
A Sustainable Synthesis of Novel 2-(3,4-Disubstituted phenyl)benzoxazole Derivatives and Their Antiproliferative and Antibacterial Evaluation

 , , ,
, , ,  and
and
Abstract
:
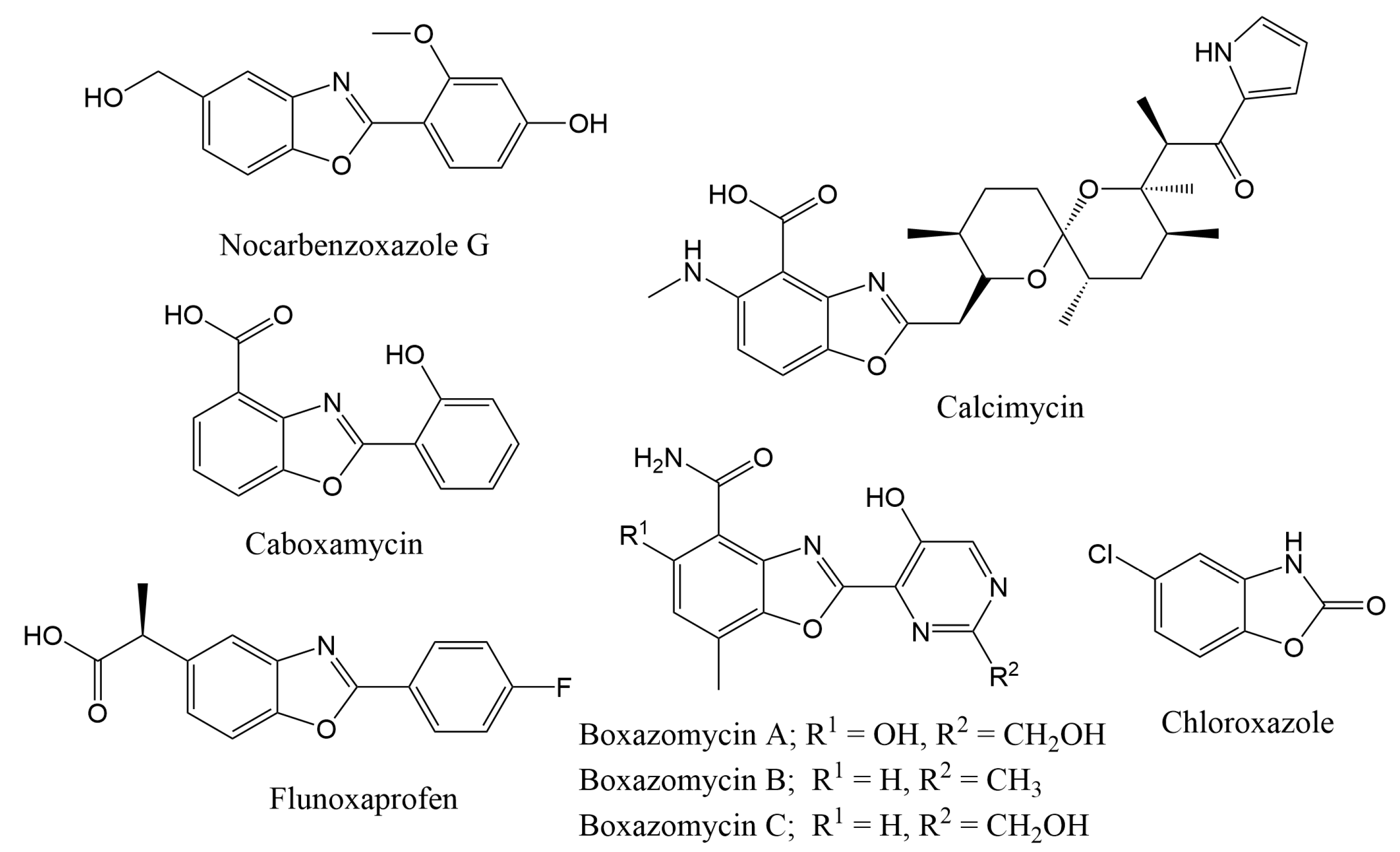
1. Introduction
2. Results and Discussion
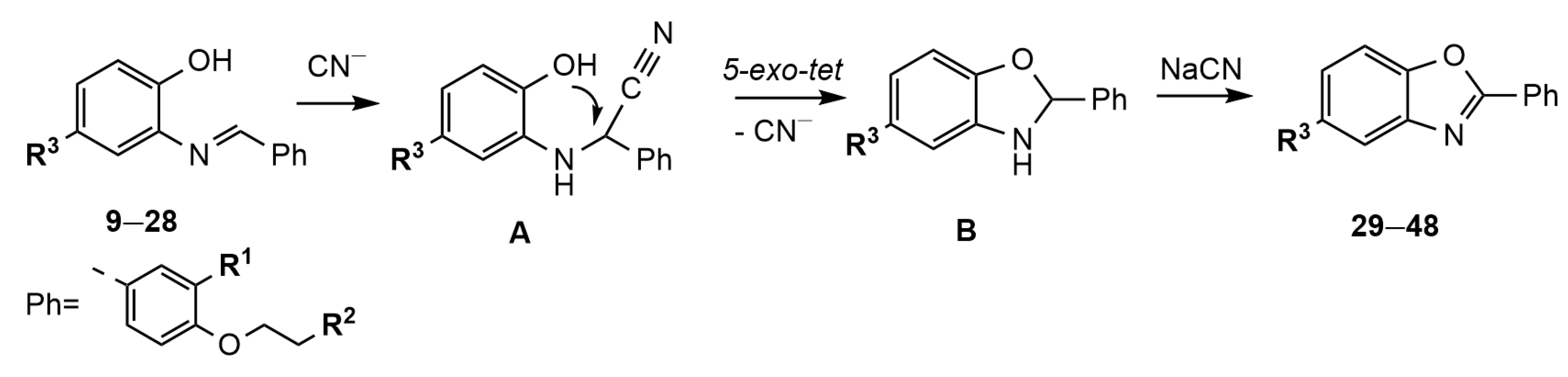
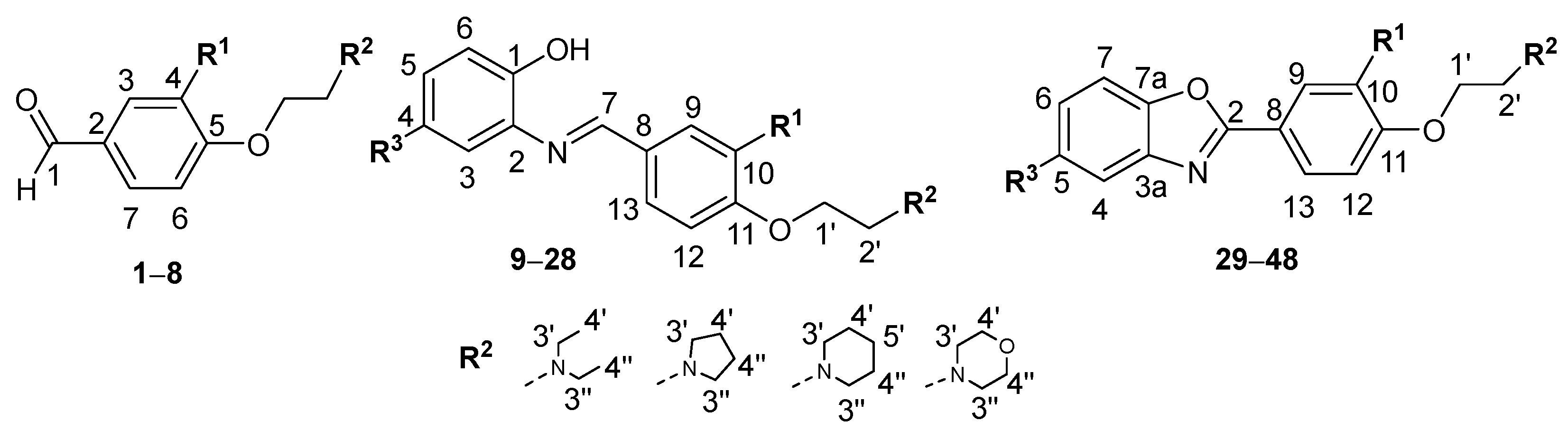
2.1. Chemistry
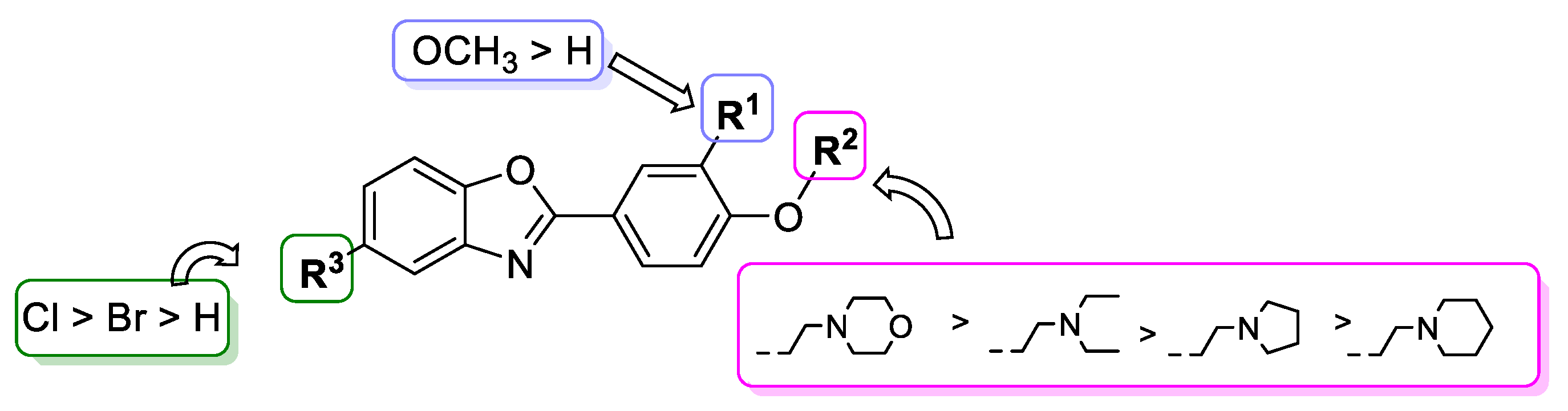
2.2. In Vitro Antiproliferative Activity of Benzoxazole Derivatives 29–48
2.3. In Vitro Antibacterial Activity of Benzoxazole Derivatives 29–48
3. Materials and Methods
3.1. Chemical Synthesis
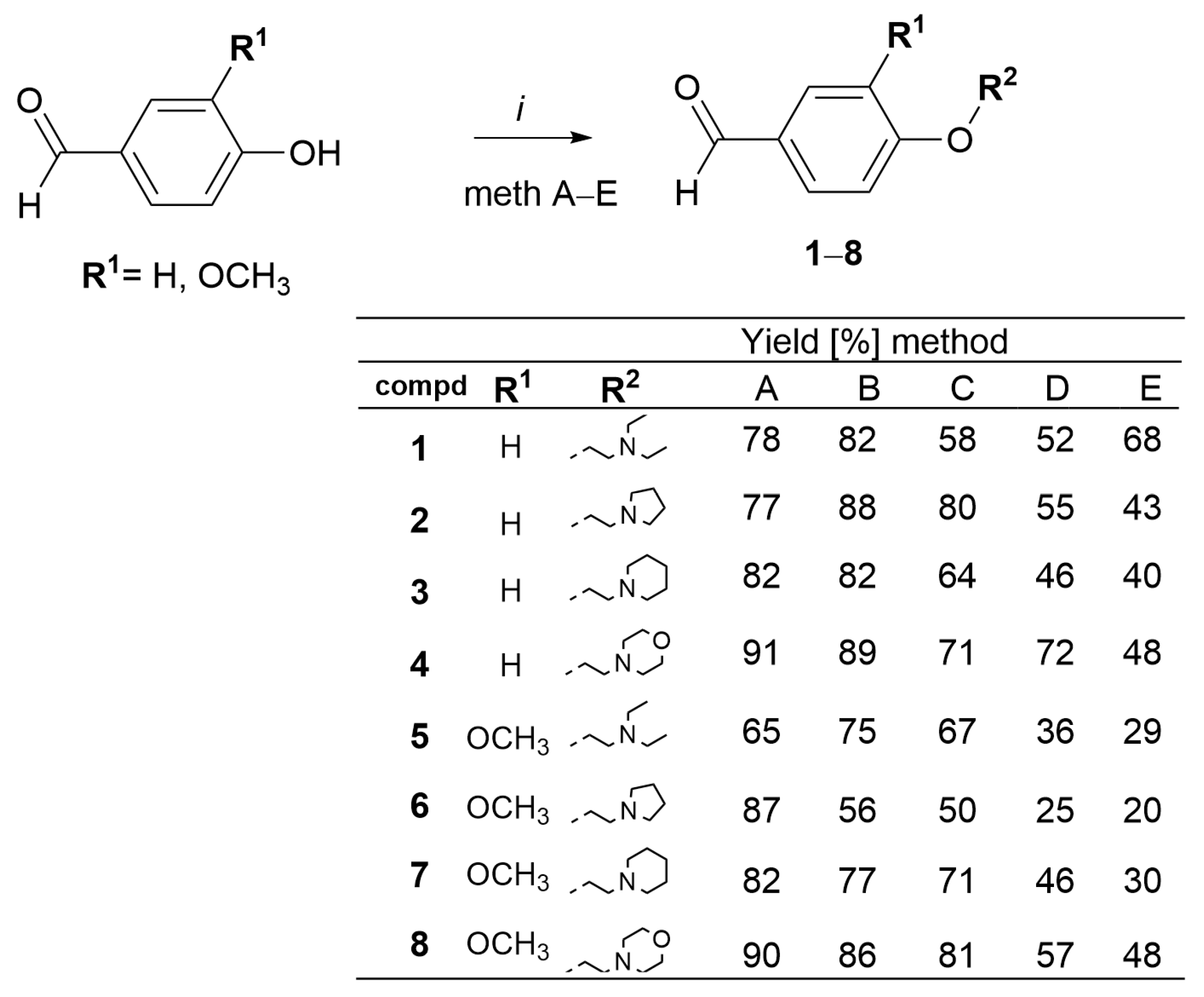
3.1.1. General Procedure for the Synthesis of 4-O-Alkylated Benzaldehyde Derivatives 1–8
- 4(4-(2-(Diethylamino)ethoxy)benzaldehyde 1. Compound 1 was synthesized according to general procedure Section 3.1.1 from 4-hydroxybenzaldehyde (200 mg, 1.64 mmol) and 2-chloro-N,N-diethylethanamine hydrochloride (236 mg, 1.64 mmol). After purification by column chromatography compound 1 was obtained as a yellowish oil. (Method A: 281 mg, 78%; Method B: 285 mg, 82%; Method C: 211 mg, 58%; Method D: 189 mg, 52%; Method E: 247 mg, 68%). 1H-NMR (600 MHz, DMSO-d6) δ 9.87 (s, 1H, H-1), 7.86 (d, J = 8.8 Hz, 2H, H-3,7), 7.13 (d, J = 8.7 Hz, 2H, H-4,6), 4.13 (t, J = 6.1 Hz, 2H, H-1′), 2.80 (t, J = 6.1 Hz, 2H, H-2′), 2.55 (q, J = 7.1 Hz, 4H, H-3′,3″), 0.97 (t, J = 7.1 Hz, 6H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 191.9, 154.0, 143.7, 130.1, 114.7, 67.1, 54.9, 46.0, 12.3.
