
Unraveling the Kinetics and Mechanism of Ethane Chlorination in the Gas Phase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
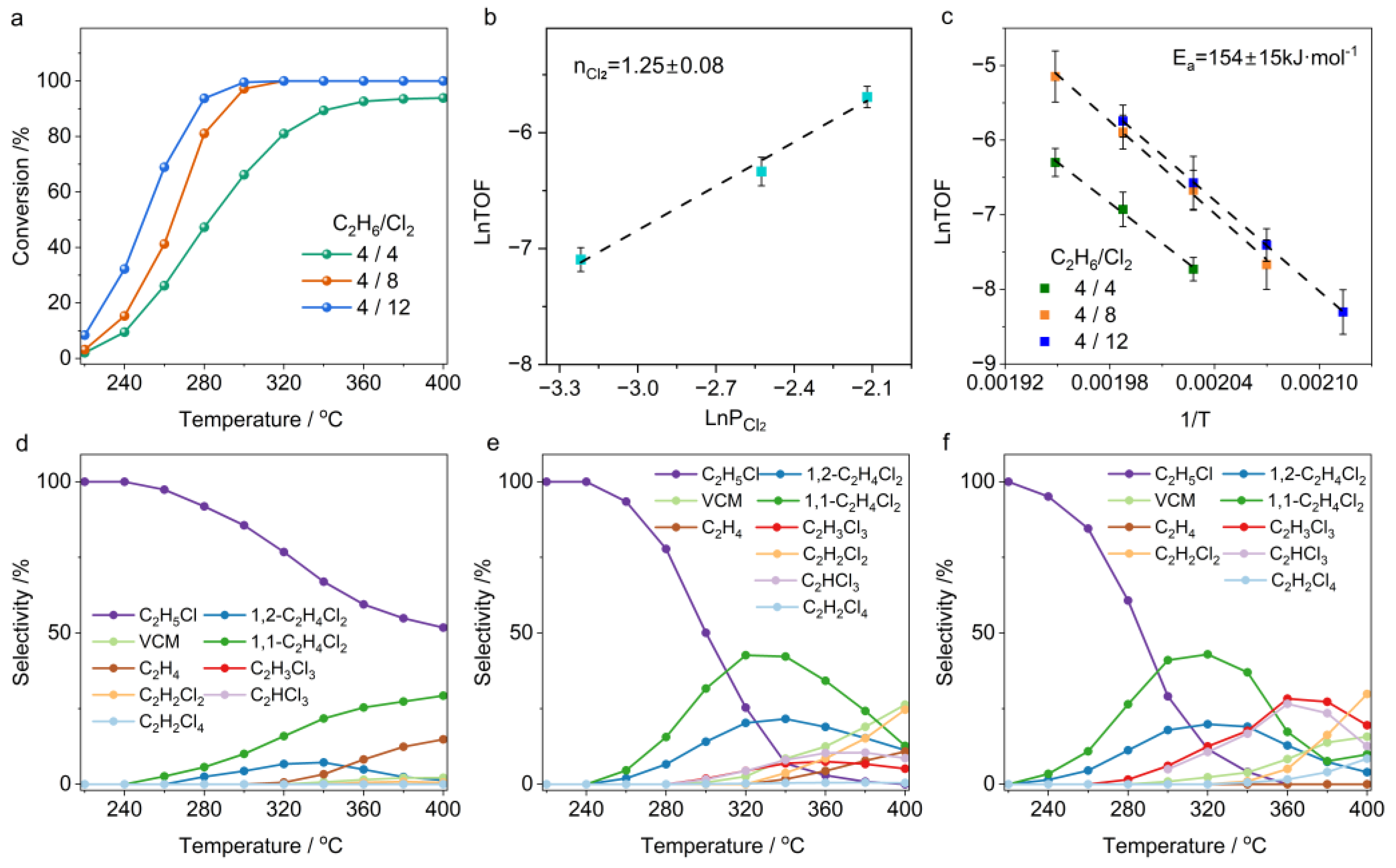
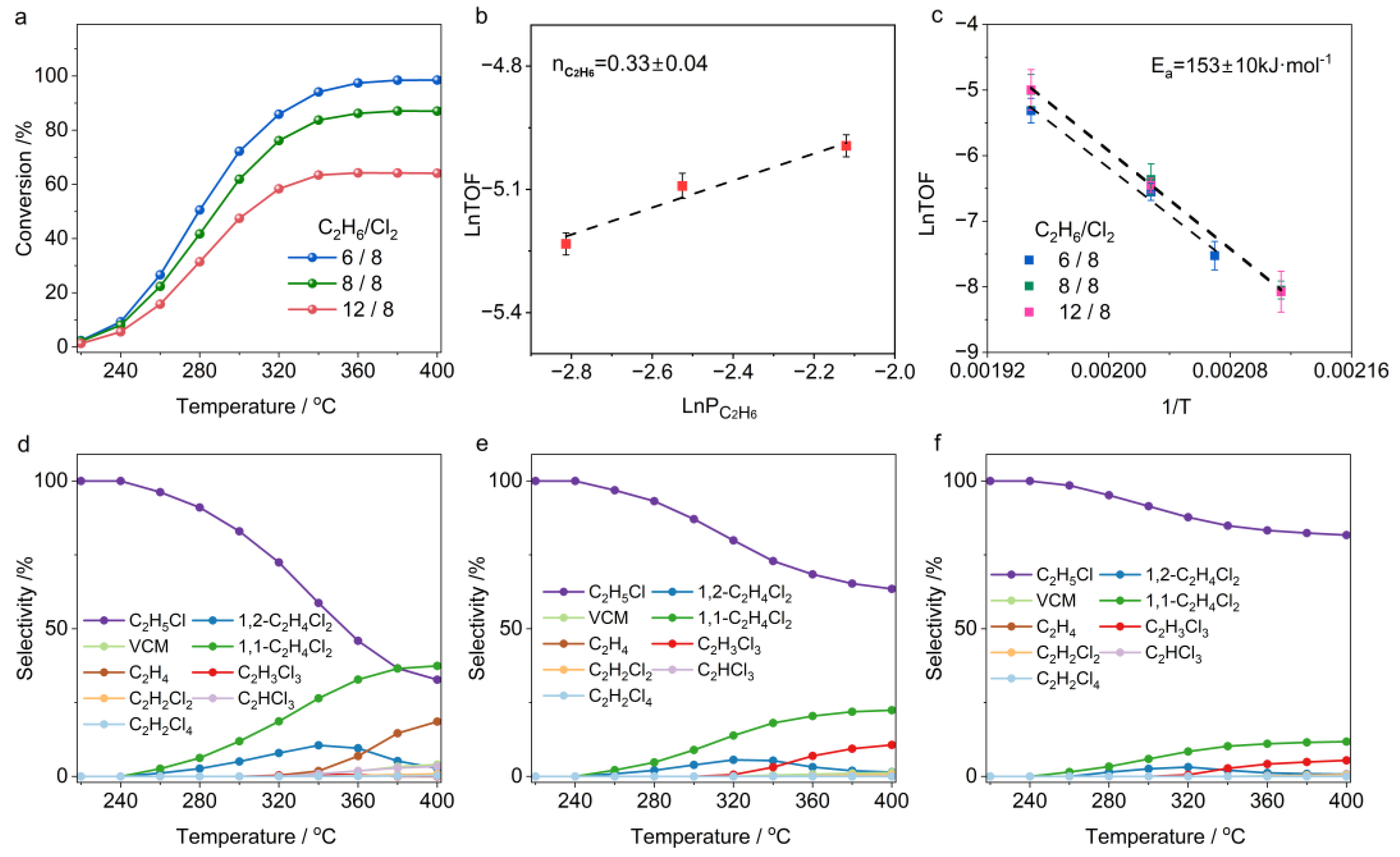
2.1. Temperature-Dependent Ethane Chlorination
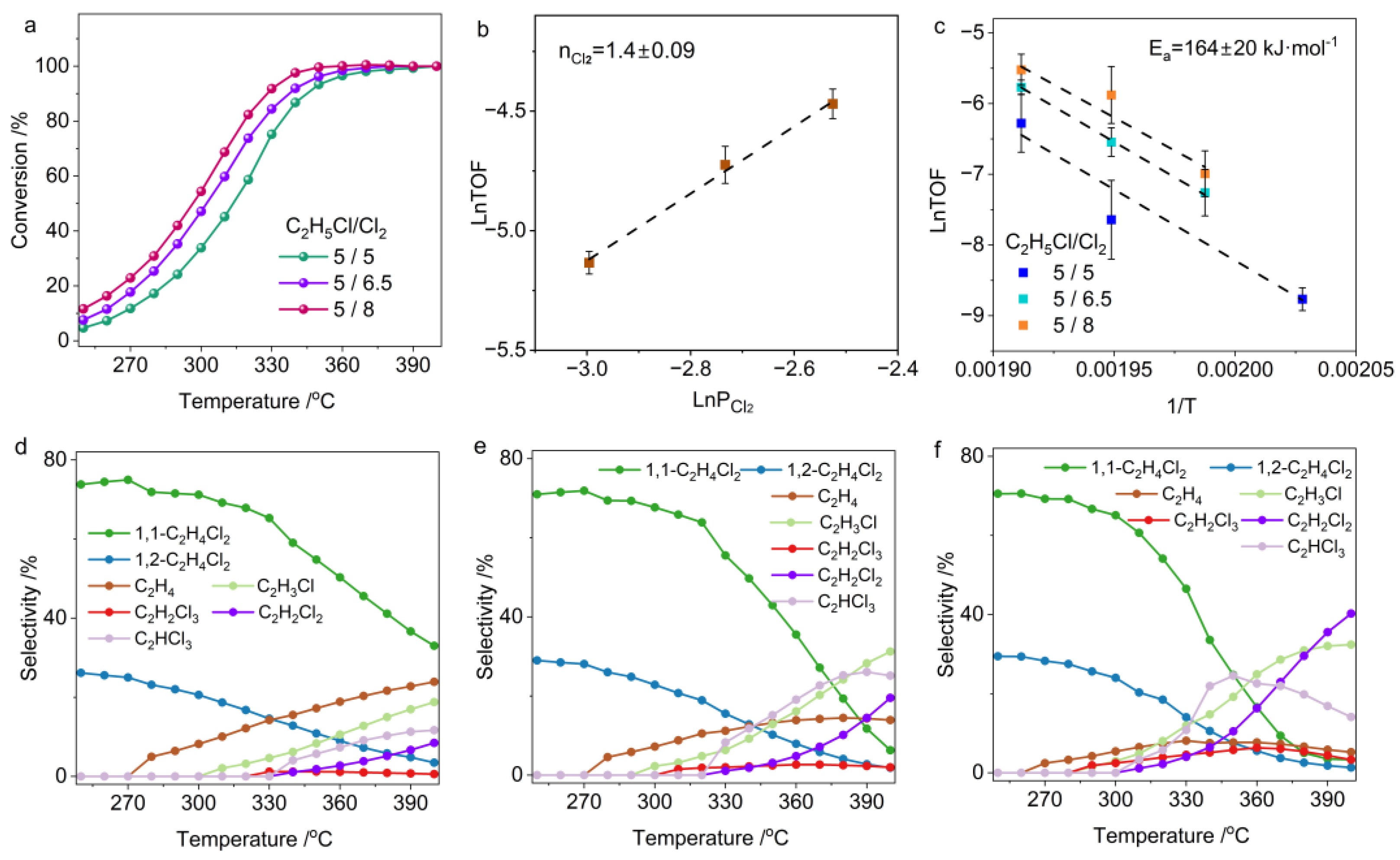
2.2. Temperature-Dependent Chloroethane Chlorination
3. Materials and Methods
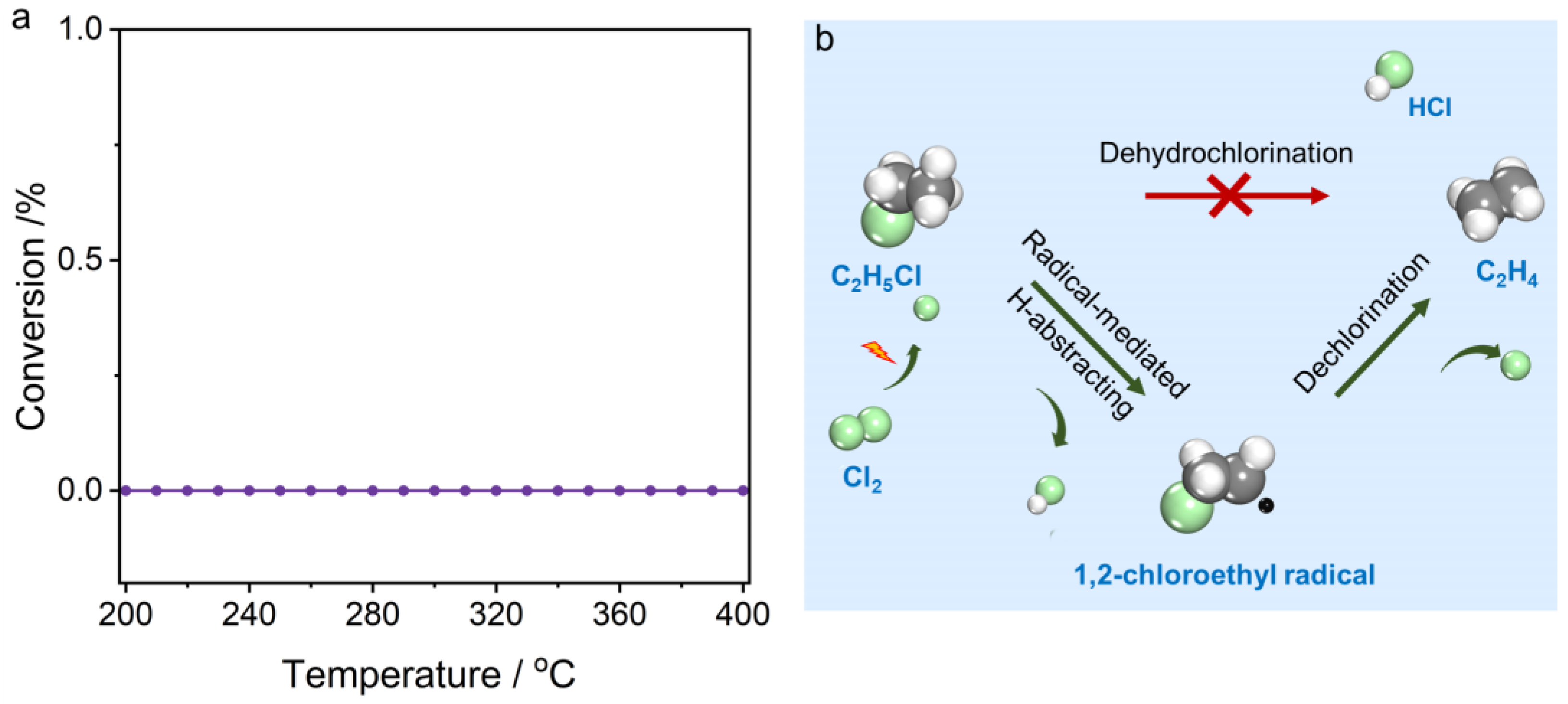
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saito, H.; Sekine, Y. Catalytic Conversion of Ethane to Valuable Products through Non-Oxidative Dehydrogenation and Dehydroaromatization. RSC Adv. 2020, 10, 21427–21453. [Google Scholar] [CrossRef] [PubMed]
- McFarland, E. Unconventional Chemistry for Unconventional Natural Gas. Science 2012, 338, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Amrute, A.P.; Pérez-Ramírez, J. Halogen-Mediated Conversion of Hydrocarbons to Commodities. Chem. Rev. 2017, 117, 4182–4247. [Google Scholar] [CrossRef]
- Cao, X.; Wu, X.; Liu, Y.; Geng, H.; Yu, S.; Liu, S. Boron and Nitrogen Co-Doped Porous Carbon Nanospheres for Oxidative Dehydrogenation of Ethane to Ethylene. Carbon 2022, 197, 120–128. [Google Scholar] [CrossRef]
- Altus, K.M.; Love, J.A. The Continuum of Carbon–Hydrogen (C–H) Activation Mechanisms and Terminology. Commun. Chem. 2021, 4, 173. [Google Scholar] [CrossRef]
