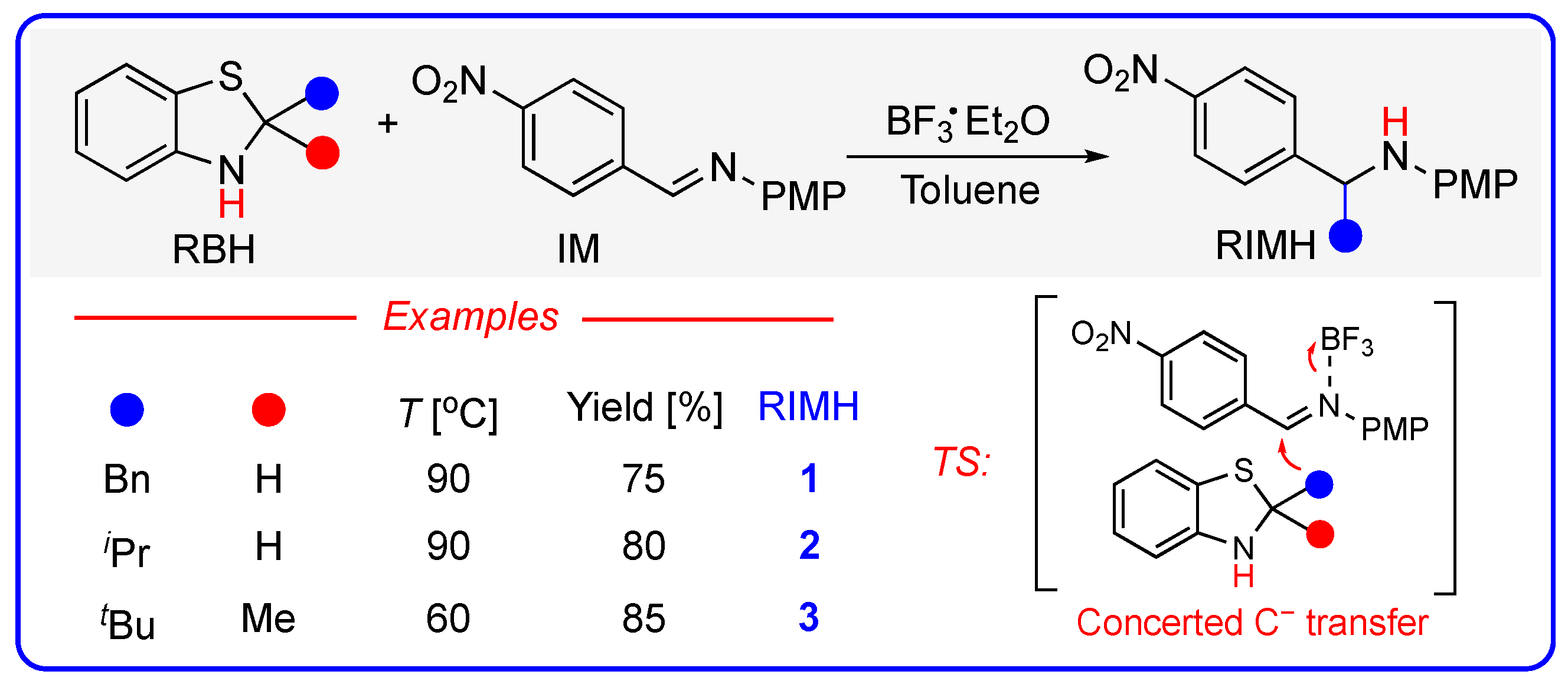
2. Mechanism 1: Benzothiazolines Directly Acting as Carbanion Transfer Reagents
3. Mechanism 2: Benzothiazolines Function as Radical Transfer Reagents Through a Sequential e + R• +H+ Mechanism with RBH•+ Serving as the Radical Precursors
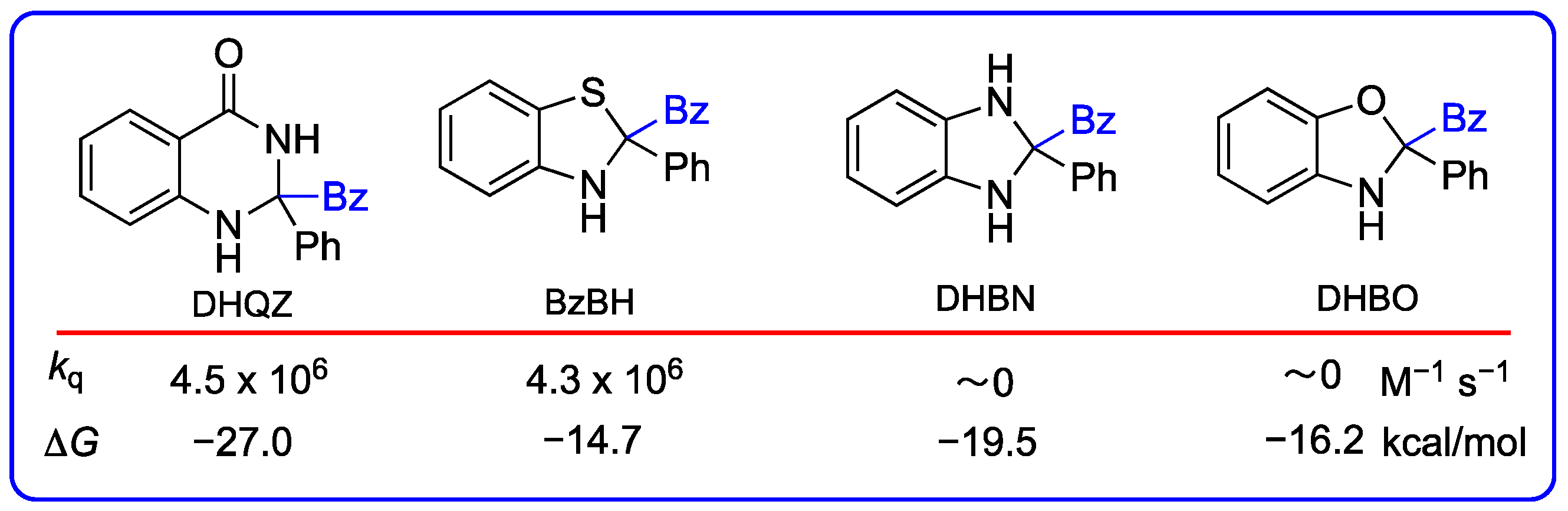
3.1. Evaluation of Benzothiazolines as Radical Transfer Reagents Through Stern–Volmer Quenching Experiments and DFT Calculations
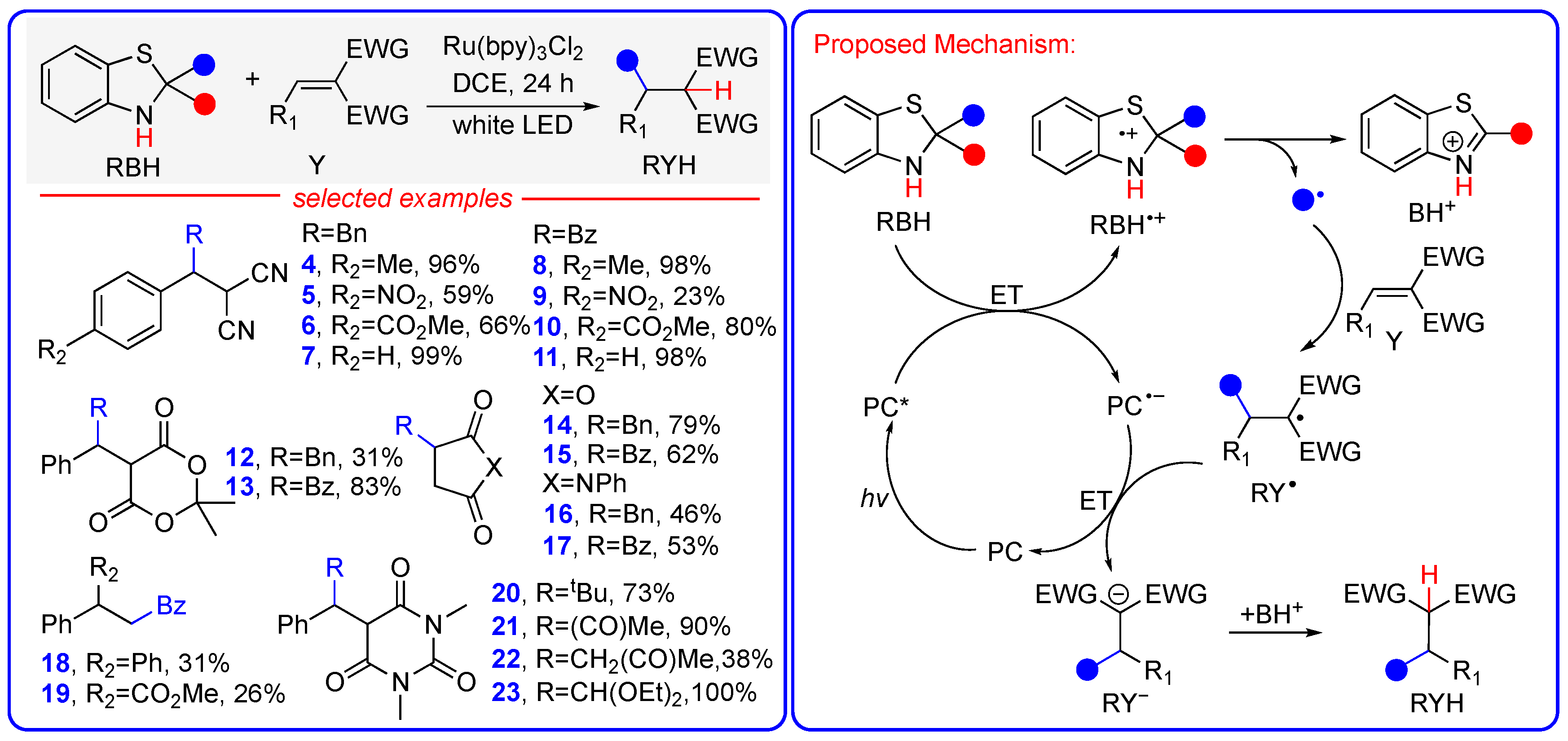
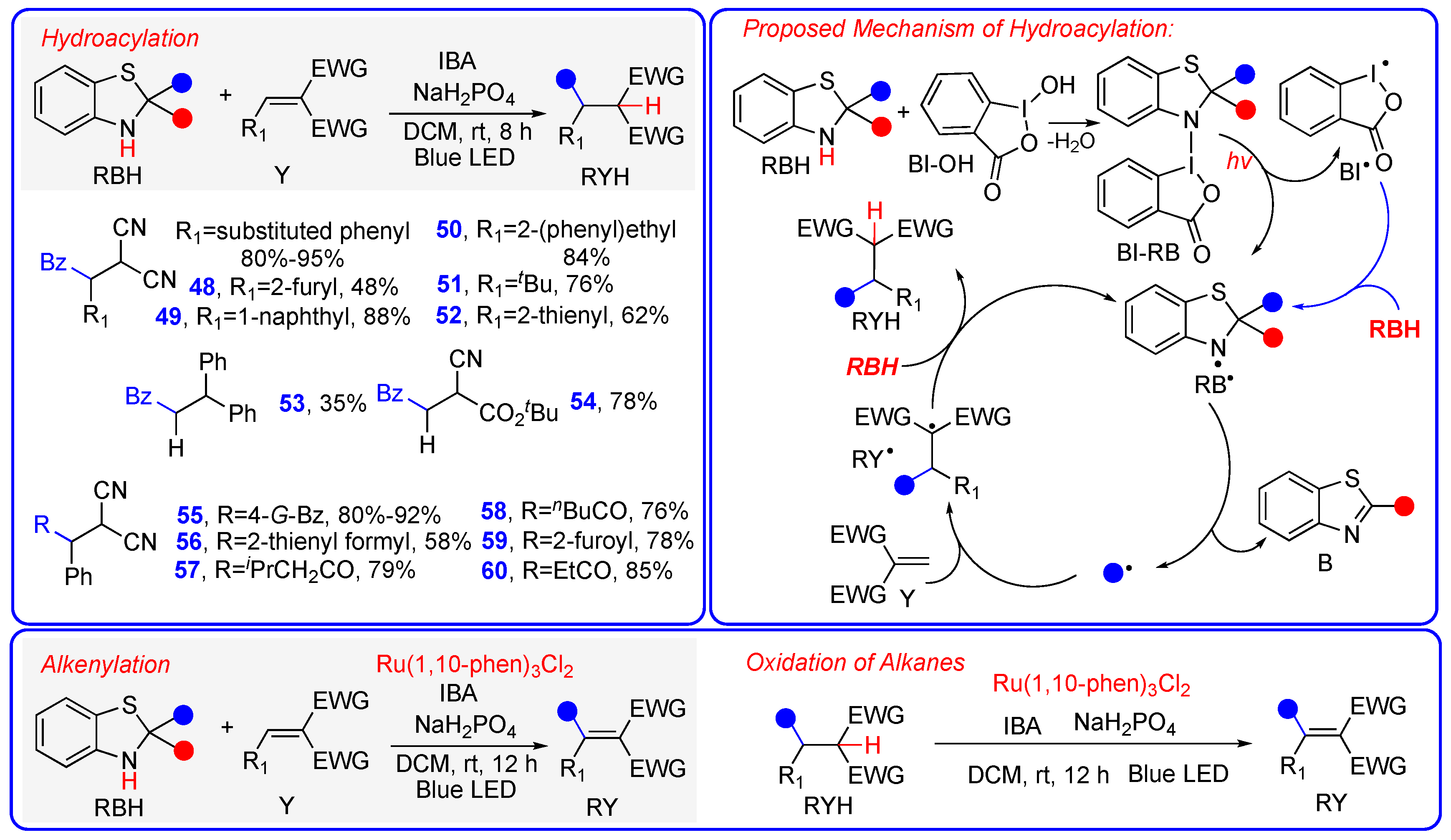
3.2. Hydroalkylation and Hydroacylation of Michael Acceptors via Photo-Promoted Radical Addition Reaction
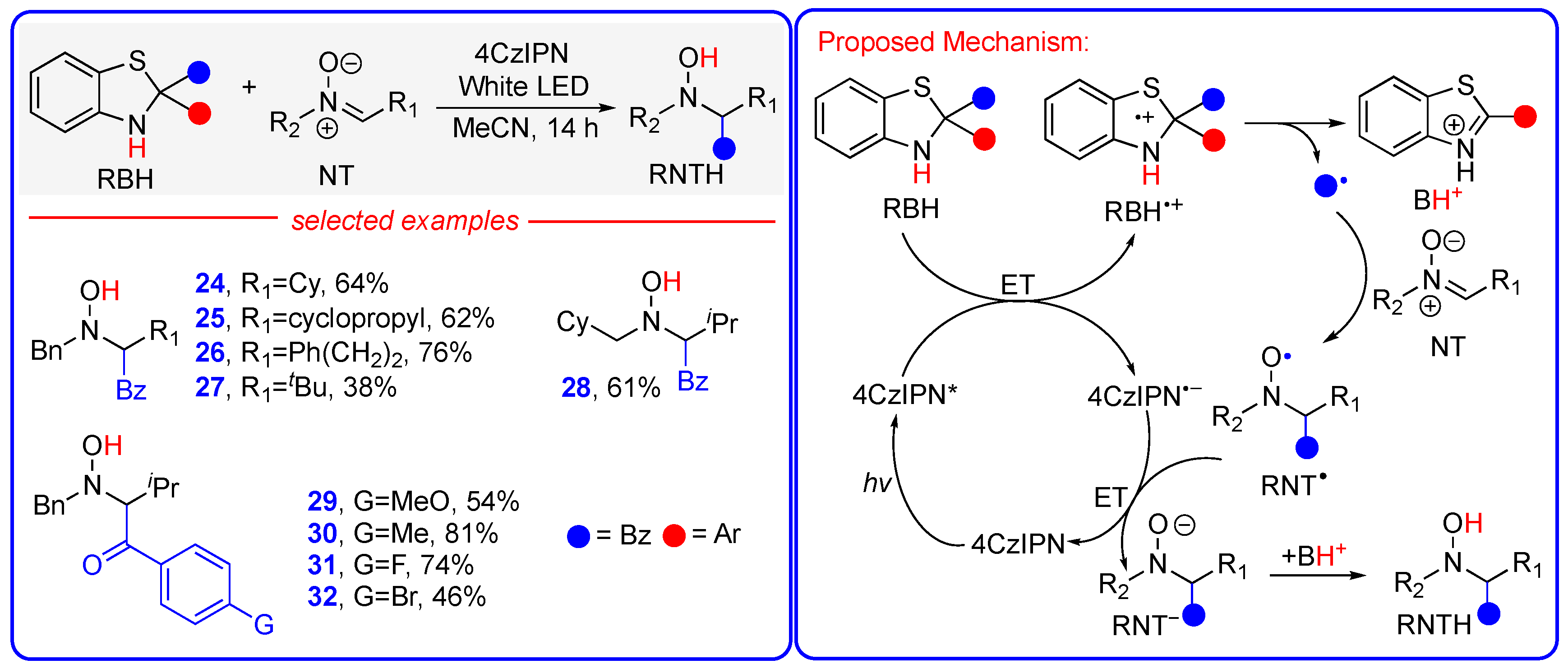
3.3. Metal- and Additive-Free Hydroacylation of Nitrones to Synthetize α-Hydroxyamino Ketones Enabled by Organic Photocatalyst
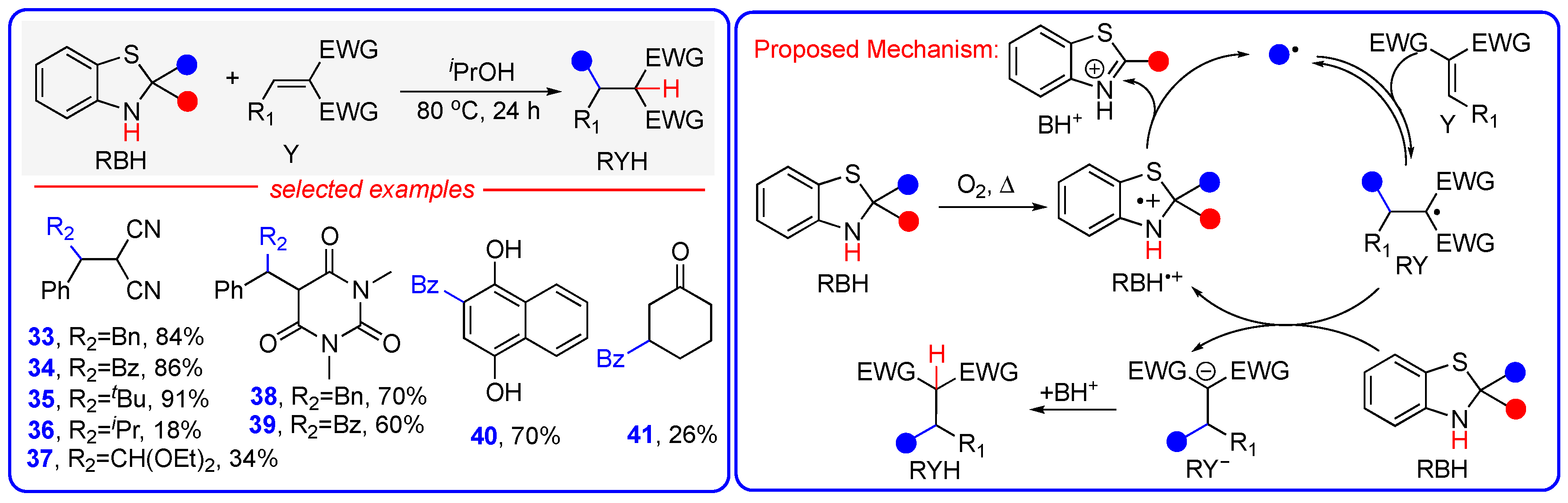
3.4. Photo- and Additive-Free Hydroalkylation and Hydroacylation of Electron-Deficient Alkenes Under Thermal Condition
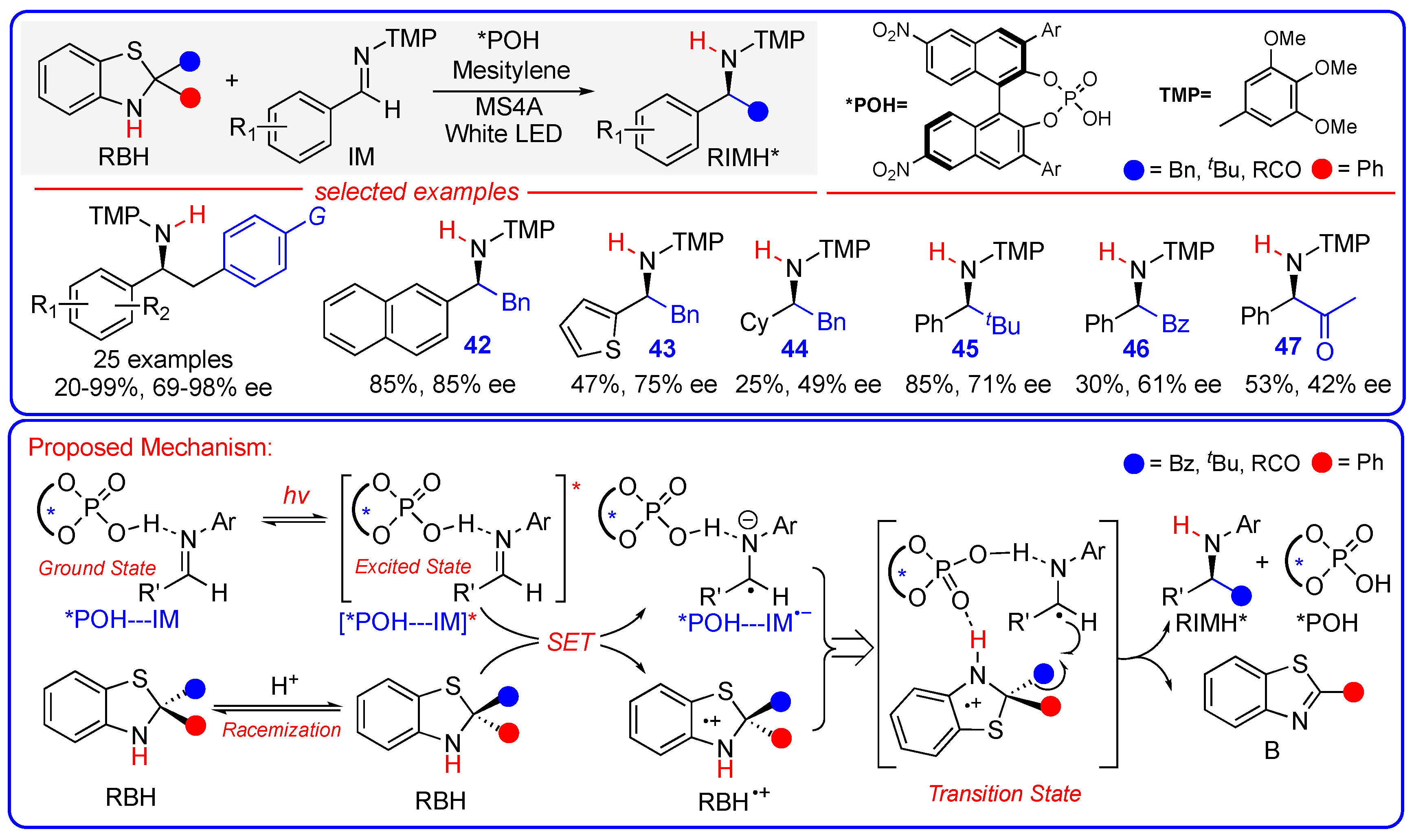
3.5. Enantioselective Radical Addition of Imines Driven by the Photoexcitation of a Chiral Acid Catalyst−Imine Complex
4. Mechanism 3: Benzothiazolines Function as Radical Transfer Reagents Through a Sequential H• + R• Mechanism with RB• Serving as the Actual Radical Precursors
Acyl Radical Alkylation, Alkenylation, and Alkynylation Driven by Visible Light
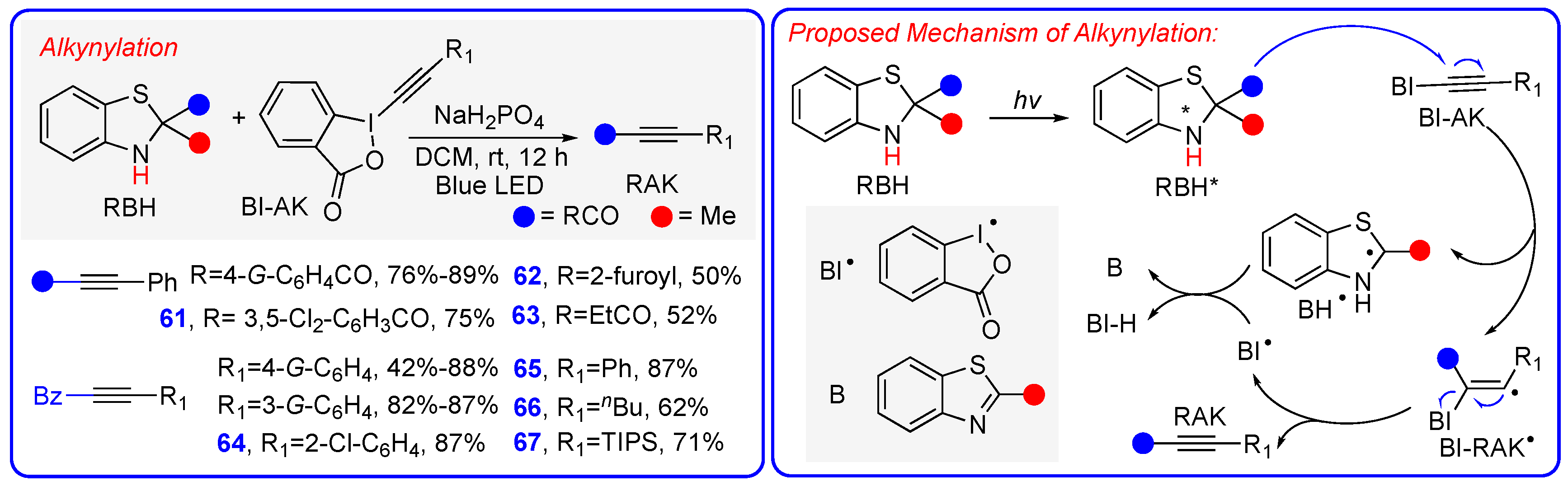
5. Mechanism 4: Photoexcited Benzothiazolines Directly Function as Radical Precursors Through a Sequential Photoactivation + R• + H• Mechanism
Radical Alkynylation Using Photoexcited Benzothiazolines as Direct Radical Precursors
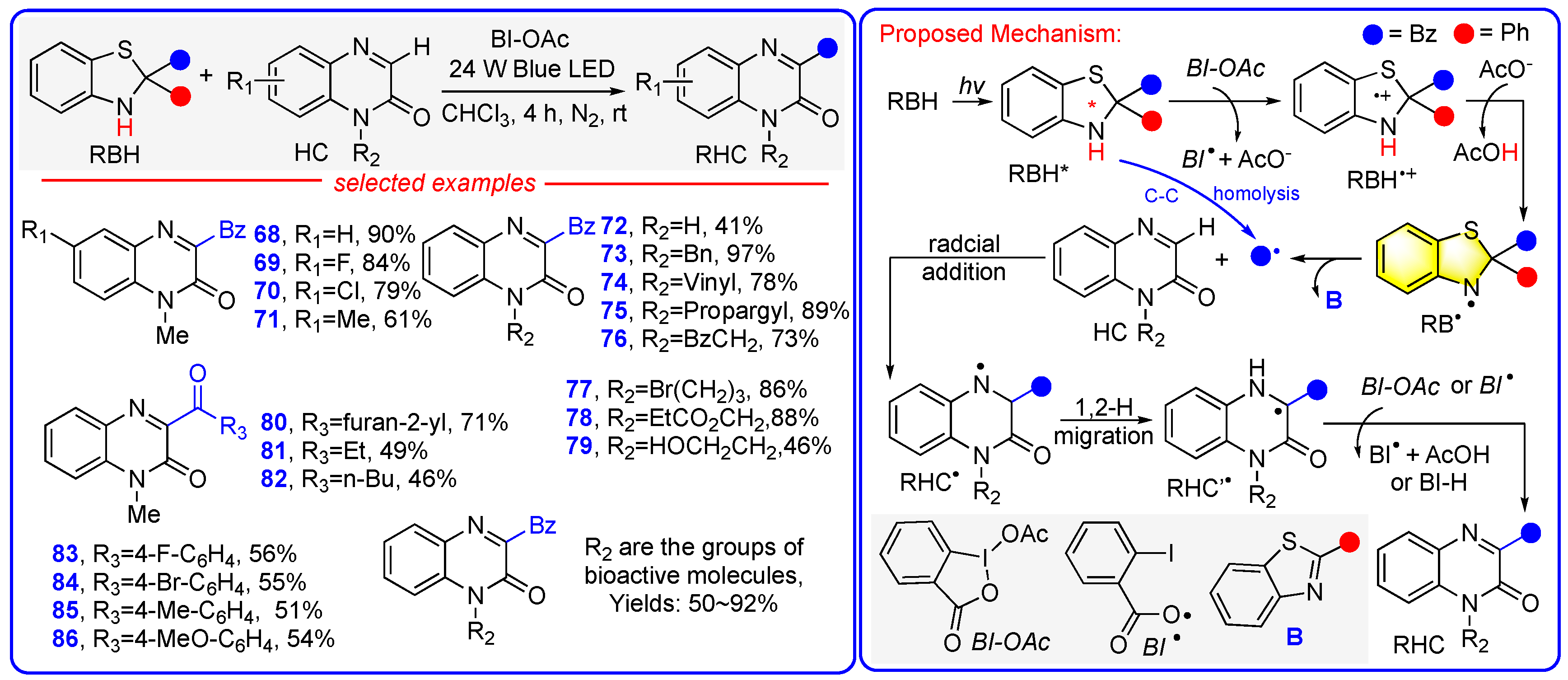
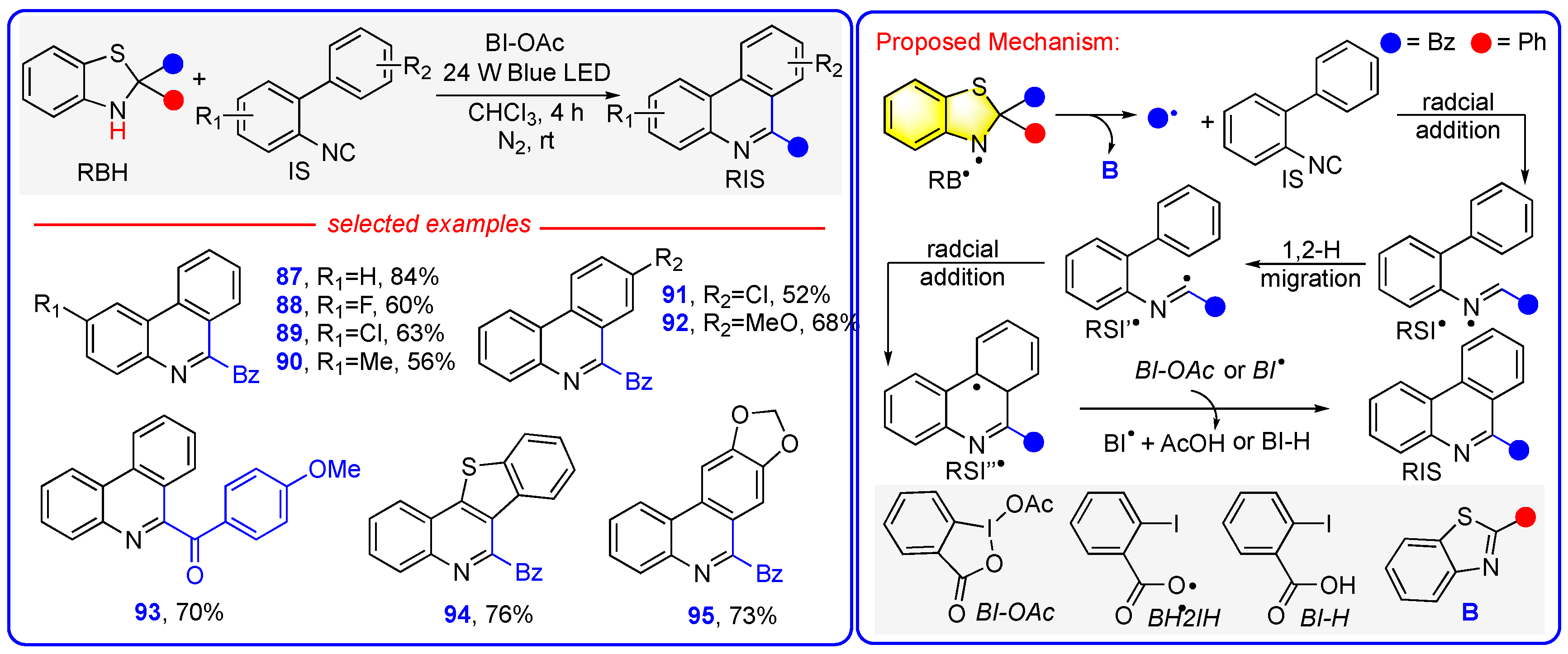
6. Mechanism 5: Benzothiazolines Function as Radical Transfer Reagents Through a Sequential Photoactivation + e + H+ + R• Mechanism with RB• Serving as the Actual Radical Precursors
Photocatalyst-Free Acylation of Quinoxaline-2-Ones and Isonitriles to Access Heterocycles Enabled by Direct Photoexcitation of Benzothiazolines
7. Limitations and Challenges in Benzothiazoline Chemistry
7.1. Mechanistic Ambiguities and Competing Pathways
7.2. Scalability and Practical Constraints
7.3. Functional Group Limitations and Substrate Scope
7.4. Environmental and Economic Considerations
7.5. Asymmetric Synthesis and Stereocontrol
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lan, J.; You, J. Oxidative C–H/C–H Coupling Reactions between Two (Hetero)arenes. Chem. Rev. 2017, 117, 8787–8863. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.K.; Guin, S.; Maiti, S.; Biswas, J.P.; Porey, S.; Maiti, D. Toolbox for Distal C-H Bond Functionalizations in Organic Molecules. Chem. Rev. 2022, 122, 5682–5841. [Google Scholar] [CrossRef] [PubMed]
- Budiman, Y.P.; Perutz, R.N.; Steel, P.G.; Radius, U.; Marder, T.B. Applications of Transition Metal-Catalyzed ortho-Fluorine-Directed C-H Functionalization of (Poly)fluoroarenes in Organic Synthesis. Chem. Rev. 2024, 124, 4822–4862. [Google Scholar] [CrossRef]