- 4-(2-(Pyrrolidin-1-yl)ethoxy)benzaldehyde 2. Compound 2 was synthesized according to general procedure Section 3.1.1 from 4-hydroxybenzaldehyde (200 mg, 1.64 mmol) and 1-(2-chloroethyl)pyrrolidine hydrochloride (279 mg, 1.64 mmol). After purification by column chromatography compound 2 was obtained as a yellowish oil. (Method A: 277 mg, 77%; Method B: 314 mg, 88%; Method C: 287 mg, 80%; Method D: 196 mg, 55%, Method E: 155 mg, 43%). 1H NMR (600 MHz, DMSO-d6) δ 9.87 (s, 1H, H-1), 7.86 (d, J = 8.8 Hz, 2H, H-3,7), 7.14 (d, J = 8.7 Hz, 2H, H-4,6), 4.19 (t, J = 5.8 Hz, 2H, H-1′), 2.81 (t, J = 5.8 Hz, 2H, H-2′), 2.53 (m, 4H, H-3′,3″), 1.68 (m, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 191.8, 164.0, 130.1, 126.6, 115.9, 67.1, 57.6, 39.9, 23.6.
- 4-(2-(Piperidin-1-yl)ethoxy)benzaldehyde 3. Compound 3 was synthesized according to general procedure Section 3.1.1 from 4-hydroxybenzaldehyde (200 mg, 1.64 mmol) and 1-(2-chloroethyl)piperidine hydrochloride (300 mg, 1.64 mmol). After purification by column chromatography compound 3 was obtained as a yellowish oil. (Method A: 315 mg, 82%; Method B: 313 mg, 82%; Method C: 246 mg, 64%, Method D: 175 mg, 46% Method E: 153 mg, 40%). 1H-NMR (600 MHz, DMSO-d6) δ 9.86 (s, 1H, H-1), 7.85 (d, J = 8.8 Hz, 2H, H-3,7), 7.12 (d, J = 8.7 Hz, 2H, H-4,6), 4.17 (t, J = 5.9 Hz, 2H, H-1′), 2.66 (t, J = 5.9 Hz, 2H, H-2′), 2.42 (t, J = 5.3 Hz, 4H, H-3′,3″), 1.48 (p, J = 5.6 Hz, 4H, H-4′,4″), 1.36 (m, 2H, H-5′). 13C-NMR (151 MHz, DMSO-d6) δ 191.2 (C-1), 163.5, 131.7, 129.5, 115.0, 65.7, 57.1, 54.3, 25.5, 23.9.
- 4-(2-Morpholinoethoxy)benzaldehyde 4. Compound 4 was synthesized according to general procedure Section 3.1.1 from 4-hydroxybenzaldehyde (200 mg, 1.64 mmol) and 4-(2-chloroethyl)morpholine hydrochloride (305 mg, 1.64 mmol). After purification by column chromatography compound 4 was obtained as a beige oil. (Method A: 285 mg, 91%, Method B: 275 mg, 89%; Method C: 218 mg, 71%, Method D: 223 mg, 72% Method E: 147 mg, 48%). 1H-NMR (300 MHz, DMSO-d6) δ 9.88 (s, 1H, H-1), 7.87 (d, J = 8.7 Hz, 2H, H-3,7), 7.14 (d, J = 8.7 Hz, 2H, H-4,6), 4.21 (t, J = 5.7 Hz, 2H, H-1′), 3.58 (t, J = 4.5 Hz, 4H, H-4′,4″), 2.72 (t, J = 5.7 Hz, 2H, H-2′), 2.52 (t, J = 4.5 Hz, 4H, H-3′,3″). 13C-NMR (151 MHz, DMSO-d6) δ 191.3, 163.4, 131.8, 129.6, 115.0, 66.1, 65.8, 56.8, 53.6.
- 4-(2-(Diethylamino)ethoxy)-3-methoxybenzaldehyde 5. Compound 5 was synthesized according to general procedure Section 3.1.1 from 4-hydroxy-3-methoxybenzaldehyde (200 mg, 1.31 mmol) and N,N-diethyl-2-chloroethan-1-amine hydrochloride (225 mg, 1.31 mmol). After purification by column chromatography compound 5 was obtained as a yellowish oil. (Method A: 216 mg, 65%; Method B: 246 mg, 75%; Method C: 222 mg, 67%; Method D: 119 mg, 36%; Method E: 96 mg, 29%). 1H-NMR (600 MHz, DMSO-d6) δ 9.83 (s, 1H, H-1), 7.52 (dd, J = 8.2, 1.9 Hz, 1H, H-7), 7.38 (d, J = 1.9 Hz, 1H, H-3), 7.18 (d, J = 8.2 Hz, 1H, H-6), 4.11 (t, J = 6.2 Hz, 2H, H-1′), 3.82 (s, 3H, OCH3), 2.81 (t, J = 6.2 Hz, 2H, H-2′,2″), 2.55 (q, J = 7.1 Hz, 4H, H-3′,3″), 0.96 (t, J = 7.1 Hz, 6H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 191.3, 153.5, 149.2, 132.7, 129.6, 112.1, 109.7, 67.3, 59.4, 55.5, 53.8, 25.1.
- 3-Methoxy-4-(2-(pyrrolidin-1-yl) ethoxy)benzaldehyde 6. Compound 6 was synthesized according to general procedure Section 3.1.1 from 4-hydroxy-3-methoxybenzaldehyde (200 mg, 1.31 mmol) and 1-(2-chloroethyl) pyrrolidine hydrochloride (222 mg, 1.31 mmol). After purification by column chromatography compound 6 was obtained as a pale-yellow oil. (Method A: 287 mg, 87%; Method B: 183 mg, 56%; Method C: 164 mg, 50%, Method D: 83 mg, 25%; Method E: 67 mg, 20%). 1H-NMR (600 MHz, DMSO-d6) δ 9.84 (s, 1H, H-1), 7.53 (dd, J = 8.2, 1.9 Hz, 1H, H-7), 7.39 (d, J = 1.9 Hz, 1H, H-3), 7.19 (d, J = 8.3 Hz, 1H, H-6), 4.17 (t, J = 6.0 Hz, 2H, H-1′), 3.83 (s, 3H, OCH3), 2.81 (t, J = 6.0 Hz, 2H, H-2′), 2.52 (m, 4H, H3′,3″), 1.68 (m, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 191.3, 153.5, 149.2, 129.6, 126.0, 111.8, 109.6, 67.7, 55.5, 54.1, 54.0, 23.1.
- 3-Methoxy-4-(2-(piperidin-1-yl) ethoxy)benzaldehyde 7. Compound 7 was synthesized according to general procedure Section 3.1.1 from 4-hydroxy-3-methoxybenzaldehyde (200 mg, 1.31 mmol) and 1-(2-chloroethyl) piperidine hydrochloride (239 mg, 1.31 mmol). After purification by column chromatography compound 7 was obtained as a light-yellow oil. (Method A: 286 g, 82%; Method B: 265 mg, 77%; Method C: 244 mg, 71%; Method D: 159 mg, 46% Method E: 103 mg, 30%). 1H-NMR (300 MHz, DMSO-d6) δ 9.84 (s, 1H, H-1), 7.53 (dd, J = 8.3, 1.9 Hz, 1H, H-7), 7.39 (d, J = 1.9 Hz, 1H, H-3), 7.20 (d, J = 8.3 Hz, 1H, H-6), 4.18 (t, J = 6.0 Hz, 2H, H-1′), 3.83 (s, 3H, OCH3), 2.70 (t, J = 6.0 Hz, 2H, H-2′), 2.45 (t, J = 5.3 Hz, 4H, H-3′,3″), 1.49 (t, J = 5.6 Hz, 4H, H-4′,4′′), 1.38 (m, 2H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 190.2, 152.4, 148.1, 128.5, 124.9, 111.0, 108.5, 66.6, 54.4, 53.0, 52.9, 24.3, 22.0.
- 3-Methoxy-4-(2-morpholinoethoxy)benzaldehyde 8. Compound 8 was synthesized according to general procedure Section 3.1.1 from 4-hydroxy-3-methoxybenzaldehyde (200 mg, 1.31 mmol) and 4-(2-chloroethyl) morpholine hydrochloride (243 mg, 1.31 mmol). After purification by column chromatography compound 8 was obtained as a brown oil. (Method A: 316 mg, 90%; Method B: 301 mg, 86%; Method C: 281 mg, 81%; Method D: 199 mg, 57% Method E: 166 mg, 48%). 1H-NMR (300 MHz, DMSO-d6) δ 9.84 (s, 1H, H-1), 7.54 (dd, J = 8.2, 1.9 Hz, 1H, H-7), 7.40 (d, J = 1.9 Hz, 1H, H-3), 7.21 (d, J = 8.2 Hz, 1H, H-6), 4.20 (t, J = 5.8 Hz, 2H, H-1′), 3.83 (s, 3H, OCH3), 3.58 (t, J = 4.6 Hz, 4H, H-4′,4″), 2.73 (t, J = 5.8 Hz, 2H, H-2′), 2.49 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 191.3, 153.6, 149.2, 129.7, 125.0, 112.3, 109.7, 66.4, 56.7, 55.6, 53.6, 53.20.
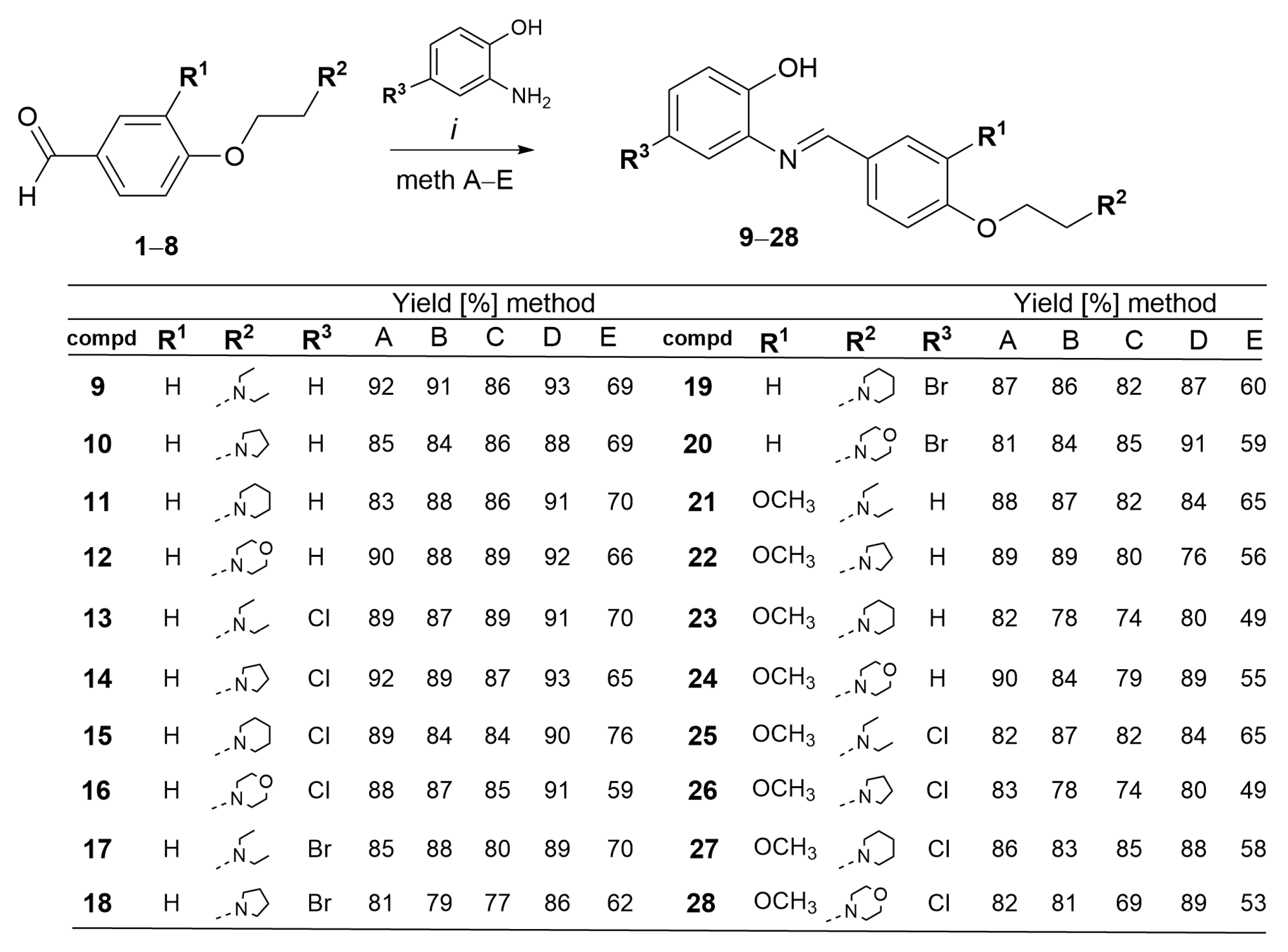
3.1.2. General Procedure for the Synthesis of Schiff Bases 9–28
- 2-((4-(2-(Diethylamino)ethoxy)benzylidene)amino)phenol 9. Compound 9 was synthesized according to general procedure Section 3.1.2 from 1 (100 mg, 0.45 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (49 mg, 0.45 mmol). Compound 9 was obtained as a brown oil. (Method A: 129 mg, 92%; Method B: 128 mg, 91%; Method C: 125 mg, 89%; Method D: 131 mg, 93%; Method E: 98 mg, 69%). 1H-NMR (600 MHz, DMSO-d6) δ 9.21 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.97 (d, J = 8.8 Hz, 2H, H-9,13), 7.86 (d, J = 8.8 Hz, 1H, H-5), 7.24 (d, J = 7.5 Hz, 2H, H-10,12), 7.14 (d, J = 8.7 Hz, 1H, H-3), 7.08 (d, J = 8.6 Hz, 1H, H-4), 6.89 (d, J = 8.6 Hz, 1H, H-6), 4.19 (t, J = 5.8 Hz, 2H, H-1′), 2.82 (t, J = 5.9 Hz, 4H, H-3′,3″), 2.54 (m, 2H, H-2′), 1.06 (t, J = 6.0 Hz, 6H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 162.9, 162.1, 150.5, 143.6, 128.5, 127.1, 125.1, 124.1, 119.6, 117.5, 115.9, 113.7, 67.4, 55.2, 50.3, 45.3, 18.8, 12.7. Calcd for C19H24N2O2: C, 73.05; H, 7.74; N, 8.97; Found: C, 73.07; H, 7.73; N, 8.95.