- Najari, S.; Saeidi, S.; Concepcion, P.; Dionysiou, D.D.; Bhargava, S.K.; Lee, A.F.; Wilson, K. Oxidative Dehydrogenation of Ethane: Catalytic and Mechanistic Aspects and Future Trends. Chem. Soc. Rev. 2021, 50, 4564–4605. [Google Scholar] [CrossRef]
- Li, X.; Pei, C.; Gong, J. Shale Gas Revolution: Catalytic Conversion of C1–C3 Light Alkanes to Value-Added Chemicals. Chem 2021, 7, 1755–1801. [Google Scholar] [CrossRef]
- Li, B.; Mu, J.; Long, G.; Song, X.; Huang, E.; Liu, S.; Wei, Y.; Sun, F.; Feng, S.; Yuan, Q.; et al. Water-Participated Mild Oxidation of Ethane to Acetaldehyde. Nat. Commun. 2024, 15, 2555. [Google Scholar] [CrossRef]
- Samanta, A.; Bai, X.; Robinson, B.; Chen, H.; Hu, J. Conversion of Light Alkane to Value-Added Chemicals over ZSM-5/Metal Promoted Catalysts. Ind. Eng. Chem. Res. 2017, 56, 11006–11012. [Google Scholar] [CrossRef]
- Fairuzov, D.; Gerzeliev, I.; Maximov, A.; Naranov, E. Catalytic Dehydrogenation of Ethane: A Mini Review of Recent Advances and Perspective of Chemical Looping Technology. Catalysts 2021, 11, 833. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, X.; Chen, X.; Liu, P.; Chen, J.G. General Descriptors for CO2-Assisted Selective C–H/C–C Bond Scission in Ethane. J. Am. Chem. Soc. 2022, 144, 4186–4195. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, H.; Zhou, H.; Wang, L.; Wang, L.; Zhu, Q.; Xiao, J.; Meng, X.; Chen, J.; Xiao, F.-S. Coking-Resistant Iron Catalyst in Ethane Dehydrogenation Achieved through Siliceous Zeolite Modulation. J. Am. Chem. Soc. 2020, 142, 16429–16436. [Google Scholar] [CrossRef]
