
Mechanochemical Sequential Deoxygenative Cross-Coupling Reactions of Phenols Under Ruthenium-Nickel Catalysis
 ,
,  , , , , , , and
, , , , , , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
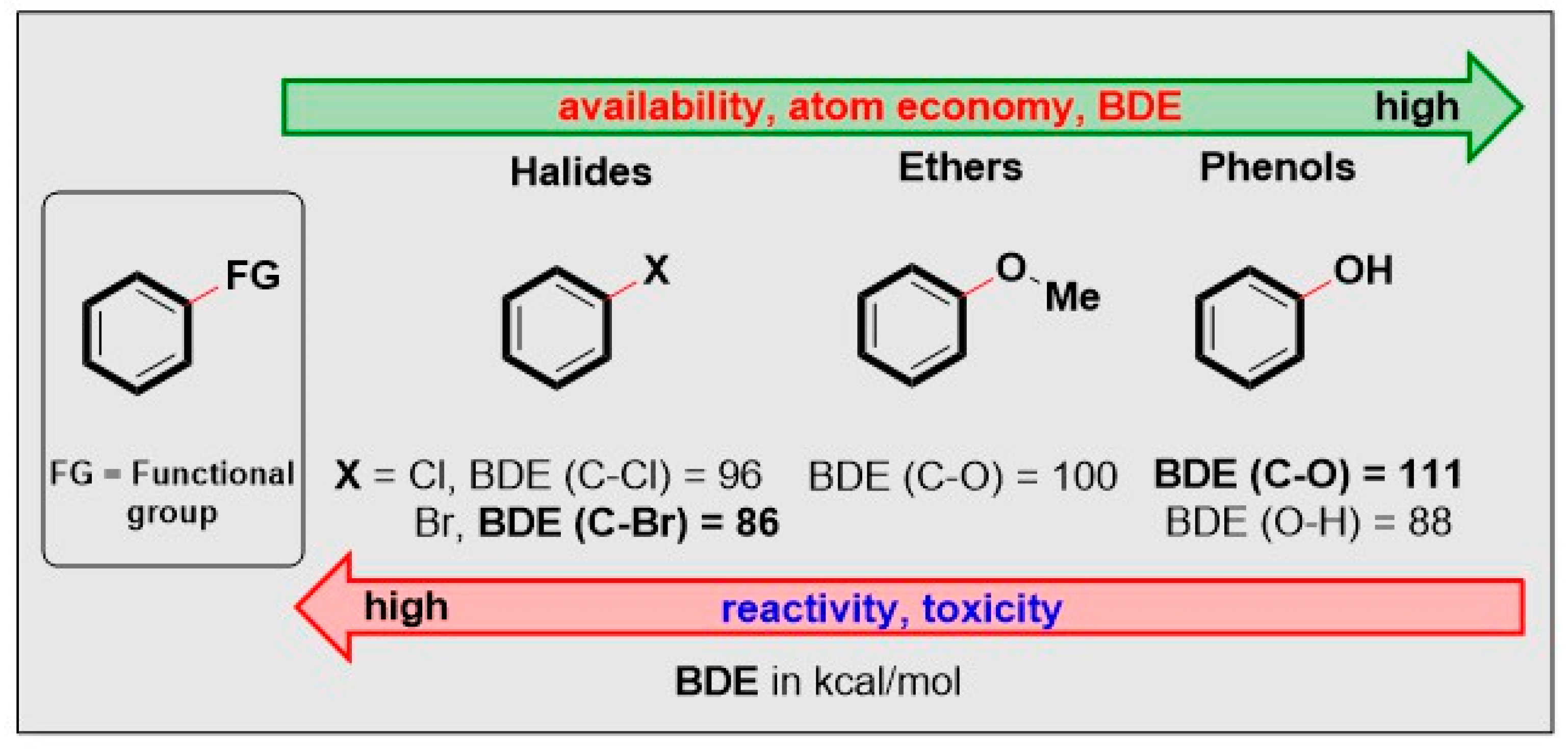
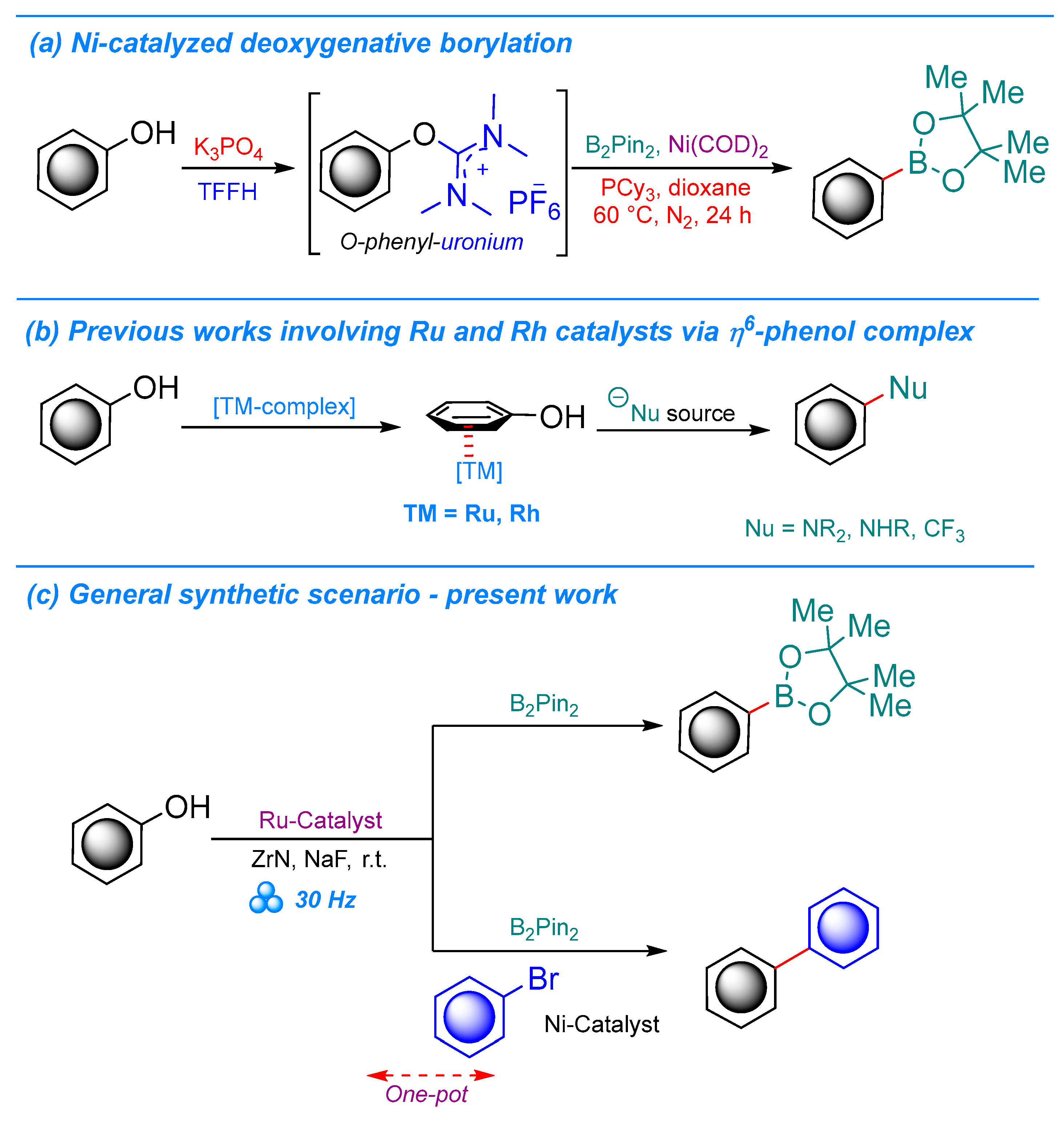
1. Introduction
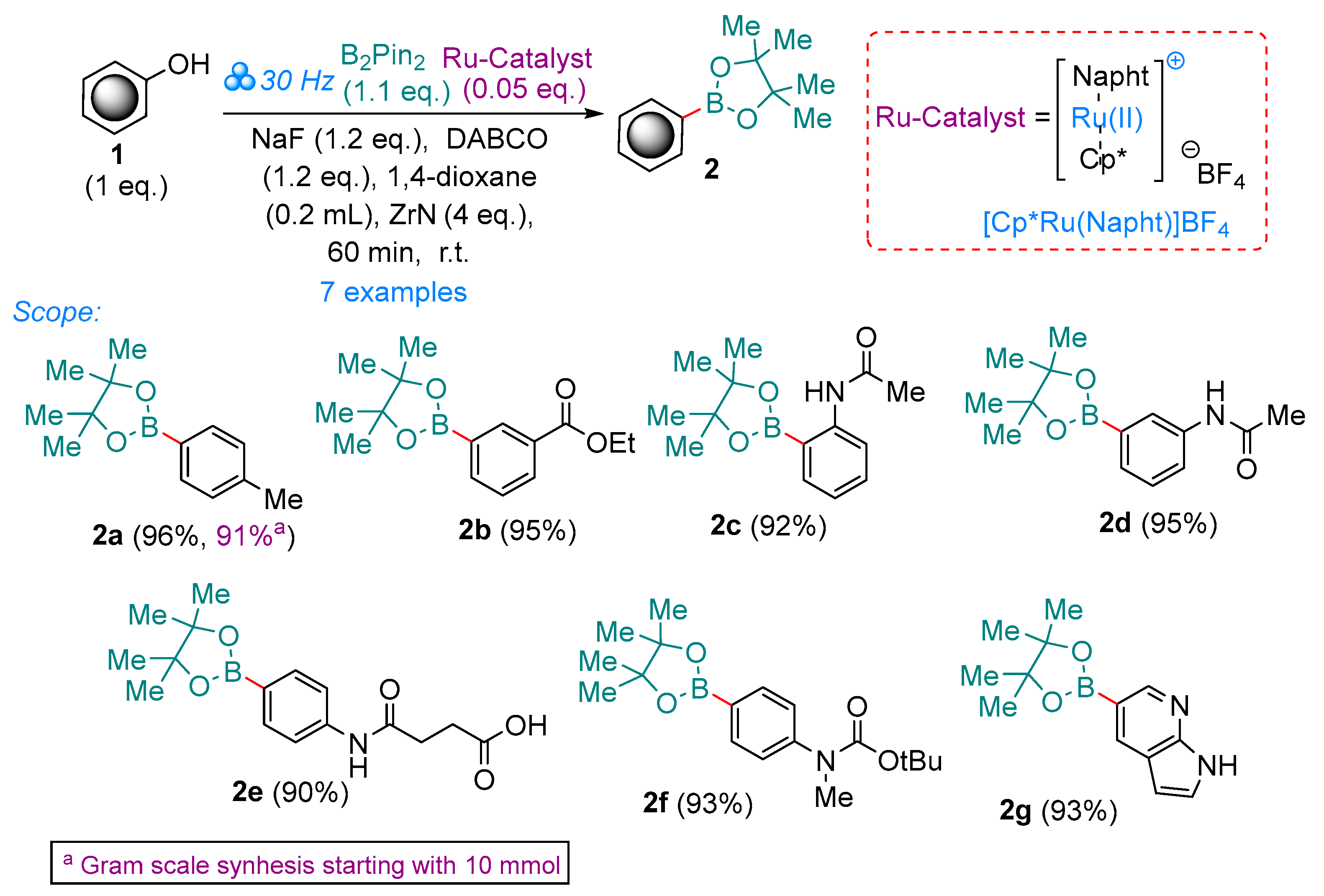
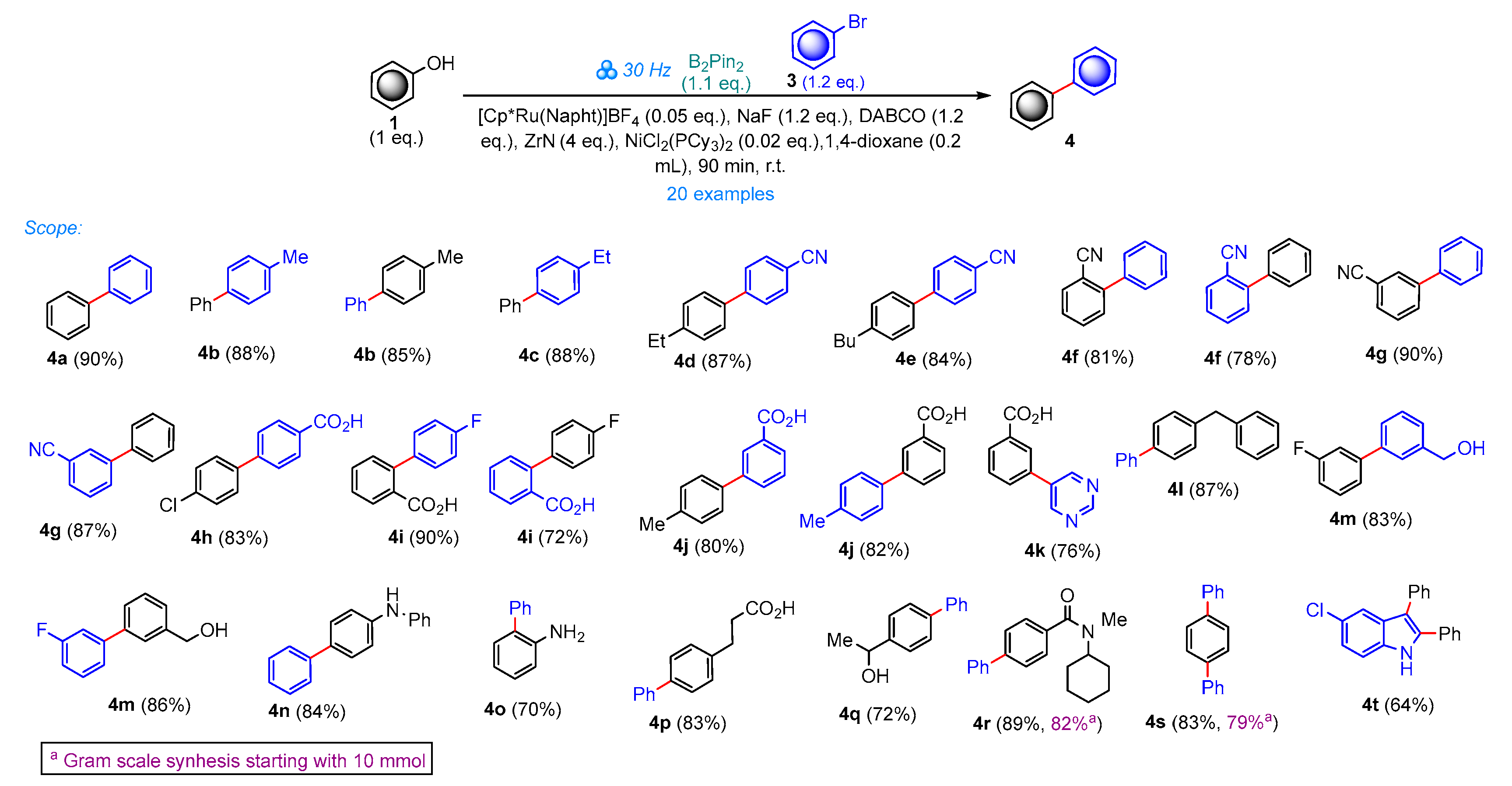
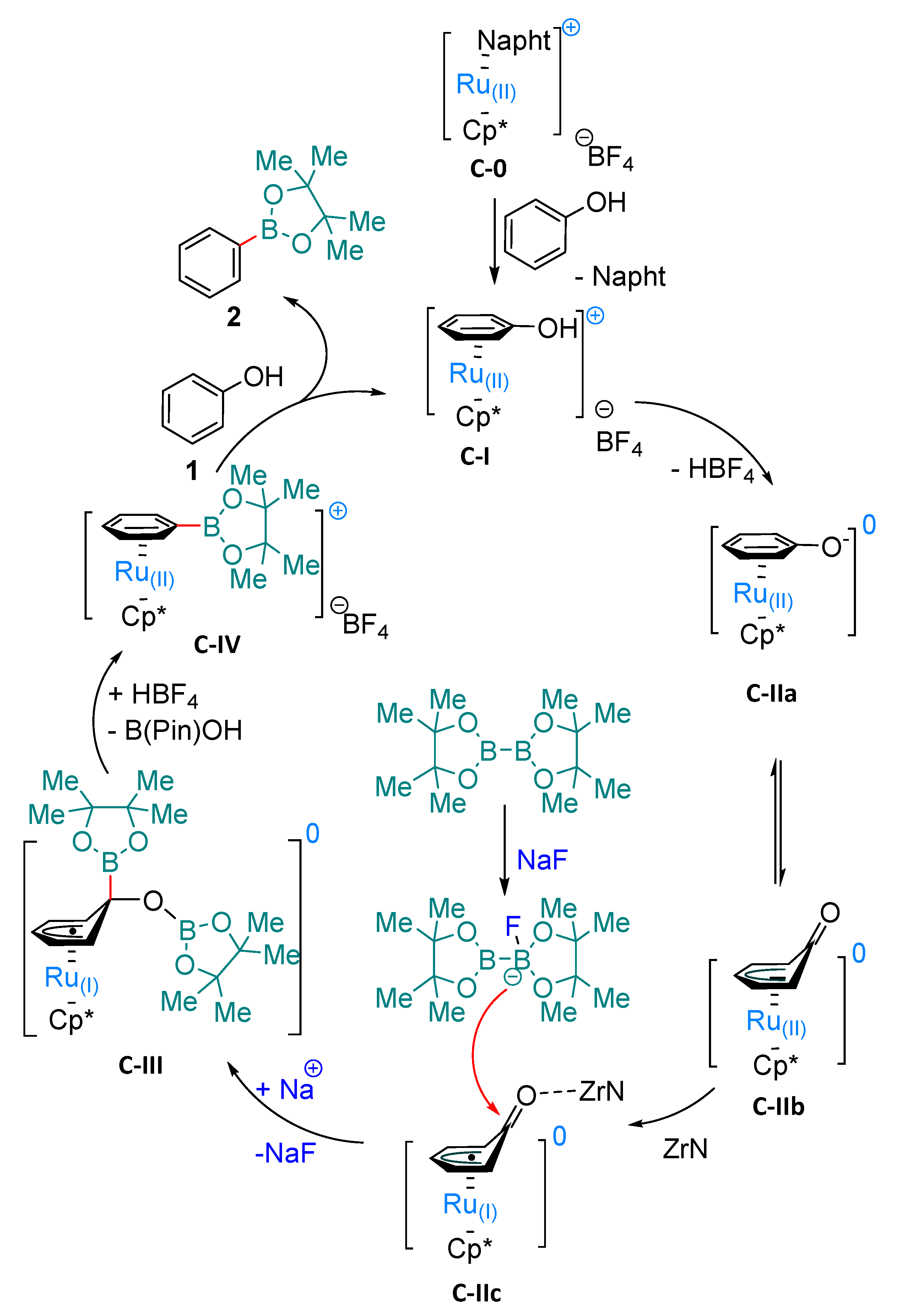
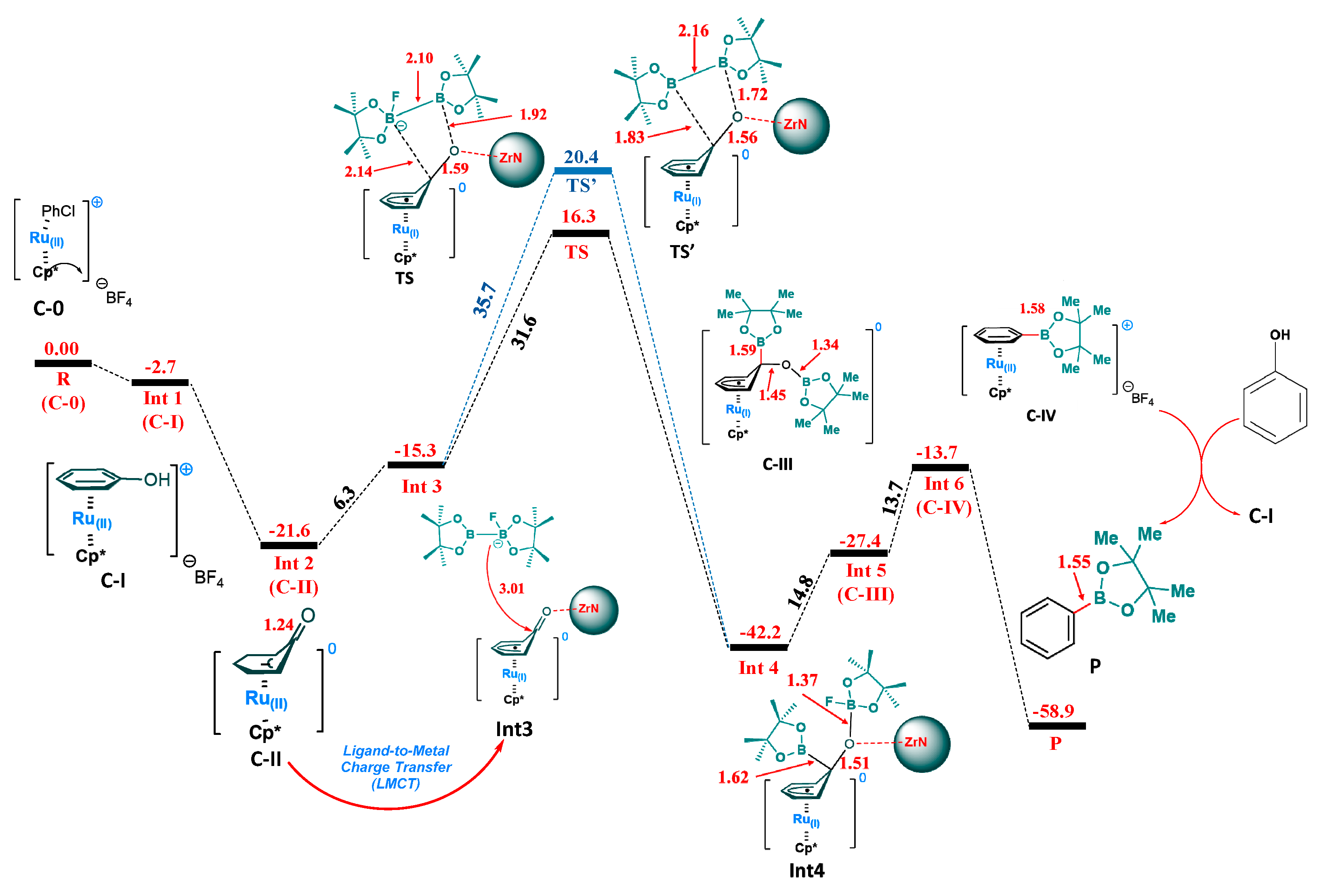
2. Results and Discussion
3. Experimental Section
3.1. Materials and Methods
3.2. Reaction Procedure with Optimised Reaction Conditions
3.2.1. General Procedure for the Synthesis of Aryl Pinacolboranes 2 Starting from Phenol 1
3.2.2. General Procedure for the Synthesis of Biphenyls 4 Starting from Phenol 1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar] [CrossRef]
- Zeng, H.; Qiu, Z.; Domínguez-Huerta, A.; Hearne, Z.; Chen, Z.; Li, C.-J. An Adventure in Sustainable Cross-Coupling of Phenols and Derivatives via Carbon–Oxygen Bond Cleavage. ACS Catal. 2017, 7, 510–519. [Google Scholar] [CrossRef]
- Qiu, Z.; Li, C.-J. Transformations of Less-Activated Phenols and Phenol Derivatives via C–O Cleavage. Chem. Rev. 2020, 120, 10454–10515. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-G.; Li, B.-J.; Shi, Z.-J. Exploration of New C–O Electrophiles in Cross-Coupling Reactions. Acc. Chem. Res. 2010, 43, 1486–1495. [Google Scholar] [CrossRef]
- Rosen, B.M.; Quasdorf, K.W.; Wilson, D.A.; Zhang, N.; Resmerita, A.-M.; Garg, N.K.; Percec, V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. [Google Scholar] [CrossRef]
- Liu, X.; Xu, B.; Su, W. Ni-Catalyzed Deoxygenative Borylation of Phenols Via O-Phenyl-uronium Activation. ACS Catal. 2022, 12, 8904–8910. [Google Scholar] [CrossRef]
- Dominguez-Huerta, A.; Dai, X.-J.; Zhou, F.; Querard, P.; Qiu, Z.; Ung, S.; Liu, W.; Li, J.; Li, C.-J. Exploration of new reaction tools for late-stage functionalization of complex chemicals. Can. J. Chem. 2019, 97, 67–85. [Google Scholar] [CrossRef]