- 2-((4-(2-(Pyrrolidin-1-yl)ethoxy)benzylidene)amino)phenol 10. Compound 10 was synthesized according to general procedure Section 3.1.2 from 2 (100 mg, 0.46 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (50 mg, 0.46 mmol). Compound 10 was obtained as an orange oil. (Method A: 120 mg, 85%; Method B: 119 mg, 84%; Method C: 122 mg, 86%; Method D: 125 mg, 88%; Method E: 96 mg, 69%). 1H-NMR (600 MHz, DMSO-d6) δ 9.87 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.97 (d, J = 8.8 Hz, 2H, H-9,13), 7.86 (d, J = 8.8 Hz, 1H, H-5), 7.24 (d, J = 7.4 Hz, 2H, H-10,12), 7.14 (d, J = 8.7 Hz, 1H, H-3), 7.08 (dd, J = 8.6, 2.9 Hz, 1H, H-4), 6.89 (d, J = 8.6 Hz, 1H, H-6), 4.19 (t, J = 5.8 Hz, 2H, H-1′), 2.82 (m, 2H, H-2′), 2.55 (m, 4H, H-3′,3″), 1.69 (m, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.59, 160.21, 151.08, 142.35, 129.81, 125.60, 120.01, 116.54, 111.52, 55.18, 23.19. Calcd for C19H22N2O2: C, 73.52; H, 7.14; N, 9.03; Found: C, 73.50; H, 7.15; N, 9.01.
- 2-((4-(2-(Piperidin-1-yl)ethoxy)benzylidene)amino)phenol 11. Compound 11 was synthesized according to general procedure Section 3.1.2 from 3 (100 mg, 0.43 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (47 mg, 0.43 mmol). Compound 11 was obtained as a brown oil. (Method A: 115 mg, 83%; Method B: 123 mg, 88%; Method C: 120 mg, 86%; Method D: 126 mg, 91%; Method E: 97 mg, 70%). 1H-NMR (600 MHz, DMSO-d6) δ 8.88 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.97 (d, J = 8.7 Hz, 1H, H-9,13), 7.17 (dd, J = 7.9, 1.6 Hz, 1H, H-5), 7.07 (d, J = 8.8 Hz, 2H, H-10,12), 7.04 (d, J = 1.6 Hz, 1H, H-3), 6.89 (d, J = 1.3 Hz, 1H, H-4), 6.82 (d, J = 1.4 Hz, 1H, H-6), 4.15 (t, J = 5.9 Hz, 2H, H-1′), 2.68 (t, J = 5.9 Hz, 2H, H-2″), 2.44 (m, 4H, H-3′,3″), 1.51 (m, 4H, H-4′,4″), 1.39 (m, 2H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.49, 158.82, 151.59, 144.36, 131.00, 127.38, 121.30, 119.30, 116.25, 66.03, 57.25, 54.72, 25.78, 24.30. Calcd for C20H24N2O2: C, 74.05; H, 7.46; N, 8.63; Found: C, 74.07; H, 7.45; N, 8.64.
- 2-((4-(2-Morpholinoethoxy)benzylidene)amino)phenol 12. Compound 12 was synthesized according to general procedure Section 3.1.2 from 4 (100 mg, 0.42 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (46 mg, 0.42 mmol). Compound 12 was obtained as a brown oil. (Method A: 125 mg, 90%; Method B: 122 mg, 88%; Method C: 124 mg, 89%; Method D: 127 mg, 92%; Method E: 91 mg, 66%). 1H-NMR (600 MHz, DMSO-d6) δ 9.23 (s, 1H, OH), 8.63 (s, 1H, N=CH-7), 7.97 (d, J = 8.8 Hz, 2H, H-9,13), 7.30 (d, J = 2.6 Hz, 1H, H-5), 7.24 (d, J = 7.6 Hz, 2H, H-10,12), 7.09 (d, J = 2.7 Hz, 1H, H-3), 7.07 (d, J = 2.6 Hz, 1H, H-4), 6.89 (dd, J = 8.6, 2.6 Hz, 1H, H-6), 4.27 (t, J = 5.7 Hz, 2H, H-1′), 3.59 (m, 4H, H-4′,4″), 2.76 (t, J = 5.7 Hz, 2H, H-2′), 2.51 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.6, 160.6, 149.2, 143.3, 131.4, 127.7, 127.1, 126.5, 119.1, 117.8, 113.3, 110.0, 66.6,66.2, 56.7, 54.1.
- 4-Chloro-2-((4-(2-(diethylamino)ethoxy)benzylidene)amino)phenol 13. Compound 13 was synthesized according to general procedure Section 3.1.2 from 1 (100 mg, 0.45 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (65 mg, 0.45 mmol). Compound 13 was obtained as a brown oil. (Method A: 139 mg, 89%; Method B: 136 mg, 87%; Method C: 140 mg, 89%; Method D: 144 mg, 91%; Method E: 109 mg, 70%). 1H-NMR (600 MHz, DMSO-d6) δ 9.25 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.96 (d, J = 8.8 Hz, 2H, H-9,13), 7.23 (d, J = 8.6 Hz, 1H, H-5), 7.08 (d, J = 2.6 Hz, 1H, H-3), 7.07 (m, 2H, H-10,12), 6.88 (d, J = 8.6 Hz, 1H, H-6), 4.11 (t, J = 6.1 Hz, 2H, H-1′), 2.81 (t, J = 6.1 Hz, 2H, H-2′), 2.57 (q, J = 7.2 Hz, 4H, H-3′,3″), 1.06 (t, J = 7.0 Hz, 6H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.4, 161.5, 150.9, 143.3, 139.6, 131.5, 127.4, 125.1, 119.7, 117.4, 115.9, 115.1, 113.2, 67.4, 57.4, 54.2, 48.3, 19.8, 12.7. Calcd for C19H23ClN2O2: C, 65.79; H, 6.68; N, 8.08; Found: C, 65.77; H, 6.69; N, 8.06.
- 4-Chloro-2-((4-(2-(pyrrolidin-1-yl)ethoxy)benzylidene)amino)phenol 14. Compound 14 was synthesized according to general procedure Section 3.1.2 from 2 (100 mg, 0.46 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (65 mg, 0.46 mmol). Compound 14 was obtained as an organic oil. (Method A: 145 mg, 92%; Method B: 140 mg, 89%; Method C: 136 mg, 87%; Method D: 147 mg, 93%; Method E: 102 mg, 65%). 1H-NMR (600 MHz, DMSO-d6) δ 8.93 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.97 (d, J = 8.8 Hz, 2H, H-9,13), 7.17 (dd, J = 7.9, 1.6 Hz, 1H, H-5), 7.07 (d, J = 8.8 Hz, 2H, H-10,12), 7.04 (d, J = 1.6 Hz, 1H, H-3), 6.88 (d, J = 8.1 Hz, 1H, H-6), 4.16 (t, J = 5.9 Hz, 2H, H-1′), 2.82 (t, J = 5.9 Hz, 2H, H-2′), 2.54 (m, 4H, H-3′,3″), 1.69 (m, 4H, H-4′,4″). Calcd for C19H21ClN2O2: C, 66.18; H, 6.14; N, 8.12; Found: C, 66.16; H, 6.15; N, 8.13.
- 4-Chloro-2-((4-(2-(piperidin-1-yl)ethoxy)benzylidene)amino)phenol 15. Compound 15 was synthesized according to general procedure Section 3.1.2 from 3 (100 mg, 0.43 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (61 mg, 0.43 mmol). Compound 15 was obtained as a light brown oil. (Method A: 137 mg, 89%; Method B: 134 mg, 84%; Method C: 129 mg, 84%; Method D: 139 mg, 90%; Method E: 117 mg, 76%). 1H-NMR (600 MHz, DMSO-d6) δ 9.25 (s, 1H, OH), 8.66 (s, 1H, N=CH-7), 8.01 (d, J = 8.4 Hz, 2H, H-9,13), 7.40 (d, J = 8.4 Hz, 2H, H-10,12), 7.32 (d, J = 7.6 Hz, 1H, H-5), 7.22 (d, J = 2.4 Hz, 1H, H-3), 6.85 (d, J = 8.6 Hz, 1H, H-6), 4.27 (t, J = 5.8 Hz, 2H, H-1′), 2.72 (m, 2H, H-2′), 2.44 (m, 4H, H-3′,3″), 1.51 (m, 4H, H-4′,4″), 1.38 (p, J = 5.9 Hz, 2H, H-5′). Calcd for C20H23ClN2O2: C, 66.94; H, 6.46; N, 7.81; Found: C, 66.96; H, 6.47; N, 7.80.
- 4-Chloro-2-((4-(2-morpholinoethoxy)benzylidene)amino)phenol 16. Compound 16 was synthesized according to general procedure Section 3.1.2 from 4 (100 mg, 0.42 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (61 mg, 0.42 mmol). Compound 16 was obtained as an orange oil. (Method A: 135 mg, 88%; Method B: 133 mg, 87%; Method C: 130 mg, 85%; Method D: 139 mg, 91%; Method E: 90 mg, 89%). 1H-NMR (600 MHz, DMSO-d6) δ 9.24 (s, 1H, OH), 8.67 (s, 1H, N=CH-7), 8.02 (d, J = 8.6 Hz, 2H, H-9,13), 7.32 (d, J = 8.5 Hz, 1H, H-5), 7.30 (d, J = 7.5 Hz, 2H, H-10,12), 7.11 (d, J = 1.8 Hz, 1H, H-3), 6.90 (d, J = 8.6 Hz, 1H, H-6), 4.26 (t, J = 5.7 Hz, 2H, H-1′), 3.59 (t, J = 4.7 Hz, 4H, H-4′,4″), 2.75 (t, J = 5.7 Hz, 2H, H-2′), 2.49 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.9, 161.7, 148.9, 143.0, 129.3, 125.8, 125.5, 119.1, 118.2, 115.8, 109.7, 66.1, 65.7, 56.7, 54.3. Calcd for C19H21ClN2O3: C, 63.24; H, 5.87; N, 7.76; Found: C, 63.23; H, 5.89; N, 7.77.
- 4-Bromo-2-((4-(2-(diethylamino)ethoxy)benzylidene)amino)phenol 17. Compound 17 was synthesized according to general procedure Section 3.1.2 from 1 (100 mg, 0.45 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-bromophenol (85 mg, 0.45 mmol). Compound 17 was obtained as a brown oil. (Method A: 151 mg, 85%; Method B: 156 mg, 88%; Method C: 143 mg, 80%; Method D: 158 mg, 89%; Method E: 124 mg, 70%). 1H-NMR (600 MHz, DMSO-d6) δ 9.25 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.96 (d, J = 8.8 Hz, 2H, H-9,13), 7.23 (d, J = 8.6 Hz, 1H, H-5), 7.07 (d, J = 7.8 Hz, 2H, H-10,12), 7.06 (d, J = 2.1 Hz, 1H, H-3), 6.88 (d, J = 8.6 Hz, 1H, H-6), 4.11 (t, J = 6.1 Hz, 2H, H-1′), 2.81 (t, J = 6.1 Hz, 2H, H-2′), 2.57 (q, J = 7.2 Hz, 4H, H-3′,3″), 0.99 (t, J = 7.1 Hz, 6H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.1, 161.3, 150.7, 143.4, 138.2, 131.7, 126.2, 124.7, 119.0, 117.7, 115.3, 114.8, 112.5, 67.7, 54.7, 51.2, 47.3, 19.4, 12.2.
- 4-Bromo-2-((4-(2-(pyrrolidin-1-yl)ethoxy)benzylidene)amino)phenol 18. Compound 18 was synthesized according to general procedure Section 3.1.2 from 2 (100 mg, 0.46 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-bromophenol (86 mg, 0.46 mmol). Compound 18 was obtained as a brown oil. (Method A: 144 mg, 81%; Method B: 141 mg, 79%; Method C: 137 mg, 77%; Method D: 152 mg, 86%; Method E: 109 mg, 62%). 1H-NMR (600 MHz, DMSO-d6) δ 8.99 (s, 1H, OH), 8.60 (s, 1H, N=CH-7), 7.76 (d, J = 6.9 Hz, 2H, H-9,13), 7.19 (d, J = 8.4 Hz, 1H, H-5), 7.10 (d, J = 7.3 Hz, 2H, H-10,12), 7.05 (d, J = 1.6 Hz, 1H, H-3), 6.89 (d, J = 8.4 Hz, 1H, H-6), 4.14 (t, J = 6.0 Hz, 2H, H-1′), 2.82 (t, J = 6.0 Hz, 2H, H-2′), 2.54 (m, 4H, H-3′,3″), 1.69 (t, J = 3.6 Hz, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 159.4, 161.9, 150.4, 148.4, 139.6, 138.1, 128.9, 127.7, 125.3, 124.2, 119.3, 117.3, 115.8, 67.3, 57.7, 54.8, 23.4. Calcd for C19H21BrN2O2: C, 58.62; H, 5.44; N, 7.20; Found: C, 58.60; H, 5.46; N, 7.22.