- Liu, B.; Romine, A.M.; Rubel, C.Z.; Engle, K.M.; Shi, B.-F. Transition-Metal-Catalyzed, Coordination-Assisted Functionalization of Nonactivated C(sp3)-H Bonds. Chem. Rev. 2021, 121, 14957–15074. [Google Scholar] [CrossRef]
- Wu, X.; Ma, Z.; Feng, T.; Zhu, C. Radical-Mediated Rearrangements: Past, Present, and Future. Chem. Soc. Rev. 2021, 50, 11577–11613. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D.; Fagnoni, M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C-H Bonds Elaboration. Chem. Rev. 2022, 122, 1875–1924. [Google Scholar] [CrossRef]
- Zaera, F. Designing Sites in Heterogeneous Catalysis: Are We Reaching Selectivities Competitive with Those of Homogeneous Catalysts? Chem. Rev. 2022, 122, 8594–8757. [Google Scholar] [CrossRef]
- Maity, B.; Dutta, S.; Cavallo, L. The Mechanism of Visible Light-Induced C-C Cross-Coupling by Csp3-H Bond Activation. Chem. Soc. Rev. 2023, 52, 5373–5387. [Google Scholar] [CrossRef]
- Holmberg-Douglas, N.; Nicewicz, D.A. Photoredox-Catalyzed C-H Functionalization Reactions. Chem. Rev. 2022, 122, 1925–2016. [Google Scholar] [CrossRef]
- Murray, P.R.D.; Cox, J.H.; Chiappini, N.D.; Roos, C.B.; McLoughlin, E.A.; Hejna, B.G.; Nguyen, S.T.; Ripberger, H.H.; Ganley, J.M.; Tsui, E.; et al. Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis. Chem. Rev. 2022, 122, 2017–2291. [Google Scholar] [CrossRef]
- Cheung, K.P.S.; Sarkar, S.; Gevorgyan, V. Visible Light-Induced Transition Metal Catalysis. Chem. Rev. 2022, 122, 1543–1625. [Google Scholar] [CrossRef]
- Xiao, W.-J.; Zhou, Q.-Q.; Zou, Y.-Q.; Lu, L.-Q. Visible-Light-Induced Organic Photochemical Reactions via Energy Transfer Pathways. Angew. Chem. Int. Ed. 2018, 58, 1586–1604. [Google Scholar]
- Genzink, M.J.; Kidd, J.B.; Swords, W.B.; Yoon, T.P. Chiral Photocatalyst Structures in Asymmetric Photochemical Synthesis. Chem. Rev. 2022, 122, 1654–1716. [Google Scholar] [CrossRef] [PubMed]
- Mai, H.; Le, T.C.; Chen, D.; Winkler, D.A.; Caruso, R.A. Machine Learning for Electrocatalyst and Photocatalyst Design and Discovery. Chem. Rev. 2022, 122, 13478–13515. [Google Scholar] [CrossRef]
- Huang, Y.; Hayashi, T. Chiral Diene Ligands in Asymmetric Catalysis. Chem. Rev. 2022, 122, 14346–14404. [Google Scholar] [CrossRef] [PubMed]
- Großkopf, J.; Kratz, T.; Rigotti, T.R.; Bach, T. Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer. Chem. Rev. 2022, 122, 1626–1653. [Google Scholar] [CrossRef]
- Imamoto, T. P-Stereogenic Phosphorus Ligands in Asymmetric Catalysis. Chem. Rev. 2024, 124, 8657–8739. [Google Scholar] [CrossRef]
- Steinlandt, P.S.; Zhang, L.; Meggers, E. Metal Stereogenicity in Asymmetric Transition Metal Catalysis. Chem. Rev. 2023, 123, 4764–4794. [Google Scholar] [CrossRef]
- Yang, H.; Yu, H.; Stolarzewicz, I.A.; Tang, W. Enantioselective Transformations in the Synthesis of Therapeutic Agents. Chem. Rev. 2023, 123, 9397–9446. [Google Scholar] [CrossRef]