- Zhang, W.; Fu, C.; Low, J.; Duan, D.; Ma, J.; Jiang, W.; Chen, Y.; Liu, H.; Qi, Z.; Long, R.; et al. High-Performance Photocatalytic Nonoxidative Conversion of Methane to Ethane and Hydrogen by Heteroatoms-Engineered TiO2. Nat. Commun. 2022, 13, 2806. [Google Scholar] [CrossRef] [PubMed]
- Venegas, J.M.; McDermott, W.P.; Hermans, I. Serendipity in Catalysis Research: Boron-Based Materials for Alkane Oxidative Dehydrogenation. Acc. Chem. Res. 2018, 51, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, X.; Shi, R.; Zhao, J.; Waterhouse, G.I.N.; Tang, J.; Zhang, T. Photocatalytic Ethylene Production by Oxidative Dehydrogenation of Ethane with Dioxygen on ZnO-Supported PdZn Intermetallic Nanoparticles. Nat. Commun. 2024, 15, 789. [Google Scholar] [CrossRef]
- Sarkar, B.; Goyal, R.; Sivakumar Konathala, L.N.; Pendem, C.; Sasaki, T.; Bal, R. MoO3 Nanoclusters Decorated on TiO2 Nanorods for Oxidative Dehydrogenation of Ethane to Ethylene. Appl. Catal. B Environ. 2017, 217, 637–649. [Google Scholar] [CrossRef]