- Zarate, C.; van Gemmeren, M.; Somerville, R.; Martin, R. Phenol derivatives: Modern electrophiles in cross-coupling reactions. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; Volume 66, pp. 143–222. [Google Scholar]
- Cornella, J.; Zarate, C.; Martin, R. Metal-catalyzed activation of ethers via C–O bond cleavage: A new strategy for molecular diversity. Chem. Soc. Rev. 2014, 43, 8081–8097. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Miyaura, N. Metal-catalyzed cross-coupling reactions of organoboron compounds with organic halides. Metal-Catalyzed Cross-Coupling React. 2004, 41–123. [Google Scholar]
- Smith, K. Organoboron Chemistry. In Organometallics in Synthesis: A Manual; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; pp. 465–533. [Google Scholar]
- Schneider, F.; Stolle, A.; Ondruschka, B.; Hopf, H. The Suzuki–Miyaura reaction under mechanochemical conditions. Org. Process Res. Dev. 2009, 13, 44–48. [Google Scholar] [CrossRef]
- Schneider, F.; Szuppa, T.; Stolle, A.; Ondruschka, B.; Hopf, H. Energetic assessment of the Suzuki–Miyaura reaction: A curtate life cycle assessment as an easily understandable and applicable tool for reaction optimization. Green Chem. 2009, 11, 1894–1899. [Google Scholar] [CrossRef]
- Seo, T.; Ishiyama, T.; Kubota, K.; Ito, H. Solid-state Suzuki–Miyaura cross-coupling reactions: Olefin-accelerated C–C coupling using mechanochemistry. Chem. Sci. 2019, 10, 8202–8210. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.G.; Grätz, S.; Lukin, S.; Halasz, I.; Etter, M.; Evans, J.D.; Borchardt, L. Direct mechanocatalysis: Palladium as milling media and catalyst in the mechanochemical Suzuki polymerization. Angew. Chem. Int. Ed. 2019, 58, 18942–18947. [Google Scholar] [CrossRef] [PubMed]
- Seo, T.; Toyoshima, N.; Kubota, K.; Ito, H. Tackling solubility issues in organic synthesis: Solid-state cross-coupling of insoluble aryl halides. J. Am. Chem. Soc. 2021, 143, 6165–6175. [Google Scholar] [CrossRef]