- Nagib, D.A. Asymmetric Catalysis in Radical Chemistry. Chem. Rev. 2022, 122, 15989–15992. [Google Scholar] [CrossRef]
- Mondal, S.; Dumur, F.; Gigmes, D.; Sibi, M.P.; Bertrand, M.P.; Nechab, M. Enantioselective Radical Reactions Using Chiral Catalysts. Chem. Rev. 2022, 122, 5842–5976. [Google Scholar] [CrossRef]
- Wang, X.; He, J.; Wang, Y.-N.; Zhao, Z.; Jiang, K.; Yang, W.; Zhang, T.; Jia, S.; Zhong, K.; Niu, L.; et al. Strategies and Mechanisms of First-Row Transition Metal-Regulated Radical C-H Functionalization. Chem. Rev. 2024, 124, 10192–10280. [Google Scholar] [CrossRef] [PubMed]
- Docherty, J.H.; Lister, T.M.; Mcarthur, G.; Findlay, M.T.; Domingo-Legarda, P.; Kenyon, J.; Choudhary, S.; Larrosa, I. Transition-Metal-Catalyzed C-H Bond Activation for the Formation of C-C Bonds in Complex Molecules. Chem. Rev. 2023, 123, 7692–7760. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Akana, M.E.; Sanders, K.M.; Weix, D.J. A Decarbonylative Approach to Alkylnickel Intermediates and C(sp3)-C(sp3) Bond Formation. Science 2024, 385, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-X.; Yin, S.-Y.; Zhao, F.; Yang, H.; Feng, Z.; Gu, Q.; You, S.-L. Rhodium-Catalyzed Asymmetric C-H Functionalization Reactions. Chem. Rev. 2023, 123, 10079–10134. [Google Scholar] [CrossRef]
- Connon, R.; Roche, B.; Rokade, B.V.; Guiry, P.J. Further Developments and Applications of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2021, 121, 6373–6521. [Google Scholar] [CrossRef]
- Bhunia, A.; Studer, A. Recent Advances in Radical Chemistry Proceeding through Pro-Aromatic Radicals. Chem 2021, 7, 2060–2100. [Google Scholar] [CrossRef]
- Carson, W.P.; Tsymbal, A.V.; Pipal, R.W.; Edwards, G.A.; Martinelli, J.R.; Cabré, A.; MacMillan, D.W.C. Free-Radical Deoxygenative Amination of Alcohols via Copper Metallaphotoredox Catalysis. J. Am. Chem. Soc. 2024, 146, 15681–15687. [Google Scholar] [CrossRef]
- Patra, T.; Bellotti, P.; Strieth-Kalthoff, F.; Glorius, F. Photosensitized Intermolecular Carboimination of Alkenes through the Persistent Radical Effect. Angew. Chem. Int. Ed. 2020, 59, 3172–3177. [Google Scholar] [CrossRef]
- Zhang, Z.; Gevorgyan, V. Visible Light-Induced Reactions of Diazo Compounds and Their Precursors. Chem. Rev. 2024, 124, 7214–7261. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; An, Q.; Duan, L.; Feng, K.; Zuo, Z. Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev. 2022, 122, 2429–2486. [Google Scholar] [CrossRef]
- Kwon, K.; Simons, R.T.; Nandakumar, M.; Roizen, J.L. Strategies to Generate Nitrogen-centered Radicals That May Rely on Photoredox Catalysis: Development in Reaction Methodology and Applications in Organic Synthesis. Chem. Rev. 2022, 122, 2353–2428. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-Q.; Ge, D.; Cui, Y.-Y.; Shen, Z.-L.; Li, C.-J. Desulfonylation via Radical Process: Recent Developments in Organic Synthesis. Chem. Rev. 2021, 121, 12548–12680. [Google Scholar] [CrossRef]