- Melzer, D.; Mestl, G.; Wanninger, K.; Zhu, Y.; Browning, N.D.; Sanchez-Sanchez, M.; Lercher, J.A. Design and Synthesis of Highly Active MoVTeNb-Oxides for Ethane Oxidative Dehydrogenation. Nat. Commun. 2019, 10, 4012. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, P.; Yang, J.; Zhu, A.; Chen, D. C–H Bond Activation in Light Alkanes: A Theoretical Perspective. Chem. Soc. Rev. 2021, 50, 4299–4358. [Google Scholar] [CrossRef]
- Zichittella, G.; Pérez-Ramírez, J. Ethane-Based Catalytic Process for Vinyl Chloride Manufacture. Angew. Chem. Int. Ed. 2021, 60, 24089–24095. [Google Scholar] [CrossRef]
- Ma, H.; Ma, G.; Qi, Y.; Wang, Y.; Chen, Q.; Rout, K.R.; Fuglerud, T.; Chen, D. Nitrogen-Doped Carbon-Assisted One-Pot Tandem Reaction for Vinyl Chloride Production via Ethylene Oxychlorination. Angew. Chem. Int. Ed. 2020, 59, 22080–22085. [Google Scholar] [CrossRef]
- Johnston, P.; Carthey, N.; Hutchings, G.J. Discovery, Development, and Commercialization of Gold Catalysts for Acetylene Hydrochlorination. J. Am. Chem. Soc. 2015, 137, 14548–14557. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, S.; Guo, M.; Zhou, D.; Shen, Z.; Wang, W.; Feng, B.; Jiang, B. Catalytic Dehydrochlorination of 1,2-Dichloroethane into Vinyl Chloride over Nitrogen-Doped Activated Carbon. ACS Omega 2019, 4, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Schirmeister, R.; Kahsnitz, J.; Träger, M. Influence of EDC Cracking Severity on the Marginal Costs of Vinyl Chloride Production. Ind. Eng. Chem. Res. 2009, 48, 2801–2809. [Google Scholar] [CrossRef]