- 4-Bromo-2-((4-(2-(piperidin-1-yl)ethoxy)benzylidene)amino)phenol 19. Compound 19 was synthesized according to general procedure Section 3.1.2 from 3 (100 mg, 0.43 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-bromophenol (81 mg, 0.43 mmol). Compound 19 was obtained as a dark brown oil. (Method A: 152 mg, 87%; Method B: 149 mg, 86%; Method C: 142 mg, 82%; Method D: 150 mg, 87%; Method E: 104 mg, 60%). 1H-NMR (600 MHz, DMSO-d6) δ 9.25 (s, 1H, OH), 8.66 (s, 1H, N=CH-7), 8.02 (d, J = 7.1 Hz, 2H, H-9,13), 7.71 (d, J = 7.5 Hz, 1H, H-5), 7.40 (d, J = 7.1 Hz, 2H, H-10,12), 7.33 (d, J = 2.5 Hz, 1H, H-3), 6.85 (d, J = 8.6 Hz, 1H, H-6), 4.24 (t, J = 5.9 Hz, 2H, H-1′), 2.71 (m, 2H, H-2′), 2.46 (m, 4H, H-3′,3″), 1.50 (q, J = 7.0 Hz, 4H, H-4′,4″), 1.38 (p, J = 6.8 Hz, 2H, H-5′). Calcd for C20H23BrN2O2: C, 59.56; H, 5.75; N, 6.95; Found: C, 59.57; H, 5.76; N, 6.94.
- 4-Bromo-2-((4-(2-morpholinoethoxy)benzylidene)amino)phenol 20. Compound 20 was synthesized according to general procedure Section 3.1.2 from 4 (100 mg, 0.42 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-bromophenol (80 mg, 0.42 mmol). Compound 21 was obtained as a dark brown oil. (Method A: 140 mg, 81%; Method B: 144 mg, 84%; Method C: 130 mg, 85%; Method D: 139 mg, 91%; Method E: 90 mg, 59%). 1H-NMR (600 MHz, DMSO-d6) δ 9.24 (s, 1H, OH), 8.67 (s, 1H, N=CH-7), 8.03 (d, J = 7.6 Hz, 2H, H-9,13), 7.73 (d, J = 7.8 Hz, 1H, H-5), 7.30 (d, J = 7.6 Hz, 2H, H-10,12), 7.12 (d, J = 2.5 Hz, 1H, H-3), 6.90 (d, J = 8.1 Hz, 1H, H-6), 4.26 (t, J = 5.7 Hz, 2H, H-1′), 3.59 (t, J = 4.7 Hz, 4H, H-4′,4″), 2.75 (t, J = 5.7 Hz, 2H, H-2′), 2.49 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.3, 161.4, 148.4, 142.4, 129.1, 128.9, 125.0, 124.2, 119.4, 118.0, 115.2, 109.9, 66.3, 65.4, 56.4, 54.0. Calcd for C19H21BrN2O3: C, 56.31; H, 5.22; N, 6.91; Found: C, 56.30; H, 5.24; N, 6.92.
- 2-((4-(2-(Diethylamino)ethoxy)-3-methoxybenzylidene)amino)phenol 21. Compound 21 was synthesized according to general procedure Section 3.1.2 from 5 (100 mg, 0.40 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (43 mg, 0.40 mmol). Compound 21 was obtained as a light brown oil. (Method A: 120 mg, 88%; Method B: 118 mg, 87%; Method C: 112 mg, 82%; Method D: 115 mg, 84%; Method E: 89 mg, 65%). 1H-NMR (300 MHz, DMSO-d6) δ 8.88 (s, 1H, OH), 8.60 (s, 1H, N=CH-7), 7.76 (d, J = 1.9 Hz, 1H, H-9), 7.46 (d, J = 1.9 Hz, 1H, H-13), 7.20 (d, J = 1.7 Hz, 1H, H-5), 7.08 (d, J = 8.3 Hz, 1H, H-4), 6.90 (d, J = 1.4 Hz, 1H, H-12), 6.88 (d, J = 1.4 Hz, 1H, H-3), 6.82 (d, J = 1.5 Hz, 1H, H-6), 4.08 (t, J = 6.3 Hz, 2H, H-1′), 3.86 (s, 3H, OCH3), 2.81 (t, J = 6.3 Hz, 2H, H-2′), 2.56 (t, J = 7.1 Hz, 4H, H-3′,3″), 0.98 (t, J = 7.1 Hz, 6H, H-4′,4″). Calcd for C20H26N2O3: C, 70.15; H, 7.65; N, 8.18; Found: C, 70.14; H, 7.64; N, 8.19.
- 2-((3-Methoxy-4-(2-(pyrrolidin-1-yl)ethoxy)benzylidene)amino)phenol 22. Compound 22 was synthesized according to general procedure Section 3.1.2 from 6 (100 mg, 0.40 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (43 mg, 0.40 mmol). Compound 22 was obtained as a light orange oil. (Method A: 122 mg, 89%; Method B: 121 mg, 89%; Method C: 108 mg, 80%; Method D: 130 mg, 78%; Method E: 93 mg, 56%). 1H-NMR (600 MHz, DMSO-d6) δ 8.88 (s, 1H, OH), 8.61 (s, 1H, N=CH-7), 7.78 (d, J = 1.9 Hz, 1H, H-9), 7.46 (d, J = 1.9 Hz, 1H, H-13), 7.21 (d, J = 1.6 Hz, 1H, H-5), 7.08 (d, J = 1.6 Hz, 1H, H-4), 6.90 (d, J = 1.4 Hz, 1H, H-12), 6.88 (d, J = 1.4 Hz, 1H, H-3), 6.83 (d, J = 1.4 Hz, 1H, H-6), 4.16 (t, J = 6.2 Hz, 2H, H-1′), 3.84 (s, 3H, OCH3), 2.70 (t, J = 6.1 Hz, 2H, H-2′), 2.45 (m, 4H, H-3′,3″), 1.51 (p, J = 5.6 Hz, 4H,H-4′,4″). 13C-NMR (151 MHz, DMSO-d6) δ 160.5, 153.9, 150.7, 149.3, 138.9, 130.1, 128.7, 126.1, 125.9, 121.3, 118.7, 112.7, 109.1, 67.5, 55.4, 54.1, 54.0, 23.1.
- 2-((3-Methoxy-4-(2-(piperidin-1-yl)ethoxy)benzylidene)amino)phenol 23. Compound 23 was synthesized according to general procedure Section 3.1.2 from 7 (100 mg, 0.38 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (41 mg, 0.38 mmol). Compound 23 was obtained as a brown oil. (Method A: 135 mg, 82%; Method B: 131 mg, 78%; Method C: 124 mg, 74%; Method D: 135 mg, 80%; Method E: 82 mg, 49%). 1H-NMR (600 MHz, DMSO-d6) δ 8.88 (s, 1H, OH), 8.61 (s, 1H, N=CH-7), 7.78 (d, J = 1.9 Hz, 1H, H-9), 7.46 (d, J = 1.9 Hz, 1H, H-5), 7.21 (d, J = 1.6 Hz, 1H, H-4), 7.05 (d, J = 1.6 Hz, 1H, H-12), 6.90 (d, J = 1.4 Hz, 1H, H-3), 6.88 (d, J = 1.4 Hz, 1H, H-6), 4.16 (t, J = 6.2 Hz, 2H, H-1′), 3.84 (s, 1H, OCH3), 2.70 (t, J = 6.1 Hz, 2H, H-2′), 2.46 (m, 4H, H-3′,3″), 1.51 (p, J = 5.6 Hz, 4H, H-4′,4″), 1.39 (p, J = 6.3 Hz, 2H, H-5′). 13C-NMR (151 MHz, DMSO-d6) δ 160.5, 153.9, 150.7, 145.3, 137.4, 130.1, 128.7, 126.1, 125.9, 121.27, 118.69, 112.74, 109.05, 66.7, 59.40, 56.85, 54.3, 53.5, 23.4. Calcd for C21H26N2O3: C, 71.16; H, 7.39; N, 7.90; Found: C, 71.15; H, 7.38; N, 7.92.
- 2-((3-Methoxy-4-(2-morpholinoethoxy)benzylidene)amino)phenol 24. Compound 24 was synthesized according to general procedure Section 3.1.2 from 8 (100 mg, 0.38 mmol), ZnONP (10 mg, 0.12 mmol) and 2-aminophenol (41 mg, 0.38 mmol). Compound 24 was obtained as a dark brown oil. (Method A: 121 mg, 90%; Method B: 113 mg, 84%; Method C: 106 mg, 79%; Method D: 120 mg, 89%; Method E: 74 mg, 55%). 1H-NMR (600 MHz, DMSO-d6) δ 8.90 (s, 1H, OH), 8.60 (s, 1H, N=CH-7), 7.75 (d, J = 1.9 Hz, 1H, H-9), 7.44 (d, J = 1.9 Hz, 1H, H-13), 7.18 (d, J = 1.6 Hz, 1H, H-5), 7.10 (d, J = 8.3 Hz, 1H, H-4), 7.04 (d, J = 1.5 Hz, 1H, H-12), 6.89 (d, J = 1.4 Hz, 1H, H-3), 6.88 (d, J = 1.4 Hz, 1H, H-6), 4.16 (t, J = 5.9 Hz, 2H, H-1′), 3.86 (s, 3H, OCH3), 3.58 (m, 4H, H-4′,4″), 2.72 (t, J = 5.9 Hz, 2H, H-2′), 2.49 (m, 4H, H-3′,3″). Calcd for C20H24N2O4: C, 67.40; H, 6.79; N, 7.86; Found: C, 67.41; H, 6.77; N, 7.85.
- 4-Chloro-2-((4-(2-(diethylamino)ethoxy)-3-methoxybenzylidene)amino)phenol 25. Compound 25 was synthesized according to general procedure Section 3.1.2 from 5 (100 mg, 0.40 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (57 mg, 0.40 mmol). Compound 25 was obtained as a dark yellow oil. (Method A: 120 mg, 82%; Method B: 118 mg, 87%; Method C: 112 mg, 82%; Method D: 115 mg, 84%; Method E: 89 mg, 65%). 1H-NMR (600 MHz, DMSO-d6) δ 8.90 (s, 1H, OH), 8.60 (s, 1H, N=CH-7), 7.75 (d, J = 1.9 Hz, 1H, H-9), 7.44 (d, J = 1.9 Hz, 1H, H-13), 7.18 (d, J = 1.6 Hz, 1H, H-5), 7.06 (d, J = 1.6 Hz, 1H, H-3), 6.83 (d, J = 1.4 Hz, 1H, H-12), 6.81 (d, J = 1.4 Hz, 1H,H-6), 4.16 (t, J = 5.9 Hz, 2H, H-1′), 3.86 (s, 3H, OCH3), 2.72 (t, J = 5.9 Hz, 2H, H-2′), 2.49 (m, 4H, H-3′,3″), 1.06 (t, J = 7.0 Hz, 6H, H-4′,4″). 13C-NMR (151 MHz, DMSO-d6) δ 161.6, 153.7, 151.8, 149.4, 138.6, 130.6, 129.0, 126.2, 125.7, 123.5, 119.3, 111.2, 110.5, 67.4, 55.3, 51.2, 46.4, 11.7. Calcd for C20H25ClN2O3: C, 63.74; H, 6.69; N, 7.43; Found: C, 63.75; H, 6.68; N, 7.41.
- 4-Chloro-2-((3-methoxy-4-(2-(pyrrolidin-1-yl)ethoxy)benzylidene)amino)phenol 26. Compound 26 was synthesized according to general procedure Section 3.1.2 from 6 (100 mg, 0.40 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (56 mg, 0.40 mmol). Compound 26 was obtained as a brown oil. (Method A: 125 mg, 83%; Method B: 131 mg, 78%; Method C: 124 mg, 74%; Method D: 135 mg, 80%; Method E: 82 mg, 49%). 1H-NMR (600 MHz, DMSO-d6) δ 8.99 (s, 1H), 8.60 (s, 1H, N=CH-7), 7.76 (d, J = 1.9 Hz, 1H, H-9), 7.45 (d, J = 1.9 Hz, 1H, H-13), 7.20 (d, J = 1.6 Hz, 1H, H-5), 7.10 (d, J = 8.3 Hz, 1H, H-12), 7.05 (d, J = 1.6 Hz, 1H, H-3), 6.89 (d, J = 8.1 Hz, 1H, H-6), 4.14 (t, J = 6.0 Hz, 2H, H-1′), 3.87 (s, 3H, OCH3), 2.82 (t, J = 6.0 Hz, 2H, H-2′), 2.54 (m, 4H, H-3′,3″), 1.69 (p, J = 3.1 Hz, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 160.4, 153.6, 150.8, 149.2, 139.4, 130.7, 129.8, 126.6, 123.3, 118.2, 114.5, 112.3, 110.6, 66.2, 57.0, 57.4, 56.6, 21.3. Calcd for C20H23ClN2O3: C, 64.08; H, 6.18; N, 7.47; Found: C, 64.09; H, 6.17; N, 7.45.