- He, F.-S.; Ye, S.; Wu, J. Recent Advances in Pyridinium Salts as Radical Reservoirs in Organic Synthesis. ACS Catal. 2019, 9, 8943–8960. [Google Scholar] [CrossRef]
- Shi, C.; Zhong, N.; Guo, L.; Yin, J.; Yang, C.; Xia, W. Light-Driven N-Heterocyclic Carbene-Catalyzed Multi-Component Reaction for the Synthesis of β-Amino Ketones. Org. Lett. 2024, 26, 8848–8853. [Google Scholar] [CrossRef]
- Wang, T.-C.; Zhang, Z.; Rao, G.; Li, J.; David, J.S.R.; Zhu, B.Q.; Yang, Y. Threonine Aldolase-Catalyzed Enantioselective α-Alkylation of Amino Acids through Unconventional Photoinduced Radical Initiation. J. Am. Chem. Soc. 2024, 146, 22476–22484. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, R.; Truong, L.; Hedouin, M.; Oulyadi, H.; Schiavi, B.; Jean, A.; Jubault, P.; Poisson, T. Electron Donor-Acceptor Complex Photoactivation for Deaminative Alkynylation, Alkenylation and Allenylation: A Comprehensive Study. Org. Chem. Front. 2024, 11, 2231–2240. [Google Scholar] [CrossRef]
- Han, G.; You, J.; Choi, J.; Kang, E.J. N-Iminopyridinium Compounds in Giese Reaction: Photoinduced Homolytic N-N and C-C Bond Cleavage for Cyanoalkyl Radical Generation. Org. Lett. 2024, 26, 2414–2419. [Google Scholar] [CrossRef]
- Lee, G.S.; Hong, S.H. Direct C(sp3)-H Acylation by Mechanistically Controlled Ni/Ir Photoredox Catalysis. Acc. Chem. Res. 2023, 56, 2170–2184. [Google Scholar] [CrossRef]
- Dash, R.; Panda, S.P.; Bhati, K.S.; Sharma, S.; Murarka, S. Electrochemical C–H Alkylation of Azauracils Using N-(Acyloxy)phthalimides. Org. Lett. 2024, 26, 7227–7232. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Cornella, J.; Li, C.; Malins, L.R.; Edwards, J.T.; Kawamura, S.; Maxwell, B.D.; Eastgate, M.D.; Baran, P.S. A general Alkyl-Alkyl Cross-Coupling Enabled by Redox-Active Esters and Alkylzinc Reagents. Science 2016, 352, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, S.; Wu, X. Iron/Photoredox Dual-Catalyzed Redox-Neutral Double Decarboxylative C(sp3)-C(sp3) Cross-Coupling. Green Chem. 2024, 26, 11334–11339. [Google Scholar] [CrossRef]
- Xie, K.A.; Bednarova, E.; Joe, C.L.; Sherwood, T.C.; Welin, E.R.; Rovis, T. A Unified Method for Oxidative and Reductive Decarboxylative Arylation with Orange Light-Driven Ir/Ni Metallaphotoredox Catalysis. J. Am. Chem. Soc. 2024, 146, 25780–25787. [Google Scholar] [CrossRef]
- Xia, H.; Jiang, X.; Lin, D.; Zhang, S.; Yu, Z.; Wu, X.; Qu, J.; Chen, Y. Catalytic Asymmetric Barbier Reaction of Ketones with Unactivated Alkyl Electrophiles. J. Am. Chem. Soc. 2024, 146, 28468–28481. [Google Scholar] [CrossRef]
- Mühlfenzl, K.S.; Enemærke, V.J.; Gahlawat, S.; Golbækdal, P.I.; Munksgaard-Ottosen, N.; Neumann, K.T.; Hopmann, K.H.; Norrby, P.-O.; Elmore, C.S.; Skrydstrup, T. Nickel Catalyzed Carbonylative Cross Coupling for Direct Access to Isotopically Labeled Alkyl Aryl Ketones. Angew. Chem. Int. Ed. 2024, 63, e202412247. [Google Scholar] [CrossRef]