- Boudewijns, T.; Piccinini, M.; Degraeve, P.; Liebens, A.; De Vos, D. Pathway to Vinyl Chloride Production via Dehydrochlorination of 1,2-Dichloroethane in Ionic Liquid Media. ACS Catal. 2015, 5, 4043–4047. [Google Scholar] [CrossRef]
- Malta, G.; Kondrat, S.A.; Freakley, S.J.; Davies, C.J.; Lu, L.; Dawson, S.; Thetford, A.; Gibson, E.K.; Morgan, D.J.; Jones, W.; et al. Identification of Single-Site Gold Catalysis in Acetylene Hydrochlorination. Science 2017, 355, 1399–1403. [Google Scholar] [CrossRef]
- Scharfe, M.; Zichittella, G.; Kondratenko, V.A.; Kondratenko, E.V.; López, N.; Pérez-Ramírez, J. Mechanistic Origin of the Diverging Selectivity Patterns in Catalyzed Ethane and Ethene Oxychlorination. J. Catal. 2019, 377, 233–244. [Google Scholar] [CrossRef]
- Quoc Pham, L.; Uspenskaya, M.V.; Olekhnovich, R.O.; Olvera Bernal, R.A. A Review on Electrospun PVC Nanofibers: Fabrication, Properties, and Application. Fibers 2021, 9, 12. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Guillén-Gosálbez, G. Catalyst: A Step Forward for PVC Manufacture from Natural Gas. Chem 2022, 8, 883–885. [Google Scholar] [CrossRef]
- Medrano-García, J.D.; Giulimondi, V.; Ceruti, A.; Zichittella, G.; Pérez-Ramírez, J.; Guillén-Gosálbez, G. Economic and Environmental Competitiveness of Ethane-Based Technologies for Vinyl Chloride Synthesis. ACS Sustain. Chem. Eng. 2023, 11, 13062–13069. [Google Scholar] [CrossRef]