- 4-Chloro-2-((3-methoxy-4-(2-(piperidin-1-yl)ethoxy)benzylidene)amino)phenol 27. Compound 27 was synthesized according to general procedure Section 3.1.2 from 7 (100 mg, 0.38 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (55 mg, 0.38 mmol). Compound 27 was obtained as a light brown oil. (Method A: 126 mg, 86%; Method B: 122 mg, 83%; Method C: 125 mg, 85%; Method D: 129 mg, 88%; Method E: 86 mg, 58%). 1H-NMR (600 MHz, DMSO-d6) δ 9.25 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.47 (d, J = 1.9 Hz, 1H, H-9), 7.26 (d, J = 2.6 Hz, 1H, H-13), 7.12 (d, J = 8.3 Hz, 1H, H-5), 7.09 (d, J = 2.6 Hz, 1H, H-12), 7.08 (d, J = 2.6 Hz, 1H, H-3), 6.90 (d, J = 8.6 Hz, 1H, H-6), 4.17 (t, J = 6.0 Hz, 2H, H-1′), 3.86 (s, 1H, OCH3), 2.76 (m, 2H, H-2′), 2.51 (m, 4H, H-3′,3″), 1.53 (p, J = 5.6 Hz, 4H, H-4′,4″), 1.38 (m, 2H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.0, 159.8 150.5, 140.1, 130.2, 129.6, 127.4, 126.5, 120.0, 119.7, 118.0, 112.4, 66.1, 66.5, 57.2, 57.6, 56.7. Calcd for C21H25ClN2O3: C, 64.86; H, 6.48; N, 7.20; Found: C, 64.84; H, 6.47; N, 7.22.
- 4-Chloro-2-((3-methoxy-4-(2-morpholinoethoxy)benzylidene)amino)phenol 28. Compound 28 was synthesized according to general procedure Section 3.1.2 from 8 (100 mg, 0.37 mmol), ZnONP (10 mg, 0.12 mmol) and 2-amino-4-chlorophenol (54 mg, 0.37 mmol). Compound 28 was obtained as an orange oil. (Method A: 121 mg, 82%; Method B: 119 mg, 80%; Method C: 101 mg, 69%; Method D: 123 mg, 89%; Method E: 77 mg, 52%). 1H-NMR (600 MHz, DMSO-d6) δ 9.25 (s, 1H, OH), 8.62 (s, 1H, N=CH-7), 7.75 (d, J = 1.9 Hz, 1H, H-9), 7.47 (d, J = 1.9 Hz, 1H, H-13), 7.27 (d, J = 2.5 Hz, 1H, H-5), 7.12 (d, J = 8.3 Hz, 1H, H-12), 7.10 (d, J = 2.6 Hz, 1H, H-3), 6.90 (d, J = 8.6 Hz, 1H, H-6), 4.37 (t, J = 5.1 Hz, 2H, H-1′), 3.86 (s, 1H, OCH3), 3.59 (d, J = 4.7 Hz, 4H, H-4′,4″), 2.73 (dd, J = 7.0, 4.5 Hz, 2H, H-2′), 2.49 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 160.5, 153.9, 150.7, 149.4, 142.1, 138.9, 130.0, 128.7, 126.1, 121.3, 118.4, 112.6, 109.3, 67.4, 65.4, 57.5, 55.1, 55.0. Calcd for C21H25ClN2O4: C, 61.46; H, 5.93; N, 7.17; Found: C, 61.44; H, 5.94; N, 7.19.
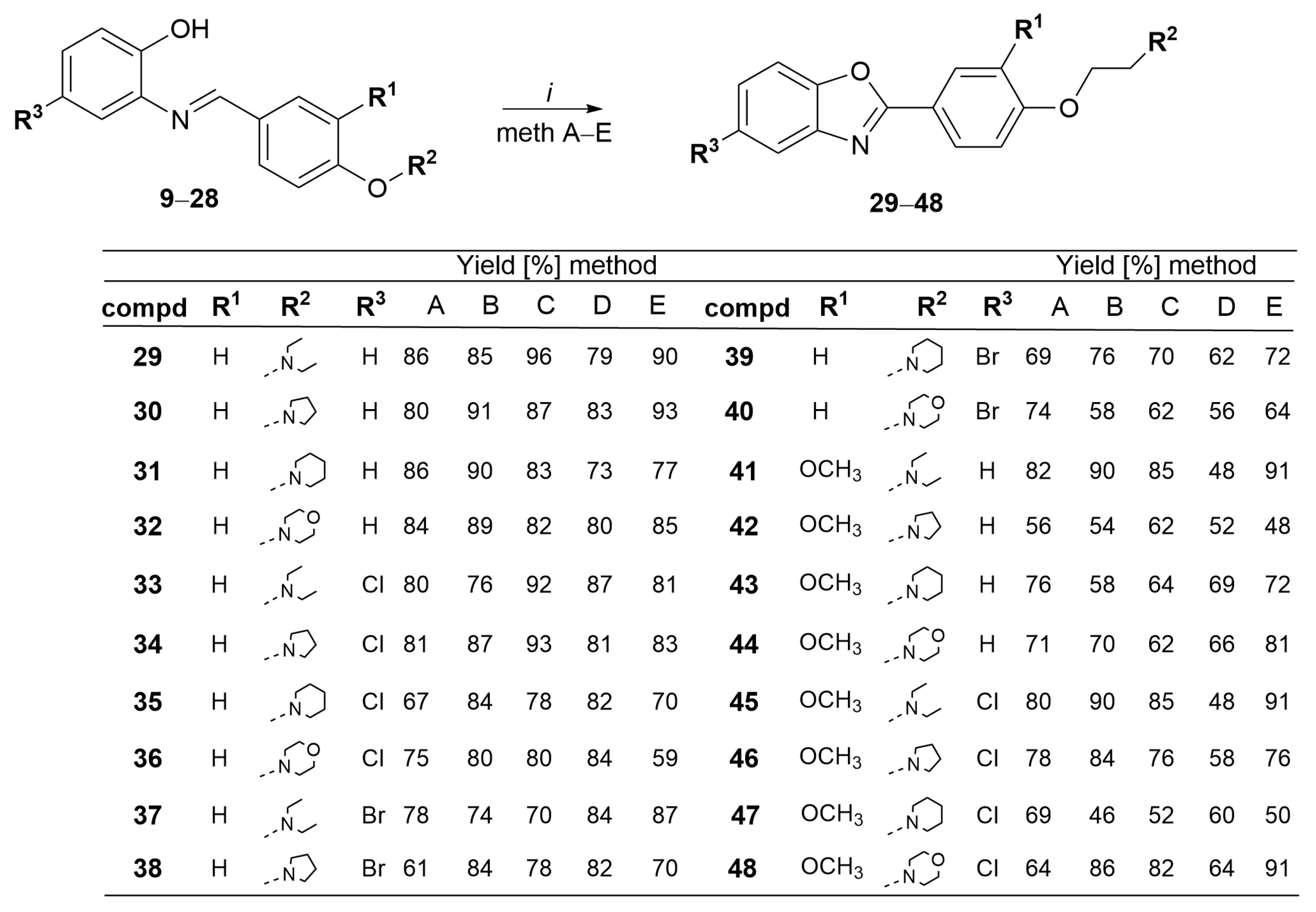
3.1.3. General Procedure for the Synthesis of Benzoxazole Derivatives 29–48
- 2-(4-(2-Diethylaminoethoxy)phenyl)benzo[d]oxazole 29. Compound 29 was synthesized according to general procedure Section 3.1.3 from 9 (50 mg, 0.16 mmol) and NaCN. After purification by column chromatography compound 29 was obtained as a white powder. (Method A: 45 mg, 86%; Method B: 42 mg, 85%; Method C: 48 mg, 96%; Method D: 39 mg, 79%; Method E: 74 mg, 96%, m.p. = 115–117 °C). 1H NMR (300 MHz, DMSO-d6) δ 8.17 (d, J = 8.8 Hz, 2H, H-9,13), 7.77 (dt, J = 6.7, 2.1 Hz, 2H, H-5,6), 7.39 (m, 2H, H-4,7), 7.21 (d, J = 8.9 Hz, 2H, H-10,12), 4.40 (s, 2H, H-1′), 3.05 (s, 2H, H-2′), 2.49 (m, 4H, H-3′,3″), 1.19 (t, J = 7.4 Hz, 6H, H-4′,4″). 13C-NMR (151 MHz, DMSO-d6) δ 161.9, 159.1, 150.34, 141.86, 129.41, 124.9, 124.1, 119.7, 119.2, 115.53, 110.4, 67.3, 51.5, 46.01, 9. EI+ mode: m/z = 310.9, [M+] (calcd for C19H22N2O2 = 310.17), Calcd for C19H22N2O2: C, 73.52; H, 7.14; N, 9.03. Found: C, 73.50; H, 7.19; N, 9.05.
- 2-(4-(2-(Pyrrolidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 30. Compound 30 was synthesized according to general procedure Section 3.1.3 from 10 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 30 was obtained as a light-yellow powder. (Method A: 40 mg, 80%; Method B: 45 mg, 91%; Method C: 43 mg, 87%; Method D: 41 mg, 83%; Method E: 46 mg, 93%, m.p. = 142–144 °C). 1H-NMR (300 MHz, DMSO-d6) δ 8.15 (d, J = 8.9 Hz, 2H, H-9,13), 7.76 (m, 2H, H-5,6), 7.39 (m, 2H, H-4,7), 7.19 (d, J = 8.8 Hz, 2H, H-10,12), 4.24 (t, J = 5.8 Hz, 2H, H-1′), 2.70 (s, 2H, H-2′), 2.52 (m, 4H, H-3′,3″), 1.75 (m, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.7, 162.1, 149.7, 143.2, 129.7, 126.2, 125.9, 121.2, 118.2, 116.6, 111.4, 67.1, 54.8, 54.2, 22.3. Calcd for C19H20N2O2: C, 74.00; H, 6.54; N, 9.08. Found: C, 73.98; H, 6.53; N, 9.06.
- 2-(4-(2-(Piperidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 31. Compound 31 was synthesized according to general procedure Section 3.1.3 from 11 (50 mg, 0.15 mmol) and NaCN. After purification by column chromatography compound 31 was obtained as a white powder. (Method A: 43 mg, 86%; Method B: 45 mg, 90%; Method C: 41 mg, 83%; Method D: 36 mg, 73%; Method E: 38 mg, 77%, m.p. = 137–139 °C). 1H-NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.8 Hz, 2H, H-9,13), 7.76 (m, 2H, H-5,6), 7.39 (m, 2H, H-4,7), 7.16 (d, J = 8.8 Hz, 2H, H-10,12), 4.18 (t, J = 5.9 Hz, 2H, H-1′), 2.69 (t, J = 5.8 Hz, 2H, H-2′), 2.45 (t, J = 5.3 Hz, 4H, H-3′,3″), 1.50 (p, J = 5.5 Hz, 2H, H-4′,4″), 1.38 (m, 2H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.8, 160.3, 149.6, 143.9, 129.8, 125.0, 124.7, 121.3, 119.5, 114.1, 113.2, 110.3, 65.2, 57.2, 54.0, 25.4, 24.0. Calcd for C20H22N2O2: C, 74.51; H, 6.88; N, 8.69. Found: C, 74.49; H, 6.87; N, 8.71.
- 2-(4-(2-Morpholinoethoxy)phenyl)benzo[d]oxazole 32. Compound 32 was synthesized according to general procedure Section 3.1.3 from 12 (50 mg, 0.15 mmol) and NaCN. After purification by column chromatography compound 32 was obtained as a white powder. (Method A: 42 mg, 84%; Method B: 44 mg, 89%; Method C: 41 mg, 82%; Method D: 38 mg, 80%; Method E: 42 mg, 85%, m.p. = 156–158 °C). 1H-NMR (600 MHz, DMSO-d6) δ 8.13 (d, J = 8.8 Hz, 2H, H-9,13), 7.75 (m, 2H, H-5,6), 7.39 (m, 2H, H-4,7), 7.16 (d, J = 8.8 Hz, 2H, H-10,12), 4.20 (t, J = 5.7 Hz, 2H, H-1′), 3.58 (t, J = 4.7 Hz, 4H, H-4′,4″), 2.72 (t, J = 5.7 Hz, 2H, H-2′), 2.48 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 162.6, 162.0, 159.6, 150.4, 142.1, 129.2, 125.9, 124.1, 120.4, 119.6, 118.2, 114.4, 110.2, 67.2, 65.2, 47.3, 44.1. EI+ mode: m/z = 324.9, [M+] (calcd for C19H20N2O3 = 324.15), Calcd for C19H20N2O3: C, 70.35; H, 6.21; N, 8.64; Found: C, 70.36; H, 6.23; N, 8.63.
- 5-Chloro-2-(4-(2-diethylaminoethoxy)phenyl)benzo[d]oxazole 33. Compound 33 was synthesized according to general procedure Section 3.1.3 from 13 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 33 was obtained as a yellowish powder. (Method A: 39 mg, 80%; Method B: 38 mg, 76%; Method C: 41 mg, 92%; Method D: 43 mg, 87%; Method E: 40 mg, 81%, m.p. = 130–132 °C). 1H-NMR (600 MHz, DMSO-d6) δ 7.86 (d, J = 2.1 Hz, 1H, H-4), 7.80 (d, J = 8.6 Hz, 2H, H-9,13), 7.67 (d, J = 2.0 Hz, 1H, H-7), 7.43 (dd, J = 8.6, 2.1 Hz, 1H, H-6), 7.21 (d, J = 8.4 Hz, 2H, H-10,12), 4.14 (t, J = 6.2 Hz, 2H, H-1′), 2.86 (t, J = 6.1 Hz, 2H, H-2′), 2.60 (q, J = 7.1 Hz, 4H, H-3′,3″), 1.00 (t, J = 7.2 Hz, 6H, H-4′,4″). 13C-NMR (151 MHz, DMSO-d6) δ 162.4, 161.2, 149.4, 143.3, 129.6, 127.7, 126.3, 122.0, 116.7, 113.1, 112.5, 64.2, 54.5, 45.6, 12.8. Calcd for C19H21ClN2O2: C, 66.18; H, 6.14; N, 8.12. Found: C, 66.19; H, 6.15; N, 8.10.