- Manoharan, K.; Bieszczad, B. Acyl-1,4-Dihydropyridines: Universal Acylation Reagents for Acyl-1,4-Dihydropyridines: Universal Acylation Reagents for Organic Synthesis. Molecules 2024, 29, 3844. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. Hantzsch Esters: An Emerging Versatile Class of Reagents in Photoredox Catalyzed Organic Synthesis. Org. Biomol. Chem. 2019, 17, 6936–6951. [Google Scholar] [CrossRef]
- Ye, S.; Wu, J. 4-Substituted Hantzsch Esters as Alkylation Reagents in Organic Synthesis. Acta Chim. Sin. 2019, 77, 814–831. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, Z.; Wang, J.; Zhu, L.; Li, C. Facile Synthesis of Alkylphosphonates from 4-Alkyl-1,4-Dihydropyridines via Photoinduced Formal Deformylative Phosphonylation. Org. Chem. Front. 2024, 11, 4502–4507. [Google Scholar] [CrossRef]
- Li, Q.; Wang, L.-C.; Bao, Z.-P.; Wu, X.-F. Photoredox-Catalyzed Carbonylative Acylation of Styrenes with Hantzsch Esters. Chem. Commun. 2024, 60, 4656–4658. [Google Scholar] [CrossRef]
- Li, Q.-Z.; He, M.-Z.; Zeng, R.; Lei, Y.-Y.; Yu, Z.-Y.; Jiang, M.; Zhang, X.; Li, J.-L. Molecular Editing of Ketones through N-Heterocyclic Carbene and Photo Dual Catalysis. J. Am. Chem. Soc. 2024, 146, 22829–22839. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.-J.; Zhang, J.-H.; Li, W.; Yang, W.; Xin, H.; Gao, P.; Duan, X.-H.; Guo, L.-N. Aromatization-driven Deconstructive Functionalization of Spiro Dihydroquinazolinones via Dual Photoredox/Nickel Catalysis. Chem. Sci. 2024, 15, 8993–8999. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Z.; Gao, F.; Yan, X. Homologation of Ketones: Direct Transformation of Alkyl Ketones to Aryl Ketones via Photoredox Catalyzed Deacylation-Aroylation Sequence. Org. Lett. 2024, 26, 6915–6920. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-P.; Miao, H.-J.; Liu, L.; Duan, X.-H.; Guo, L.-N. Visible Light-Promoted Aromatization-Driven Deconstructive Fluorination of Spiro Carbocycles. Org. Lett. 2024, 26, 7442–7446. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Miao, H.-J.; Zhang, J.-H.; Duan, X.-H.; Guo, L.-N. Copper-Catalyzed Aromatization-Driven Ring-Opening Amination and Oxygenation of Spiro Dihydroquinazolinones. Chem. Eur. J. 2024, 30, e202402602. [Google Scholar] [CrossRef]
- Li, G.; Chen, R.; Wu, L.; Fu, Q.; Zhang, X.; Tang, Z. Alkyl Transfer from C-C Cleavage. Angew. Chem. Int. Ed. 2013, 52, 8432–8436. [Google Scholar] [CrossRef]
- Uchikura, T.; Moriyama, K.; Toda, M.; Mouri, T.; Ibáñez, I.; Akiyama, T. Benzothiazolines as Radical Transfer Reagents: Hydroalkylation and Hydroacylation of Alkenes by Radical Generation Under Photoirradiation Conditions. Chem. Commun. 2019, 55, 11171–11174. [Google Scholar] [CrossRef]