- Hickman, D.A.; Jones, M.E.; Jovanovic, Z.R.; Olken, M.M.; Podkolzin, S.G.; Stangland, E.E.; Thompson, R.K. Reactor Scale-up for Fluidized Bed Conversion of Ethane to Vinyl Chloride. Ind. Eng. Chem. Res. 2010, 49, 10674–10681. [Google Scholar] [CrossRef]
- Podkolzin, S.G.; Stangland, E.E.; Jones, M.E.; Peringer, E.; Lercher, J.A. Methyl Chloride Production from Methane over Lanthanum-Based Catalysts. J. Am. Chem. Soc. 2007, 129, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Scharfe, M.; Lira-Parada, P.A.; Amrute, A.P.; Mitchell, S.; Pérez-Ramírez, J. Lanthanide Compounds as Catalysts for the One-Step Synthesis of Vinyl Chloride from Ethylene. J. Catal. 2016, 344, 524–534. [Google Scholar] [CrossRef]
- Gaviría, J.P.; Navarro, L.G.; Bohé, A.E. Chlorination of Lanthanum Oxide. J. Phys. Chem. A 2012, 116, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Hey, D.H.; Waters, W.A. Free Radicals in Solution. Nature 1937, 140, 934–935. [Google Scholar] [CrossRef]
- Dahl, I.M.; Myhrvold, E.M.; Olsbye, U.; Rohr, F.; Rokstad, O.A.; Swang, O. On the Gas-Phase Chlorination of Ethane. Ind. Eng. Chem. Res. 2001, 40, 2226–2235. [Google Scholar] [CrossRef]
- Goryachev, V.V.; Treger, Y.A.; Flid, R.M.; Krishtal’, N.F.; Fedorova, V.M. Kinetics of Gaseous-Phase Catalytic Chlorination of Ethane. Pet. Chem. U.S.S.R. 1976, 16, 228–234. [Google Scholar] [CrossRef]