- 5-Chloro-2-(4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 34. Compound 34 was synthesized according to general procedure Section 3.1.3 from 14 (50 mg, 0.15 mmol) and NaCN. After purification by column chromatography compound 34 was obtained as a light-yellow powder. (Method A: 41 mg, 81%; Method B: 43 mg, 87%; Method C: 46 mg, 93%; Method D: 40 mg, 81%; Method E: 43 mg, 83%, m.p. = 146–148 °C). 1H-NMR (300 MHz, DMSO-d6) δ 8.12 (d, J = 8.9 Hz, 2H, H-9,13), 7.99 (d, J = 2.0 Hz, 1H, H-4), 7.74 (d, J = 8.6 Hz, 1H, H-7), 7.55 (dd, J = 8.6, 2.0 Hz, 1H, H-6), 7.17 (d, J = 8.9 Hz, 2H, H-10,12), 4.18 (t, J = 5.9 Hz, 2H, H-1′), 2.68 (t, J = 5.9 Hz, 2H, H-2′), 2.42 (m, 4H, H-3′,3″), 1.50 (p, J = 5.5 Hz, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.9, 159.9, 153.6, 143.5, 129.8, 125.6, 124.8, 122.0, 121.1, 114.6, 109.3, 66.2, 59.5, 54.3, 26.5. Calcd for C19H19ClN2O2: C, 66.57; H, 5.59; N, 8.17;. Found: C, 66.55; H, 5.61; N, 8.15.
- 5-Chloro-2-(4-(2-(piperidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 35. Compound 35 was synthesized according to general procedure Section 3.1.3 from 15 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 35 was obtained as a white powder. (Method A: 30 mg, 67%; Method B: 42 mg, 84%; Method C: 39 mg, 78%; Method D: 41 mg, 82%; Method E: 35 mg, 70%, m.p. = 141–143 °C). 1H-NMR (600 MHz, DMSO-d6) δ 7.76 (m, 3H, H-7,9,13), 7.70 (d, J = 2.0 Hz, 1H, H-4), 7.40 (d, J = 3.7 Hz, 1H, H-6), 7.21 (d, J = 8.5 Hz, 2H, H-10,12), 4.18 (t, J = 5.9 Hz, 2H, H-1′), 2.74 (t, J = 4.7 Hz, 2H, H-2′), 2.48 (m, 4H, H-3′,3″), 1.52 (t, J = 5.6 Hz, 4H, H-4′,4″), 1.39 (m, 2H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 162.9, 159.7, 149.2, 143.6, 131.6, 128.7, 126.7, 121.9, 116.5, 113.1, 110.2, 64.3, 57.6 54.2, 38.0. Calcd for C20H21ClN2O2: C, 67.32; H, 5.93; N, 7.85. Found: C, 67.30; H, 5.94; N, 7.84.
- 5-Chloro-2-(4-(2-morpholinoethoxy)phenyl)benzo[d]oxazole 36. Compound 36 was synthesized according to general procedure Section 3.1.3 from 16 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 36 was obtained as a yellowish powder. (Method A: 35 mg, 75%; Method B: 39 mg, 80%; Method C: 37 mg, 80%; Method D: 42 mg, 84%; Method E: 30 mg, 59%, m.p. = 141–143 °C). 1H-NMR (300 MHz, DMSO-d6) δ 8.12 (d, J = 8.9 Hz, 2H, H-9,13), 7.85 (d, J = 2.2 Hz, 1H, H-4), 7.78 (d, J = 8.6 Hz, 1H, H-7), 7.42 (dd, J = 8.7, 2.2 Hz, 1H, H-6), 7.17 (d, J = 8.9 Hz, 2H, H-10,12), 4.21 (t, J = 5.7 Hz, 2H, H-1′), 3.59 (t, J = 4.6 Hz, 4H, H-4′,4″), 2.75 (t, J = 4.4 Hz, 2H, H-2′), 2.49 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 162.0, 159.1, 149.3, 143.1, 129.8, 129.1, 127.7, 121.1, 118.2, 116.6, 100.02, 66.1, 65.8, 56.8, 53.6. Calcd for C19H19ClN2O3: C, 63.60; H, 5.34; N, 7.81. Found: C, 63.61; H, 5.32; N, 7.80.
- 5-Bromo-2-(4-(2-diethylaminoethoxy)phenyl)benzo[d]oxazole 37. Compound 37 was synthesized according to general procedure Section 3.1.3 from 17 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 37 was obtained as a beige powder. (Method A: 39 mg, 78%; Method B: 37 mg, 74%; Method C: 35 mg, 70%; Method D: 42 mg, 84%; Method E: 44 mg, 87%, m.p. = 139–141 °C). 1H-NMR (600 MHz, DMSO-d6) δ 7.86 (s, 1H, H-4), 7.80 (d, J = 8.6 Hz, 2H, H-9,13), 7.77 (dd, J = 8.4, 2.0 Hz, 1H, H-7), 7.43 (dd, J = 8.6, 2.1 Hz, 1H, H-6), 7.21 (d, J = 8.4 Hz, 2H, H-10,12), 4.14 (s, 2H, H-1′), 2.86 (t, J = 6.1 Hz, 2H, H-2′), 2.60 (q, J = 7.1 Hz, 4H, H-3′,3″), 1.00 (t, J = 7.2 Hz, 6H, H-4′,4″). 13C-NMR (151 MHz, DMSO-d6) δ 162.4, 160.1, 151.2, 143.9, 130.2, 127.2, 126.5, 121.3, 118.2, 115.5, 100.4, 67.1, 54.6, 44.2, 13.3. Calcd for C19H21BrN2O2: C, 58.62; H, 5.44; N, 7.20. Found: C, 58.62; H, 5.43; N, 7.22.
- 5-Bromo-2-(4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 38. Compound 38 was synthesized according to general procedure Section 3.1.3 from 18 (50 mg, 0.13 mmol) and NaCN. After purification by column chromatography compound 38 was obtained as a beige powder. (Method A: 31 mg, 61%; Method B: 42 mg, 84%; Method C: 39 mg, 78%; Method D: 41 mg, 82%; Method E: 35 mg, 70%, m.p. = 157–159 °C). 1H-NMR (600 MHz, DMSO-d6) δ 8.12 (d, J = 8.9 Hz, 2H, H-9,13), 7.99 (d, J = 2.0 Hz, 1H, H-4), 7.74 (d, J = 8.7 Hz, 1H, H-7), 7.55 (dd, J = 8.6, 2.0 Hz, 1H, H-6), 7.17 (d, J = 8.9 Hz, 2H, H-10,12), 4.18 (t, J = 5.9 Hz, 2H, H-1′), 2.68 (t, J = 5.9 Hz, 2H, H-2′), 2.44 (t, J = 5.2 Hz, 4H, H-3′,3″), 1.49 (d, J = 5.4 Hz, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 162.4, 160.1, 151.5, 143.5, 129.8, 126.5, 125.7, 121.7, 120.3, 117.1, 101.3, 67.8, 57.8, 39.8, 23.6. Calcd for C19H19BrN2O2: C, 58.93; H, 4.95; N, 7.23. Found: C, 58.95; H, 4.97; N, 7.25.
- 5-Bromo-2-(4-(2-(piperidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 39. Compound 39 was synthesized according to general procedure Section 3.1.3 from 19 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 39 was obtained as a yellowish powder. (Method A: 34 mg, 69%; Method B: 38 mg, 76%; Method C: 35 mg, 70%; Method D: 31 mg, 62%; Method E: 36 mg, 72%, m.p. = 155–157 °C). 1H-NMR (300 MHz, DMSO-d6) δ 8.12 (d, J = 8.4 Hz, 2H, H-9,13), 8.00 (s, 1H, H-4), 7.75 (d, J = 8.5 Hz, 1H, H-7), 7.55 (d, J = 8.6 Hz, 1H, H-6), 7.18 (d, J = 8.5 Hz, 2H, H-10,12), 4.19 (t, J = 5.8 Hz, 2H, H-1′), 2.69 (t, J = 5.9 Hz, 2H, H-2′), 2.44 (m, 4H, H-3′,3″), 1.55 (m, 4H, H-4′,4″), 1.39 (m, 1H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.7, 162.2, 149.2, 143.7, 129.5, 126.2, 125.7, 120.6, 118.2, 116.6, 113.1, 65.1, 57.1, 39.5, 23.9. Calcd for C20H21BrN2O2: C, 59.86; H, 5.27; N, 6.98. Found: C, 59.83; H, 5.28; N, 6.96.
- 5-Bromo-2-(4-(2-morpholinoethoxy)phenyl)benzo[d]oxazole 40. Compound 40 was synthesized according to general procedure Section 3.1.3 from 20 (50 mg, 0.12 mmol) and NaCN. After purification by column chromatography compound 40 was obtained as a light-yellow powder. (Method A: 37 mg, 74%; Method B: 29 mg, 58%; Method C: 31 mg, 62%; Method D: 28 mg, 56%; Method E: 32 mg, 64%, m.p. = 173–175 °C). 1H-NMR (300 MHz, DMSO-d6) δ 7.76 (m, 3H, H-7,9,13), 7.70 (d, J = 2.0 Hz, 1H, H-4), 7.39 (m, 1H, H-6), 7.22 (d, J = 8.5 Hz, 2H, H-10,12), 4.19 (t, J = 5.9 Hz, 2H, H-1′), 3.59 (t, J = 4.7 Hz, 4H, H-4′,4″), 2.74 (t, J = 5.8 Hz, 2H, H-2′), 2.48 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.6, 159.1, 149.8, 143.1, 132.4, 130.2, 127.2, 122.0, 118.9, 117.0, 113.2, 112.4, 111.5, 66.3, 54.3, 51.7, 39.5. EI+ mode: m/z = 404.6, [M+] (calcd for C19H19BrN2O3 = 404.06), Calcd for C19H19BrN2O3: C, 56.59; H, 4.75; N, 6.95. Found: C, 56.60; H, 4.74; N, 6.94.
- 2-(4-(2-Diethylaminoethoxy)-3-methoxyphenyl)benzo[d]oxazole 41. Compound 41 was synthesized according to general procedure Section 3.1.3 from 21 (50 mg, 0.15 mmol) and NaCN. After purification by column chromatography compound 41 was obtained as a yellow powder. (Method A: 41 mg, 82%; Method B: 45 mg, 90%; Method C: 42 mg, 85%; Method D: 48 mg, 48%; Method E: 45 mg, 91%, m.p. = 160–162 °C). 1H-NMR (600 MHz, DMSO-d6) δ 7.78 (m, 3H, H-5,6,13), 7.70 (d, J = 2.0 Hz, 1H, H-9), 7.39 (m, 2H, H-4,7), 7.21 (d, J = 8.4 Hz, 1H, H-12), 4.13 (t, J = 6.2 Hz, 2H, H-1′), 3.90 (s, 3H, OCH3), 2.60 (m, 2H, H-2′), 2.51 (m, 2H, H-3′,3″), 1.00 (t, J = 7.1 Hz, 6H). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.7, 152.0, 150.7, 149.7, 143.9, 127.5, 122.2, 121.9, 118.3, 116.4, 113.2, 112.5, 100.9, 67.3, 55.5, 51.0, 47.0, 20.7. Calcd for C20H24N2O3: C, 70.57; H, 7.11; N, 8.23. Found: C, C, 70.59; H, 7.13; N, 8.21.
- 2-(3-Methoxy-4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 42. Compound 42 was synthesized according to general procedure Section 3.1.3 from 22 (50 mg, 0.15 mmol) and NaCN. After purification by column chromatography compound 42 was obtained as a white powder. (Method A: 28 mg, 56%; Method B: 27 mg, 54%; Method C: 31 mg, 62%; Method D: 26 mg, 52%; Method E: 24 mg, 48%, m.p. = 167–169 °C). 1H-NMR (600 MHz, DMSO-d6) δ 7.88 (d, J = 2.1 Hz, 1H, H-9), 7.81 (d, J = 8.6 Hz, 2H, H-5,6), 7.78 (dd, J = 8.4, 2.0 Hz, 1H, H-13), 7.68 (d, J = 2.0 Hz, 1H, H-7), 7.44 (dd, J = 8.6, 2.2 Hz, 1H, H-4), 7.23 (d, J = 8.5 Hz, 1H, H-12), 4.18 (t, J = 6.0 Hz, 2H, H-1′), 3.90 (s, 1H, OCH3), 2.70 (t, J = 6.0 Hz, 2H, H-2′), 2.45 (s, 4H, H-3′,3″), 1.50 (p, J = 5.6 Hz, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.1, 151.3, 149.4, 149.3, 141.28, 127.7, 122.0, 122.4, 118.4, 116.7, 113.2, 112.0, 100.1, 67.7, 55.5, 54.1, 54.0, 22.1. Calcd for C20H22N2O3: C, 70.99; H, 6.55; N, 8.28;. Found: C, 70.97; H, 6.54; N, 8.29.
- 2-(3-Methoxy-4-(2-(piperidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 43. Compound 43 was synthesized according to general procedure 3.1.3 from 23 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 43 was obtained as a white powder. (Method A: 41 mg, 76%; Method B: 35 mg, 58%; Method C: 39 mg, 64%; Method D: 42 mg, 69%; Method E: 47 mg, 72%, m.p. = 167–169 °C). 1H-NMR (300 MHz, DMSO-d6) δ 7.78 (m, 3H, H-5,6,13), 7.41 (m, 2H, H-9,12), 7.26 (d, J = 8.5 Hz, 2H, H-4,7), 4.40 (s, 2H, H-1′), 3.92 (s, 3H, OCH3), 3.02 (s, 1H, H-2′), 2.48 (m, 4H, H-3′,3″), 1.68 (m, 4H, H-4′,4″), 1.50 (s, 2H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.8, 152.3, 150.3, 149.5, 141.4, 124.3, 122.1, 117.8, 116.0, 113.5, 111.4, 112.1, 110.7, 67.2, 55.4, 54.2, 53.9. Calcd for C21H24N2O3: C, 71.57; H, 6.86; N, 7.95. Found: C, 71.59; H, 6.85; N, 7.97.