- Li, L.; Guo, S.; Wang, Q.; Zhu, J. Acyl Radicals from Benzothiazolines: Synthons for Alkylation, Alkenylation, and Alkynylation Reactions. Org. Lett. 2019, 21, 5462–5466. [Google Scholar] [CrossRef]
- Uchikura, T.; Toda, M.; Mouri, T.; Fujii, T.; Moriyama, K.; Ibáñez, I.; Akiyama, T. Radical Hydroalkylation and Hydroacylation of Alkenes by Use of Benzothiazoline Under Thermal Conditions. J. Org. Chem. 2020, 85, 12715–12723. [Google Scholar] [CrossRef]
- He, X.-K.; Lu, J.; Ye, H.-B.; Li, L.; Xuan, J. Direct Photoexcitation of Benzothiazolines: Acyl Radical Generation and Application to Access Heterocycles. Molecules 2021, 26, 6843. [Google Scholar] [CrossRef]
- Uchikura, T.; Kamiyama, N.; Mouri, T.; Akiyama, T. Visible-Light-Driven Enantioselective Radical Addition to Imines Enabled by the Excitation of a Chiral Phosphoric Acid−Imine Complex. ACS Catal. 2022, 12, 5209–5216. [Google Scholar] [CrossRef]
- Lee, S.-C.; Li, L.-Y.; Tsai, Z.-N.; Lee, Y.-H.; Tsao, Y.-T.; Huang, P.-G.; Cheng, C.-K.; Lin, H.-B.; Chen, T.-W.; Yang, C.-H.; et al. Aromatization as an Impetus to Harness Ketones for Metallaphotoredox-Catalyzed Benzoylation/Benzylation of (Hetero)arenes. Org. Lett. 2022, 24, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Yamanomoto, K.; Matsudaira, T.; Akiyama, T. Metal- and Additive-Free Synthesis of α-Hydroxyamino Ketones Enabled by Organophotocatalyst. Synlett 2023, 34, 1391–1394. [Google Scholar]
- Zhu, C.; Saito, K.; Yamanaka, M.; Akiyama, T. Benzothiazoline: Versatile Hydrogen Donor for Organocatalytic Transfer Hydrogenation. Acc. Chem. Res. 2015, 48, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Miñoza, S.; Librando, I.L.; Liao, H.-H. Ketone-Derived Pro-aromatic Reagents for Radical Group Transfer Reactions and Deconstructive Functionalizations. Synlett 2024, 35, 1072–1088. [Google Scholar]
- Phillips, A.M.F.; Pombeiro, A.J.L. Applications of Hantzsch Esters in Organocatalytic Enantioselective Synthesis. Catalysts 2023, 13, 419. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors. Chem. Soc. Rev. 2012, 41, 2498–2518. [Google Scholar] [CrossRef]
- Rueping, M.; Dufour, J.; Schoepke, F.R. Advances in Catalytic Metal-free Reductions: From Bio-inspired Concepts to Applications in the Organocatalytic Synthesis of Pharmaceuticals and Natural Products. Green Chem. 2011, 13, 1084–1105. [Google Scholar] [CrossRef]
- Lan, H.; Liu, Y.; Ackermann, L.; Wang, L.; Wang, D. Ruthenium(II)-Catalyzed Remote C−H Alkylation of Arenes Using Diverse N-Directing Groups through Aziridine Ring Opening. Org. Lett. 2024, 26, 7993–7998. [Google Scholar] [CrossRef]
- Wang, Z.-J.; Chen, J.-J.; Huang, H.-M. Site-Selective Pyridine Carbamoylation Enabled by Consecutive Photoinduced Electron Transfer. ACS Catal. 2024, 14, 15521–15527. [Google Scholar] [CrossRef]