- Wang, M.; Ma, D. Reaction: Direct Chlorination of Ethane to Dichloroethane. Chem 2022, 8, 886–887. [Google Scholar] [CrossRef]
- Bryukov, M.G.; Slagle, I.R.; Knyazev, V.D. Kinetics of Reactions of Cl Atoms with Ethane, Chloroethane, and 1,1-Dichloroethane. J. Phys. Chem. A 2003, 107, 6565–6573. [Google Scholar] [CrossRef]
- Sutradhar, D.; Chandra, A.K. Cl⋅⋅⋅Cl Halogen Bonding: Nature and Effect of Substituent at Electron Donor Cl Atom. ChemistrySelect 2020, 5, 554–563. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.; Li, Y.; Wu, X.; Xu, J.; Sun, X.; Liu, Q. Unraveling the Kinetics and Mechanism of Ethane Chlorination in the Gas Phase. Molecules 2025, 30, 1756. https://doi.org/10.3390/molecules30081756
Zhu Z, Li Y, Wu X, Xu J, Sun X, Liu Q. Unraveling the Kinetics and Mechanism of Ethane Chlorination in the Gas Phase. Molecules. 2025; 30(8):1756. https://doi.org/10.3390/molecules30081756
Chicago/Turabian StyleZhu, Zihan, Yuting Li, Xia Wu, Jinming Xu, Xiaohui Sun, and Qinggang Liu. 2025. "Unraveling the Kinetics and Mechanism of Ethane Chlorination in the Gas Phase" Molecules 30, no. 8: 1756. https://doi.org/10.3390/molecules30081756
APA StyleZhu, Z., Li, Y., Wu, X., Xu, J., Sun, X., & Liu, Q. (2025). Unraveling the Kinetics and Mechanism of Ethane Chlorination in the Gas Phase. Molecules, 30(8), 1756. https://doi.org/10.3390/molecules30081756