- 2-(3-Methoxy-4-(2-morpholinoethoxy)phenyl)benzo[d]oxazole 44. Compound 44 was synthesized according to general procedure Section 3.1.3 from 24 (50 mg, 0.14 mmol) and NaCN. After purification by column chromatography compound 44 was obtained as a white powder. (Method A: 36 mg, 71%; Method B: 35 mg, 70%; Method C: 31 mg, 62%; Method D: 33 mg, 66%; Method E: 40 mg, 81%, m.p. = 190–192 °C). 1H-NMR (300 MHz, DMSO-d6) δ 7.80 (m, 2H, H-5,6), 7.70 (d, J = 2.0 Hz, 1H, H-9), 7.38 (m, 2H, H-12,13), 7.22 (d, J = 8.5 Hz, 2H, H-4,7), 4.19 (t, J = 5.9 Hz, 2H, H-1′), 3.90 (s, 1H, OCH3), 3.59 (t, J = 4.7 Hz, 2H, H-4′,4″), 2.74 (t, J = 5.8 Hz, 2H, H-2′), 2.48 (m, 2H, H-3′,3″). 13C-NMR (151 MHz, DMSO-d6) δ 161.7, 152.9, 150.3, 149.7, 143.6, 127.6, 126.9, 121.1, 117.6, 116.0, 113.1, 112.2, 111.6, 66.5, 56.2, 54.2, 52.4, 39.5. EI+ mode: m/z = 354.9, [M+] (calcd for C20H22N2O4 = 354.16), Calcd for C20H22N2O4: C, 67.78; H, 6.26; N, 7.80. Found: C, 67.90; H, 6.25; N, 7.91.
- 5-Chloro-2-(4-(2-diethylaminoethoxy)-3-methoxyphenyl)benzo[d]oxazole 45. Compound 45 was synthesized according to general procedure Section 3.1.3 from 25 (50 mg, 0.15 mmol) and NaCN. After purification by column chromatography compound 45 was obtained as a white powder. (Method A: 40 mg, 80%; Method B: 45 mg, 90%; Method C: 42 mg, 85%; Method D: 48 mg, 48%; Method E: 45 mg, 91%, m.p. = 165–167 °C). 1H-NMR (300 MHz, DMSO-d6) δ 7.83 (m, 1H, H-6), 7.74 (m, 1H, H-13), 7.65 (d, J = 2.0 Hz, 1H, H-9), 7.43 (m, 2H, H-4,7), 7.18 (d, J = 8.5 Hz, 1H, H-12), 4.10 (t, J = 6.2 Hz, 2H, H-1′), 3.89 (s, 1H, OCH3), 2.83 (t, J = 6.2 Hz, 2H, H-2′), 2.58 (m, 1H, H-3′,3″), 0.99 (t, J = 7.1 Hz, 6H, H-4′,4″). 13C-NMR (151 MHz, DMSO-d6) δ 162.6, 152.4, 150.9, 149.5, 143.6, 127.7, 125.1, 121.9, 118.2, 116.6, 113.4, 112.5, 111.2, 67.3, 55.5, 51.7, 46.2, 11.3. Calcd for C20H23ClN2O3: C, 64.08; H, 6.18; N, 7.47. Found: C, 64.05; H, 6.20; N, 7.49.
- 5-Chloro-2-(3-methoxy-4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 46. Compound 46 was synthesized according to general procedure Section 3.1.3 from 26 (50 mg, 0.13 mmol) and NaCN. After purification by column chromatography compound 46 was obtained as a beige powder. (Method A: 39 mg, 78%; Method B: 42 mg, 84%; Method C: 38 mg, 76%; Method D: 29 mg, 58%; Method E: 38 mg, 76%, m.p. = 163–165 °C). 1H-NMR (300 MHz, DMSO-d6) δ 7.88 (d, J = 2.1 Hz, 1H, H-4), 7.82 (s, 1H, H-9), 7.78 (m, 1H, H-6), 7.68 (d, J = 2.0 Hz, 1H, H-7), 7.44 (dd, J = 8.6, 2.2 Hz, 2H, H-13,12), 4.18 (t, J = 6.0 Hz, 2H, H-2′), 3.90 (s, 1H,OCH3), 2.70 (t, J = 6.0 Hz, 2H, H-2′), 2.45 (s, 4H, H-3′,3″), 1.50 (p, J = 5.6 Hz, 4H, H-4′,4″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 161.2, 152.2, 150.4, 149.7, 143.6, 130.2, 127.6, 123.4, 121.1, 119.5, 113.1, 112.2, 110.6, 108.8, 66.5, 58.1.2, 56.7, 54.4, 39.5. Calcd for C20H21ClN2O3: C, 64.43; H, 5.68; N, 7.51;. Found: C, 64.45; H, 5.70; N, 7.49.
- 5-Chloro-2-(3-methoxy-4-(2-(piperidin-1-yl)ethoxy)phenyl)benzo[d]oxazole 47. Compound 47 was synthesized according to general procedure Section 3.1.3 from 27 (50 mg, 0.12 mmol) and NaCN. After purification by column chromatography compound 47 was obtained as a white powder. (Method A: 34 mg, 69%; Method B: 23 mg, 46%; Method C: 26 mg, 52%; Method D: 30 mg, 60%; Method E: 25 mg, 50%, m.p. = 172–174 °C). 1H-NMR (600 MHz, DMSO-d6) δ 7.88 (d, J = 2.1 Hz, 1H, H-4), 7.82 (s, 1H, H-9), 7.78 (dd, J = 8.4, 2.0 Hz, 1H, H-13), 7.68 (d, J = 2.0 Hz, 1H, H-6), 7.44 (dd, J = 8.6, 2.2 Hz, 1H, H-12), 7.23 (d, J = 8.5 Hz, 1H, H-7), 4.18 (t, J = 6.0 Hz, 2H, H-1′), 3.90 (s, 3H, OCH3), 2.70 (t, J = 6.0 Hz, 2H, H-2′), 2.45 (m, 4H, H-3′,3″), 1.50 (m, 4H, H-4′,4″), 1.39 (m, 2H, H-5′). 13C-NMR/APT (151 MHz, DMSO-d6) δ 162.1, 152.9, 151.7, 149.1, 143.3, 130.2, 126.4, 121.1, 117.9, 116.5, 114.3, 112.5, 111.8, 67.0, 58.1, 56.2, 55.1, 22.4, 21.3. Calcd for C21H23ClN2O3: C, 65.20; H, 5.99; N, 7.24; Found: C, 65.21; H, 6.01; N, 7.22.
- 5-Chloro-2-(3-methoxy-4-(2-morpholinoethoxy)phenyl)benzo[d]oxazole 48. Compound 48 was synthesized according to general procedure Section 3.1.3 from 28 (50 mg, 0.13 mmol) and NaCN. After purification by column chromatography compound 48 was obtained as a white powder. (Method A: 32 mg, 64%; Method B: 44 mg, 88%; Method C: 41 mg, 82%; Method D: 32 mg, 64%; Method E: 45 mg, 91%, m.p. = 166–168 °C). 1H-NMR (600 MHz, DMSO-d6) δ 7.87 (d, J = 2.1 Hz, 1H, H-4), 7.79 (s, 1H, H-9), 7.77 (dd, J = 8.4, 2.1 Hz, 1H, H-13), 7.68 (d, J = 2.1 Hz, 1H, H-6), 7.44 (dd, J = 8.6, 2.2 Hz, 1H, H-12), 7.22 (d, J = 8.5 Hz, 1H, H-7), 4.20 (t, J = 5.9 Hz, 2H, H-1′), 3.90 (s, 3H, OCH3), 3.59 (t, J = 4.7 Hz, 4H, H-4′,4″), 2.74 (t, J = 5.8 Hz, 2H, H-2′), 2.52 (m, 4H, H-3′,3″). 13C-NMR/APT (151 MHz, DMSO-d6) δ 163.3, 152.3, 151.8, 148.3, 143.7, 129.6, 127.8, 121.6, 118.1, 116.1, 113.1, 112.8, 111.0, 66.4, 56.8, 56.4, 55.8. Calcd for C20H21ClN2O4: C, 61.78; H, 5.44; N, 7.20. Found: C, 61.76; H, 5.46; N, 7.23.
3.2. Biological Evaluations
3.2.1. In Vitro Antiproliferative Evaluation
3.2.2. In Vitro Antibacterial Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, X.K.; Yeong, K.Y. A Patent Review on the Current Developments of Benzoxazoles in Drug Discovery. ChemMedChem 2021, 16, 3237–3262. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, T.; Patel, T.M. Anticancer activity of benzoxazole derivative (2015 onwards): A Review. Future J. Pharm. Sci. 2020, 6, 94–113. [Google Scholar] [CrossRef]
- Erol, M.; Celik, I.; Kuyucuklu, G. Synthesis, antimicrobial and in silico studies of new 2.5-disubstituted benzoxazole derivative. Med. Sci. 2021, 10, 400–408. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, X.; Xue, Y.; Zhou, H.; Huang, C.; Zhu, M.; Zhuang, T.; Chen, Y.; Huang, L. Discovery of a novel class of benzoxazole derivatives as histamine H3 receptor ligands for the treatment of neuropathic pain. Bioorg. Chem. 2022, 127, 106039. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.L.; Gaikwad, S.S.; Gaikwad, N.J. Anti-nociceptive and Anti-inflammatory Activity of Synthesized Novel Benzoxazole Derivatives. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2021, 20, 333–343. [Google Scholar] [CrossRef]
- Seth, K.; Garg, S.K.; Kumar, R.; Purohit, P.; Meena, V.S.; Goyal, R.; Banerjee, U.C.; Chakraborti, A.K. 2-(2-Arylphenyl) benzoxazole as a novel anti-inflammatory scaffold: Synthesis and biological evaluation. ACS Med. Chem. Lett. 2014, 5, 512–516. [Google Scholar] [CrossRef]
- Liu, Z.; Bian, M.; Ma, Q.-Q.; Zhang, Z.; Du, H.-H.; Wei, C.-X. Design and synthesis of new benzo[d]oxazole-based derivatives and their neuroprotective effects on β-amyloid-induced PC12 Cells. Molecules 2020, 25, 5391. [Google Scholar] [CrossRef]
- Di Martino, S.; De Rosa, M. The Benzoxazole Heterocycle: A Comprehensive Review of the Most Recent Medicinal Chemistry Developments of Antiproliferative, Brain-Penetrant, and Anti-inflammatory Agents. Top. Curr. Chem. 2024, 382, 33. [Google Scholar] [CrossRef]
- Kim, T.; Lee, S.-A.; Noh, T.; Choi, P.; Choi, S.-J.; Song, B.G.; Kim, Y.; Park, Y.-T.; Huh, G.; Kim, Y.-J.; et al. Synthesis, Structure Revision, and Cytotoxicity of Nocarbenzoxazole G. J. Nat. Prod. 2019, 82, 1325–1330. [Google Scholar] [CrossRef]
- Pal, S.; Manjunath, B.; Ghorai, S.; Sasmal, S. Benzoxazole Alkaloids: Occurrence, Chemistry, and Biology in The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 79, pp. 71–137. [Google Scholar]
- Hohmann, C.; Schneider, K.; Bruntner, C.; Irran, E.; Nicholson, G.; Bull, A.T.; Jones, A.L.; Brown, R.; Stach, J.E.M.; Goodfellow, M.; et al. Caboxamycin, a new antibiotic of the benzoxazole family produced by the deep-sea strain Streptomyces sp. NTK 937. J. Antibiot. 2009, 62, 99–104. [Google Scholar] [CrossRef]
- Kusumi, T.; Ooi, T.; Walchli, M.R.; Kakisawa, H. Structure of the novel antibiotics boxazomycins A, B, and C. J. Am. Chem. Soc. 1988, 110, 2954–2958. [Google Scholar] [CrossRef]
- Sacchi, C.; Magni, F.; Toia, A.; Cazzaniga, F.; Galli, G.; Berti, F. Flunoxaprofen, a new non-steroidal anti-inflammatory drug, does not interfere with prostaglandin synthesis in rat gastric mucosa. Pharmacol. Res. 1989, 21, 177–182. [Google Scholar] [CrossRef]
- Kakkar, S.; Tahlan, S.; Lim, S.M.; Ramasamy, K.; Mani, V.; Shah, S.A.A.; Narasimhan, B. Benzoxazole derivatives: Design, synthesis and biological evaluation. Chem. Cent. J. 2018, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, A.; Yeong, K.Y. Targeting disease with benzoxazoles: A comprehensive review of recent developments. Med. Chem. Res. 2024, 33, 406–438. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, X.; Dou, W.; Zhang, H.; Liu, W.; Wang, C.; Zheng, J. Synthesis and characterization of the ligand based on benzoxazole and its transition metal complexes: DNA-binding and antitumor activity. J. Inorg. Biochem. 2010, 104, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Grasel, F.d.S.; de Oliveira, T.E.; Netz, P.A. Investigation of the Interaction of 2-(2′-Hydroxyphenyl)-benzoxazoles and their Derivatives with B-DNA by Docking and Molecular Dynamics. J. Braz. Chem. Soc. 2015, 26, 420–433. [Google Scholar] [CrossRef]
- Kolbeck, P.J.; Vanderlinden, W.; Gemmecker, G.; Gebhardt, C.; Lehmann, M.; Lak, A.; Nicolaus, T.; Cordes, T.; Lipfert, J. Molecular structure, DNA binding mode, photophysical properties and recommendations for use of SYBR Gold. Nucleic Acids Res. 2021, 49, 5143–5158. [Google Scholar] [CrossRef]
- El-Helby, A.G.A.; Sakr, H.; Eissa, I.H.; Abulkhair, H.; Al-Karmalawy, A.A.; El-Adl, K. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors. Arch. Pharm. Chem. Life Sci. 2019, 352, 1900113. [Google Scholar] [CrossRef]
- Elwan, A.; Abdallah, A.E.; Mahdy, H.A.; Dahab, M.A.; Taghour, M.S.; Elkaeed, E.B.; Mehany, A.B.M.; Nabeeh, A.; Adel, M.; Alsfouk, A.A.; et al. Modified Benzoxazole-Based VEGFR-2 Inhibitors and Apoptosis Inducers: Design, Synthesis, and Anti-Proliferative Evaluation. Molecules 2022, 27, 5047. [Google Scholar] [CrossRef]