- Laudadio, G.; Deng, Y.; van der Wal, K.; Ravelli, D.; Nuño, M.; Fagnoni, M.; Guthrie, D.; Sun, Y.; Noël, T. C(sp3)-H Functionalizations of Light Hydrocarbons Using Decatungstate Photocatalysis in Flow. Science 2020, 369, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-C.; Zhang, L.; Zhang, G.-S.; Fu, Y.-H.; Shen, G.-B. Thermodynamic Evaluation on Alkoxyamines of TEMPO Derivatives, Stable Alkoxyamines or Potential Radical Donors? ChemistrySelect 2022, 7, e202204144. [Google Scholar] [CrossRef]
- Bryden, M.A.; Zysman-Colman, E. Organic Thermally Activated Delayed Fluorescence (TADF) Compounds Used in Photocatalysis. Chem. Soc. Rev. 2021, 50, 7587–7680. [Google Scholar] [CrossRef]
- Guo, Y.; Lin, G.; Zhang, M.; Xu, J.; Song, Q. Photo-induced Decarboxylative C-S Bond Formation to Access Sterically Hindered Unsymmetric S-Alkyl Thiosulfonates and SS-Alkyl Thiosulfonates. Nat. Commun. 2024, 15, 7313. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xue, G.-H.; He, Z.; Yue, J.-P.; Pan, M.; Song, L.; Zhang, W.; Ye, J.-H.; Yu, D.-G. Visible-Light Photoredox-Catalyzed Direct Carboxylation of Tertiary C(sp3)−H Bonds with CO2: Facile Synthesis of All-Carbon Quaternary Carboxylic Acids. J. Am. Chem. Soc. 2024, 146, 28350–28359. [Google Scholar] [CrossRef]
- Shen, G.-B.; Qian, B.-C.; Fu, Y.-H.; Zhu, X.-Q. Thermodynamics of the Elementary Steps of Organic Hydride Chemistry Determined in Acetonitrile and their Applications. Org. Chem. Front. 2022, 9, 6001–6062. [Google Scholar] [CrossRef]
- Wu, Y.; Kim, D.; Teets, T.S. Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations. Synlett 2022, 33, 1154–1179. [Google Scholar]
- Murahashi, S.-I.; Imada, Y. Synthesis and Transformations of Nitrones for Organic Synthesis. Chem. Rev. 2019, 119, 4684–4716. [Google Scholar] [CrossRef]
- Cabré, A.; Verdaguer, X.; Riera, A. Recent Advances in the Enantioselective Synthesis of Chiral Amines via Transition Metal-Catalyzed Asymmetric Hydrogenation. Chem. Rev. 2022, 122, 269–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wen, J.; Zhang, X. Chiral Tridentate Ligands in Transition Metal-Catalyzed Asymmetric Hydrogenation. Chem. Rev. 2021, 121, 7530–7567. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, G.-S.; Gao, Y.-D.; Yang, L.-C. Application of Imine Reductase in Bioactive Chiral Amine Synthesis. Org. Process Res. Dev. 2024, 28, 3035–3054. [Google Scholar] [CrossRef]
- 84 Silvi, M.; Verrier, C.; Rey, Y.P.; Buzzetti, L.; Melchiorre, P. Visible-Light Excitation of Iminium Ions Enables the Enantioselective Catalytic Beta-Alkylation of Enals. Nat. Chem. 2017, 9, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Swierk, J.R. The Cost of Quantum Yield. Org. Process Res. Dev. 2023, 27, 1411–1419. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Recent Progress in Synthetic Applications of Hypervalent Iodine(III) Reagents. Chem. Rev. 2024, 124, 11108–11186. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Dohi, T.; Zhdankin, V.V. Organohypervalent Heterocycles. Chem. Soc. Rev. 2024, 53, 4786–4827. [Google Scholar] [CrossRef]