- Negishi, E.I.; Idacavage, M.J. Formation of Carbon-Carbon and Carbon-Heteroatom Bonds via Organoboranes and Organoborates. Organic Reactions 2004, 33, 1–246. [Google Scholar]
- Chow, W.K.; Yuen, O.Y.; Choy, P.Y.; So, C.M.; Lau, C.P.; Wong, W.T.; Kwong, F.Y. A decade advancement of transition metal-catalyzed borylation of aryl halides and sulfonates. RSC Adv. 2013, 3, 12518–12539. [Google Scholar] [CrossRef]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Ishiyama, T.; Itoh, Y.; Kitano, T.; Miyaura, N. Synthesis of arylboronates via the palladium(0)-catalyzed cross-coupling reaction of tetra(alkoxo)diborons with aryl triflates. Tetrahedron Lett. 1997, 38, 3447–3450. [Google Scholar] [CrossRef]
- Takahashi, K.; Takagi, J.; Ishiyama, T.; Miyaura, N. Synthesis of 1-Alkenylboronic Esters via Palladium-Catalyzed Cross-Coupling Reaction of Bis(pinacolato)diboron with 1-Alkenyl Halides and Triflates. Chem. Lett. 2003, 29, 126–127. [Google Scholar] [CrossRef]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C–H Activation for the Construction of C–B Bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Baba, E.; Seo, T.; Ishiyama, T.; Ito, H. Palladium-catalyzed solid-state borylation of aryl halides using mechanochemistry. Beilstein J. Org. Chem. 2022, 18, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Merino, P.; Tejero, T. Expanding the Limits of Organoboron Chemistry: Synthesis of Functionalized Arylboronates. Angew. Chem. Int. Ed. 2010, 49, 7164–7165. [Google Scholar] [CrossRef]
- Yu, D.-G.; Shi, Z.-J. Mutual activation: Suzuki–Miyaura coupling through direct cleavage of the sp2 C-O bond of naphtholate. Angew. Chem. Int. Ed. 2011, 50, 7097–7100. [Google Scholar] [CrossRef]
- Cao, Z.-C.; Fei-Xian Luo, F.-X.; Shi, W.-J.; Shi, Z.-J. Direct borylation of benzyl alcohol and its analogues in the absence of bases. Org. Chem. Front. 2015, 2, 1505–1510. [Google Scholar] [CrossRef]
- Chen, K.; Kang, Q.-K.; Li, Y.; Wu, W.-Q.; Zhu, H.; Shi, H. Catalytic Amination of Phenols with Amines. J. Am. Chem. Soc. 2022, 144, 1144–1151. [Google Scholar] [CrossRef]