- Taghour, M.S.; Mahdy, H.A.; Gomaa, M.H.; Aglan, A.; Eldeib, M.G.; Elwan, A.; Dahab, M.A.; Elkaeed, E.B.; Alsfouk, A.A.; Khalifa, M.M.; et al. Benzoxazole derivatives as new VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and antiproliferative evaluation. J. Enzym. Inhib. Med. Chem. 2022, 37, 2063–2077. [Google Scholar] [CrossRef]
- Elkady, H.; Elwan, A.; El-Mahdy, H.A.; Doghish, A.S.; Ismail, A.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Dahab, M.A.; Mahdya, H.A.; et al. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, M.; Gupta, S.; Bansal, Y.; Bansal, G. A critical analysis of design, binding pattern and SAR of benzo-fused heteronuclear compounds as VEGFR-2 inhibitors. Bioorg. Med. Chem. 2024, 115, 117966. [Google Scholar] [CrossRef]
- Ozturk, O.; Aki-Yalcin, E.; Yalcin, I.; Grifitth, R. Insight into Mechanism of Action of Anticancer Benzazoles. Curr. Top. Med. Chem. 2020, 20, 2056–2069. [Google Scholar] [CrossRef] [PubMed]
- Zilifdara, F.; Fotoa, E.; Ertan-Bolelli, T.; Yildiz, İ.; Aki-Yalcin, E.; Diril, N. Inhibition of DNA Topoisomerases by a Series of Benzoxazoles and their Possible Metabolites. Lett. Drug. Des. Discov. 2018, 15, 1155–1162. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, F.; Min, W.; Yuan, K.; Kuang, W.; Wang, X.; Zhu, Y.; Sun, C.; Xia, F.; Wang, Y.; et al. Discovery of novel phosphoinositide-3-kinase α inhibitors with high selectivity, excellent bioavailability, and long-acting efficacy for gastric cancer. J. Med. Chem. 2022, 65, 9873–9892. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.M.; Chen, Y.S.; Ban, Y.J.; Wang, Y.J.; Dong, Y.X.; Lei, L.; Guo, B.; Wang, J.T.; Tang, L.; Li, H.L.; et al. Design, synthesis and bioevaluation of PI3Kα-selective inhibitors as potential colorectal cancer drugs. Eur. J. Med. Chem. 2023, 260, 115754. [Google Scholar] [CrossRef]
- Demmer, C.S.; Bunch, L. Benzoxazoles and oxazolopyridines in medicinal chemistry studies. Eur. J. Med. Chem. 2015, 97, 778–785. [Google Scholar] [CrossRef]
- Sattar, R.; Mukhtar, R.; Atif, M.; Hasnain, M.; Irfan, A. Synthetic transformations and biological screening of benzoxazole derivatives: A review. J. Heterocycl. Chem. 2020, 57, 2079–2107. [Google Scholar] [CrossRef]
- Jangale, A.D.; Dalal, D.S. Green synthetic approaches for biologically relevant organic compounds. Synth. Commun. 2017, 47, 2139–2173. [Google Scholar] [CrossRef]
- Selka, A.; Abidli, A.; Schiavo, L.; Jeanmart, L.; Hanquet, G.; Lubell, W.D. Recent Advances in Sustainable Total Synthesis and Chiral Pool Strategies with Emphasis on (-)-Sclareol in Natural Products Synthesis. Eur. J. Org. Chem. 2025, 28, e202400983. [Google Scholar] [CrossRef]
- Chang, W.; Sun, Y.; Huang, Y. One-pot green synthesis of benzoxazole derivatives through molecular sieve-catalyzed oxidative cyclization reaction. Heteroat. Chem. 2017, 28, 21360. [Google Scholar] [CrossRef]
- Soni, S.; Sahiba, N.; Teli, S.; Teli, P.; Agarwal, L.K.; Agarwal, S. Advances in the synthetic strategies of benzoxazoles using 2-aminophenol as a precursor: An up-to-date review. RSC Adv. 2023, 13, 24093. [Google Scholar] [CrossRef] [PubMed]
- Nikpassand, M.; Fekri, L.Z.; Farokhian, P. An efficient and green synthesis of novel benzoxazole under ultrasound irradiation. Ultrason. Sonochem. 2016, 28, 341–345. [Google Scholar] [CrossRef]
- Alter, N.; Link, S.; Heuser, S. Microwave-Assisted One-Pot Synthesis of 2-Substituted Benzoxazoles from Nitrophenol and Carboxylic Acids. Results Chem. 2022, 4, 100396. [Google Scholar] [CrossRef]
- Pham, P.T.; Nguyen, H.T.; Nguyen, T.T.; Nguyen, L.H.T.; Dang, M.-H.D.; Doan, T.L.H.; Pham, D.D.; Nguyen, C.T.; Tran, P.H. Rapid and Simple Microwave-Assisted Synthesis of Benzoxazoles Catalyzed by [CholineCl][Oxalic Acid]. Catalysts 2022, 12, 1394. [Google Scholar] [CrossRef]
- Ozil, M.; Menteşe, E. Microwave-assisted Synthesis of Benzoxazoles Derivatives. Curr. Microw. Chem. 2020, 7, 183–195. [Google Scholar] [CrossRef]
- Aboonajmi, J.; Panahi, F.; Hosseini, M.A.; Aberi, M.; Shargh, H. Iodine-catalyzed synthesis of benzoxazoles using catechols, ammonium acetate, and alkenes/alkynes/ketones via C–C and C–O bond cleavage. RSC Adv. 2022, 12, 20968. [Google Scholar] [CrossRef]
- Singh, L.P.; Chawla, V.; Chawla, P.; Saraf, S.K. Synthesis and antimicrobial activity of some 2-phenyl-benzoxazole derivatives. Pharm. Chem. 2010, 2, 206–212. [Google Scholar]
- Banerjee, S.; Payra, S.; Saha, A.; Sereda, G. ZnO nanoparticles: A green efficient catalyst for the room temperature synthesis of biologically active 2-aryl-1,3-benzothiazole and 1,3-benzoxazole derivatives. Tetrahedron Lett. 2014, 55, 5515–5520. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Lee, C.-Y.; Cheon, C.-H. Cyanide as a powerful catalyst for facile synthesis of benzofused heteroaromatic compounds via aerobic oxidation. Tetrahedron 2013, 69, 6565–6573. [Google Scholar] [CrossRef]
- Heakal, M.; Elena, M.; Yuriy, A.; Sergey, L. Bacterial Outer Membrane Permeability Increase Underlies the Bactericidal Effect of Fatty Acids from Hermetia illucens (Black Soldier Fly) Larvae Fat Against Hypermucoviscous Isolates of Klebsiella pneumoniae. Front. Microbiol. 2022, 13, 844811. [Google Scholar]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [PubMed]
- Erol, M.; Celik, I.; Kuyucuklu, G. Synthesis, Molecular Docking, Molecular Dynamics, DFT and Antimicrobial Activity Studies of 5-substituted-2-(p-methylphenyl)benzoxazole Derivatives. J. Mol. Struct. 2021, 15, 130151. [Google Scholar] [CrossRef]
- Perin, N.; Hok, L.; Beč, A.; Persoons, L.; Vanstreels, E.; Daelemans, D.; Vianello, R.; Hranjec, M. N-substituted benzimidazole acrylonitriles as in vitro tubulin polymerization inhibitors: Synthesis, biological activity and computational analysis. Eur. J. Med. Chem. 2021, 211, 113003. [Google Scholar] [CrossRef]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grows Aerobically, 9th ed.; CLSI Document MO7-A9, Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Capan-1 | HCT-116 | LN229 | NCI-H460 | DND-41 | HL-60 | K562 | Z138 |
| 29 | 74.6 ± 4.9 | >100 | >100 | 41.6 ± 3.4 | >100 | >100 | >100 | >100 |
| 30 | 70.1 ± 1.0 | 62.0 ± 0.8 | 33.9 ± 3.0 | 1.7 ± 0.2 | >100 | 74.3 ± 4.4 | >100 | >100 |
| 31 | 55.3 ± 7.6 | >100 | ≥59.7 | 62.8 ± 8.6 | ≥98.8 | ≥80.7 | >100 | ≥66.1 |
| 32 | 35.2 ± 0.7 | 36.0 ± 2.4 | 39.0 ± 2.3 | 9.4 ± 0.8 | 84.3 ± 0.8 | 44.1 ± 7.3 | 64.2 ± 5.8 | 71.3 ± 2.3 |
| 33 | ≥96.7 | >100 | ≥81.1 | 1.1 ± 0.0 | >100 | >100 | >100 | ≥56.4 |
| 34 | >100 | >100 | >100 | 73.2 ± 0.2 | >100 | >100 | >100 | >100 |
| 35 | ≥71.5 | >100 | >100 | 55.5 | >100 | >100 | >100 | ≥70.8 |
| 36 | 2.0 ± 0.0 | 5.7 ± 1.2 | 2.2 ± 0.7 | 1.3 ± 0.1 | 60.4 ± 4.0 | 52.4 ± 5.8 | 45.9 ± 1.4 | 86.2 ± 4.7 |
| 37 | >100 | >100 | >100 | 33.1 ± 4.7 | >100 | >100 | >100 | ≥87.6 |
| 38 | 51.7 ± 4.1 | >100 | >100 | 30.7 ± 7.0 | 79.4 ± 1.4 | 59.0 ± 1.5 | >100 | 56.0 ± 3.1 |
| 39 | ≥76.6 | >100 | >100 | 3.8 ± 1.5 | >100 | >100 | >100 | >100 |
| 40 | 15.7 ± 0.1 | >100 | >100 | 0.4 ± 0.0 | 77.6 ± 0.6 | 60.6 ± 4.7 | >100 | 11.2 ± 1.3 |
| 41 | 46.2 ± 8.8 | 16.3 ± 3.0 | >100 | 17.2 ± 0.1 | >100 | ≥51.8 | >100 | 14.4 ± 0.4 |
| 42 | 45.8 ± 1.3 | >100 | >100 | 14.4 ± 2.8 | 78.0 ± 1.1 | 39.9 ± 4.0 | >100 | >100 |
| 43 | 15.4 ± 4.7 | 4.7 ± 3.1 | 5.6 ± 5.1 | 1.8 ± 0.4 | >100 | 53.0 ± 5.1 | >100 | >100 |
| 44 | 74.2 ± 9.5 | 15.6 ± 0.3 | 20.3 ± 2.9 | 16.5 ± 6.0 | >100 | 17.6 ± 5.0 | >100 | 26.4 ± 2.3 |
| 45 | 23.6 ± 1.5 | 2.4 ± 1.6 | 14.3 ± 3.0 | 0.9 ± 0.1 | >100 | 73.1 ± 4.1 | >100 | ≥53.4 |
| 46 | 12.8 ± 2.5 | 6.0 ± 5.1 | 3.8 ± 2.4 | 1.1 ± 0.0 | 76.4 ± 9.6 | 24.2 ± 1.6 | >100 | 11.7 ± 3.9 |
| 47 | 43.4 ± 9.2 | >100 | >100 | 1.3 ± 0.4 | >100 | 14.8 ± 0.4 | >100 | 16.0 ± 3.9 |
| 48 | ≥39.8 | >100 | ≥50.0 | 15.2 ± 5.2 | >100 | >100 | >100 | ≥41.9 |
| Eto | 0.03 ± 0.0 | 3.4 ± 0.1 | 3.7 ± 0.1 | 6.1 ± 0.4 | 1.0 ± 0.1 | 0.8 ± 0.1 | 4.0 ± 0.6 | 0.7 ± 0.1 |
| Noco | 0.02 ± 0.0 | 0.04 ± 0.0 | 0.4 ± 0.3 | 0.5 ± 0.1 | 0.7 ± 0.1 | 0.04 ± 0.0 | 0.04 ± 0.0 | 0.04 ± 0.0 |
| MIC (μg/mL) | |||||
|---|---|---|---|---|---|
| Compound | E. coli | P. aeruginosa | K. pneumoniae | S. aureus | E. faecalis |
| 29 | >256 | >256 | >256 | >256 | 8 |
| 30, 31 | >256 | >256 | >256 | >256 | >256 |
| 32 | 256 | >256 | >256 | >256 | >256 |
| 33–38 | >256 | >256 | >256 | >256 | >256 |
| 39 | >256 | >256 | 256 | >256 | >256 |
| 40 | >256 | >256 | 128 | >256 | >256 |
| 41–46 | >256 | >256 | >256 | >256 | >256 |
| 47 | 256 | 0.25 | >256 | >256 | 0.5 |
| 48 | >256 | >256 | >256 | >256 | >256 |
| CAZ | 0.5 | 2 | 256 | 64 | 256 |
| CIP | <0.125 | 0.5 | >256 | 0.5 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakas, A.; Persoons, L.; Daelemans, D.; Grgić, D.K.; Kraljević, T.G. A Sustainable Synthesis of Novel 2-(3,4-Disubstituted phenyl)benzoxazole Derivatives and Their Antiproliferative and Antibacterial Evaluation. Molecules 2025, 30, 1767. https://doi.org/10.3390/molecules30081767
Rakas A, Persoons L, Daelemans D, Grgić DK, Kraljević TG. A Sustainable Synthesis of Novel 2-(3,4-Disubstituted phenyl)benzoxazole Derivatives and Their Antiproliferative and Antibacterial Evaluation. Molecules. 2025; 30(8):1767. https://doi.org/10.3390/molecules30081767
Chicago/Turabian StyleRakas, Anja, Leentje Persoons, Dirk Daelemans, Dajana Kučić Grgić, and Tatjana Gazivoda Kraljević. 2025. "A Sustainable Synthesis of Novel 2-(3,4-Disubstituted phenyl)benzoxazole Derivatives and Their Antiproliferative and Antibacterial Evaluation" Molecules 30, no. 8: 1767. https://doi.org/10.3390/molecules30081767
APA StyleRakas, A., Persoons, L., Daelemans, D., Grgić, D. K., & Kraljević, T. G. (2025). A Sustainable Synthesis of Novel 2-(3,4-Disubstituted phenyl)benzoxazole Derivatives and Their Antiproliferative and Antibacterial Evaluation. Molecules, 30(8), 1767. https://doi.org/10.3390/molecules30081767