- Mironova, I.A.; Noskov, D.M.; Yoshimura, A.; Yusubov, M.S.; Zhdankin, V.V. Aryl-, Akynyl-, and Alkenylbenziodoxoles: Synthesis and Synthetic Applications. Molecules 2023, 28, 2136. [Google Scholar] [CrossRef]
- Du, E.L.; Waser, J. Recent Progress in Alkynylation with Hypervalent Iodine Reagents. Chem. Commun. 2023, 59, 1589–1604. [Google Scholar]
- Luo, X.; Wang, P. Ynonylation of Acyl Radicals by Electroinduced Homolysis of 4-Acyl-1,4-dihydropyridines. Org. Lett. 2021, 23, 4960–4965. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Sakamoto, R.; Maruoka, K. Practical Synthesis of α,β-Alkynyl Ketones by Oxidative Alkynylation of Aldehydes with Hypervalent Alkynyliodine Reagents. Chem. Lett. 2020, 49, 633–636. [Google Scholar] [CrossRef]
- Ye, N.; Chen, H.; Wold, E.A.; Shi, P.-Y.; Zhou, J. Therapeutic Potential of Spirooxindoles as Antiviral Agents. ACS Infect. Dis. 2016, 2, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Smolyar, I.V.; Yudin, A.K.; Nenajdenko, V.G. Heteroaryl Rings in Peptide Macrocycles. Chem. Rev. 2019, 119, 10032–10240. [Google Scholar] [CrossRef]
- Oliva, M.; Pillitteri, S.; Schörgenhumer, J.; Saito, R.; Van der Eycken, E.V.; Sharma, U.K. Bromine Radical Release from Nickel-Complex Facilitates the Activation of Alkyl Boronic Acids: A Boron Selective Suzuki-Miyaura Cross Coupling. Chem. Sci. 2024, 15, 17490–17497. [Google Scholar] [CrossRef]
- Murakami, K.; Yamada, S.; Kaneda, T.; Itami, K. C-H Functionalization of Azines. Chem. Rev. 2017, 117, 9302–9332. [Google Scholar] [CrossRef] [PubMed]
- Escolano, M.; Gaviña, D.; Alzuet-Piña, G.; Díaz-Oltra, S.; Sánchez-Roselló, M.; del Pozo, C. Recent Strategies in the Nucleophilic Dearomatization of Pyridines, Quinolines, and Isoquinolines. Chem. Rev. 2024, 124, 1122–1246. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Ohmiya, H. Direct Excitation Strategy for Radical Generation in Organic Synthesis. Chem. Soc. Rev. 2021, 50, 6320–6332. [Google Scholar] [CrossRef]
- Protti, S.; Ravelli, D.; Fagnoni, M. Designing Radical Chemistry by Visible Light-Promoted Homolysis. Trends Chem. 2022, 4, 305–317. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Qian, B.-C. Benzothiazolines Acting as Carbanion and Radical Transfer Reagents in Carbon–Carbon Bond Construction. Molecules 2025, 30, 1711. https://doi.org/10.3390/molecules30081711
Chen X, Qian B-C. Benzothiazolines Acting as Carbanion and Radical Transfer Reagents in Carbon–Carbon Bond Construction. Molecules. 2025; 30(8):1711. https://doi.org/10.3390/molecules30081711
Chicago/Turabian StyleChen, Xiaotang, and Bao-Chen Qian. 2025. "Benzothiazolines Acting as Carbanion and Radical Transfer Reagents in Carbon–Carbon Bond Construction" Molecules 30, no. 8: 1711. https://doi.org/10.3390/molecules30081711
APA StyleChen, X., & Qian, B.-C. (2025). Benzothiazolines Acting as Carbanion and Radical Transfer Reagents in Carbon–Carbon Bond Construction. Molecules, 30(8), 1711. https://doi.org/10.3390/molecules30081711