- Chen, K.; Ma, Y.; Lin, Y.; Li, J.-Y.; Shi, H. Ruthenium/η5-Phenoxo-Catalyzed Amination of Phenols with Amines. J. Am. Chem. Soc. 2024, 146, 15833–15842. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Shalimov, O.; Garcia, M.G.; Zapletal, J.; Iaroshenko, V.O. Mechanochemical synthesis of aromatic ketones: Pyrylium tetrafluoroborate mediated deaminative arylation of amides. Chem. Sci. 2024, 15, 9155–9163. [Google Scholar] [CrossRef]
- Jakubczyk, M.; Mkrtchyan, S.; Shkoor, M.; Lanka, S.; Budzák, Š.; Iliaš, M.; Skoršepa, M.; Iaroshenko, V.O. Mechanochemical Conversion of Aromatic Amines to Aryl Trifluoromethyl Ethers. J. Am. Chem. Soc. 2022, 144, 10438–10445. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Iaroshenko, V.O. Mechanochemical synthesis of aromatic sulfonamides. Chem. Commun. 2021, 57, 11029–11032. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, S.; Jakubczyk, M.; Budzák, Š.; Benická, B.; Iaroshenko, V.O. Introducing Trifluoromethoxyarenes as Halide Surrogates in Mechanochemical Realizations of Ni-catalyzed Cross-coupling Reactions. Asian J. Org. Chem. 2023, 12, e202300094. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Jakubczyk, M.; Lanka, S.; Yar, M.; Ayub, K.; Shkoor, M.; Pittelkow, M.; Iaroshenko, V.O. Mechanochemical Transformation of CF3 Group: Synthesis of Amides and Schiff Bases. Adv. Synth. Catal. 2021, 363, 5448–5460. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Jakubczyk, M.; Lanka, S.; Yar, M.; Mahmood, T.; Ayub, K.; Sillanpää, M.; Thomas, C.M.; Iaroshenko, V.O. Mechanochemical Ni-Catalysed Arylation of ortho-Hydroxyarylenaminones: Synthesis of Isoflavones. Adv. Synth. Catal. 2022, 364, 3512–3521. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Purohit, V.B.; Iaroshenko, V.O. Nanocellulose as Convenient Reaction Media for the FeCl3 Mediated Mechanochemical Synthesis of 3-Acylchromones. ACS Sustain. Chem. Eng. 2023, 11, 13877–13884. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Purohit, V.B.; Khutsishvili, S.; Nociarová, J.; Yar, M.; Mahmood, T.; Ayub, K.; Budzák, Š.; Skoršepa, M.; Iaroshenko, V.O. Mechanochemical Defluorinative Acylation of ortho-Hydroxyarylenaminones by CF3-Compounds: Synthesis of 3-Acylchromones. Adv. Synth. Catal. 2023, 365, 2026–2035. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Purohit, V.B.; Sarfaraz, S.; Yar, M.; Ayub, K.; Iaroshenko, V.O. Metal-Free Supramolecular Reduction of Nitro Compounds into the Cucurbit [7]uril Cavity: Testing the Enabling Technique in Aqueous Media. ACS Sustain. Chem. Eng. 2023, 11, 8406–8412. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Purohit, V.B.; Zapletal, J.; Shalimov, O.O.; Iaroshenko, V.O. Nanocellulose as a Reaction Media and Stoichiometric Reagent for FeCl3-Mediated Reductive Functionalization of Nitro Compounds. ACS Sustain. Chem. Eng. 2024, 12, 1–9. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Shkoor, M.; Phanindrudu, M.; Medved′, M.; Sevastyanova, O.; Iaroshenko, V.O. Mechanochemical Defluorinative Arylation of Trifluoroacetamides: An Entry to Aromatic Amides. J. Org. Chem. 2023, 88, 863–870. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Shkoor, M.; Sarfaraz, S.; Ayub, K.; Iaroshenko, V.O. Mechanochemical arylative detrifluoromethylation of trifluoromethylarenes. Org. Biomol. Chem. 2023, 21, 6549–6555. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Purohit, V.B.; Shalimov, O.; Zapletal, J.; Sarfaraz, S.; Ayub, K.; Filo, J.; Sillanpää, M.; Skoršepa, M.; Iaroshenko, V.O. Mechanochemical Synthesis of Trifluoromethyl Arenes: Nanocellulose-Supported Deaminative Trifluoromethylation of Aromatic Amines. ACS Sustain. Chem. Eng. 2024, 12, 8980–8989. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Shalimov, O.; Purohit, V.; Zapletal, J.; Prajapati, V.; Prajapati, R.; Elumalai, D.; Garcia, M.; Filo, J.; Addova, G. Nanocellulose as Reaction Media for FeCl3-mediated Mechanochemical Deaminative Fluorination of (Hetero) aromatic Amines. Adv. Synth. Catal. 2024, 366, 3269–3276. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Purohit, V.B.; Zapletal, J.; Shalimov, O.; Nociarová, J.; Addová, G.; Filo, J.; Garcia, M.G.; Kupcová, E.; Benická, B. Mechanochemical trifluoromethoxylation of aryltrimethylammonium triflates, aryldiazonium tetrafluoroborates, and aryl pinacolboranes. Cell Rep. Phys. Sci. 2024, 5, 102118. [Google Scholar] [CrossRef]
- Patra, S.; Nandasana, B.N.; Valsamidou, V.; Katayev, D. Mechanochemistry Drives Alkene Difunctionalization via Radical Ligand Transfer and Electron Catalysis. Adv. Sci. 2024, 11, 2402970. [Google Scholar] [CrossRef] [PubMed]
- Kane-Maguire, L.A.; Honig, E.D.; Sweigart, D.A. Nucleophilic addition to coordinated cyclic.pi.-hydrocarbons: Mechanistic and synthetic studies. Chem. Rev. 1984, 84, 525–543. [Google Scholar] [CrossRef]
- Rose-Munch, F.; Gagliardini, V.; Renard, C.; Rose, E. (η6-Arene)tricarbonylchromium and (η5-cyclohexadienyl) tricarbonylmanganese complexes: Indirect nucleophilic substitutions. Coord. Chem. Rev. 1998, 178–180, 249–268. [Google Scholar] [CrossRef]
- Pike, R.D.; Sweigart, D.A. Electrophilic reactivity of coordinated cyclic π-hydrocarbons. Coord. Chem. Rev. 1999, 187, 183–222. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Chlenov, A. (Arene)Cr(CO)3 Complexes: Aromatic Nucleophilic Substitution. In Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis; Springer: Berlin/Heidelberg, Germany, 2004; pp. 43–69. [Google Scholar]
- Konovalov, A.I.; Gorbacheva, E.O.; Miloserdov, F.M.; Grushin, V.V. Ruthenium-catalyzed nucleophilic fluorination of halobenzenes. Chem. Commun. 2015, 51, 13527–13530. [Google Scholar] [CrossRef]
- Igau, A. η5-Oxocyclohexadienyl ligands in transition metal chemistry: Neglected (Brønsted) base ligands in cooperative catalysis. Coord. Chem. Rev. 2017, 344, 299–322. [Google Scholar] [CrossRef]
- Beyzavi, H.; Mandal, D.; Strebl, M.G.; Neumann, C.N.; D’Amato, E.M.; Chen, J.; Hooker, J.M.; Ritter, T. 18F-Deoxyfluorination of Phenols via Ru π-Complexes. ACS Cent. Sci. 2017, 3, 944–948. [Google Scholar] [CrossRef]
- Mkrtchyan, S.; Jakubczyk, M.; Sarfaraz, S.; Ayub, K.; Purohit, V.B.; Shalimov, O.; Iaroshenko, V.O. One-step Ru-catalyzed conversion of phenolic OH groups to trifluoromethyl under mechanochemical conditions. Cell Rep. Phys. Sci. 2024, 5, 102062. [Google Scholar] [CrossRef]
- Perekalin, D.S.; Karslyan, E.E.; Petrovskii, P.V.; Borissova, A.O.; Lyssenko, K.A.; Kudinov, A.R. Arene Exchange in the Ruthenium–Naphthalene Complex [CpRu(C10H8)]+. Eur. J. Inorg. Chem. 2012, 2012, 1485–1492. [Google Scholar] [CrossRef]
- Guo, X.; Dang, H.; Wisniewski, S.R.; Simmons, E.M. Nickel-Catalyzed Suzuki–Miyaura Cross-Coupling Facilitated by a Weak Amine Base with Water as a Cosolvent. Organometallics 2022, 41, 1269–1274. [Google Scholar] [CrossRef]
- Hazari, N.; Melvin, P.R.; Beromi, M.M. Well-defined nickel and palladium precatalysts for cross-coupling. Nat. Rev. Chem. 2017, 1, 0025. [Google Scholar] [CrossRef]
- Payard, P.-A.; Perego, L.A.; Ciofini, I.; Grimaud, L. Taming Nickel-Catalyzed Suzuki-Miyaura Coupling: A Mechanistic Focus on Boron-to-Nickel Transmetalation. ACS Catal. 2018, 8, 4812–4823. [Google Scholar] [CrossRef]
- Hernandez-Tamargo, C.; Roldan, A.; de Leeuw, N.H. Tautomerization of Phenol at the External Lewis Acid Sites of Scandium-, Iron- and Gallium-Substituted Zeolite MFI. J. Phys. Chem. C. 2019, 123, 7604–7614. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, J.; Adimi, S.; Shen, H.; Thomas, T.; Ma, R.; Attfield, J.P.; Yang, M. Zirconium nitride catalysts surpass platinum for oxygen reduction. Nature Mater. 2020, 19, 282–286. [Google Scholar] [CrossRef]
- Frisch, M. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bibi, S.; Sarfaraz, S.; Yar, M.; Zaman, M.I.; Niaz, A.; Khan, A.; Hashmi, M.A.; Ayub, K. Structure and Electronic Characterization of Pristine and Functionalized Single Wall Carbon Nanotube Interacting with Sulfide Ion: A Density Functional Theory Approach. J. Mol. Liq. 2022, 366, 120144. [Google Scholar] [CrossRef]
- Danish, M.; Raza, M.A.; Iftikhar, S.; Mumtaz, M.W.; Tahir, M.N.; Rashid, U.; Ayub, K. Synthesis, Single-Crystal X-ray Diffraction, and In Vitro Biological Evaluation of Sodium, Cobalt, and Tin Complexes of o-Nitro-/o-Methoxyphenylacetic Acid: Experimental and Theoretical Investigation. Monatsh. Chem. 2020, 151, 1727–1736. [Google Scholar] [CrossRef]
- Mukhtar, A.; Sarfaraz, S.; Ayub, K. Organic Transformations in the Confined Space of Porous Organic Cage CC2; Catalysis or Inhibition. RSC Adv. 2022, 12, 24397–24411. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mkrtchyan, S.; Purohit, V.B.; Jakubczyk, M.; Prajapati, V.D.; Prajapati, R.V.; Garcia, M.G.; Karpun, E.; Yepishev, V.; Saini, M.K.; Sarfaraz, S.; et al. Mechanochemical Sequential Deoxygenative Cross-Coupling Reactions of Phenols Under Ruthenium-Nickel Catalysis. Molecules 2025, 30, 1835. https://doi.org/10.3390/molecules30081835
Mkrtchyan S, Purohit VB, Jakubczyk M, Prajapati VD, Prajapati RV, Garcia MG, Karpun E, Yepishev V, Saini MK, Sarfaraz S, et al. Mechanochemical Sequential Deoxygenative Cross-Coupling Reactions of Phenols Under Ruthenium-Nickel Catalysis. Molecules. 2025; 30(8):1835. https://doi.org/10.3390/molecules30081835
Chicago/Turabian StyleMkrtchyan, Satenik, Vishal B. Purohit, Michał Jakubczyk, Vaibhav D. Prajapati, Ronak V. Prajapati, Michael G. Garcia, Eugene Karpun, Vitaliy Yepishev, Manoj K. Saini, Sehrish Sarfaraz, and et al. 2025. "Mechanochemical Sequential Deoxygenative Cross-Coupling Reactions of Phenols Under Ruthenium-Nickel Catalysis" Molecules 30, no. 8: 1835. https://doi.org/10.3390/molecules30081835
APA StyleMkrtchyan, S., Purohit, V. B., Jakubczyk, M., Prajapati, V. D., Prajapati, R. V., Garcia, M. G., Karpun, E., Yepishev, V., Saini, M. K., Sarfaraz, S., Ayub, K., Addová, G., Filo, J., & Iaroshenko, V. O. (2025). Mechanochemical Sequential Deoxygenative Cross-Coupling Reactions of Phenols Under Ruthenium-Nickel Catalysis. Molecules, 30(8), 1835. https://doi.org/10.3390/molecules30081835