
Insights into the Factors Controlling the Origin of Activation Barriers in the [2 + 2] Cycloaddition Reactions of Heavy Imine-like Molecules Featuring a Ge=Group 15 Double Bond with Heterocumulenes


Abstract
:1. Introduction
2. Results and Discussion
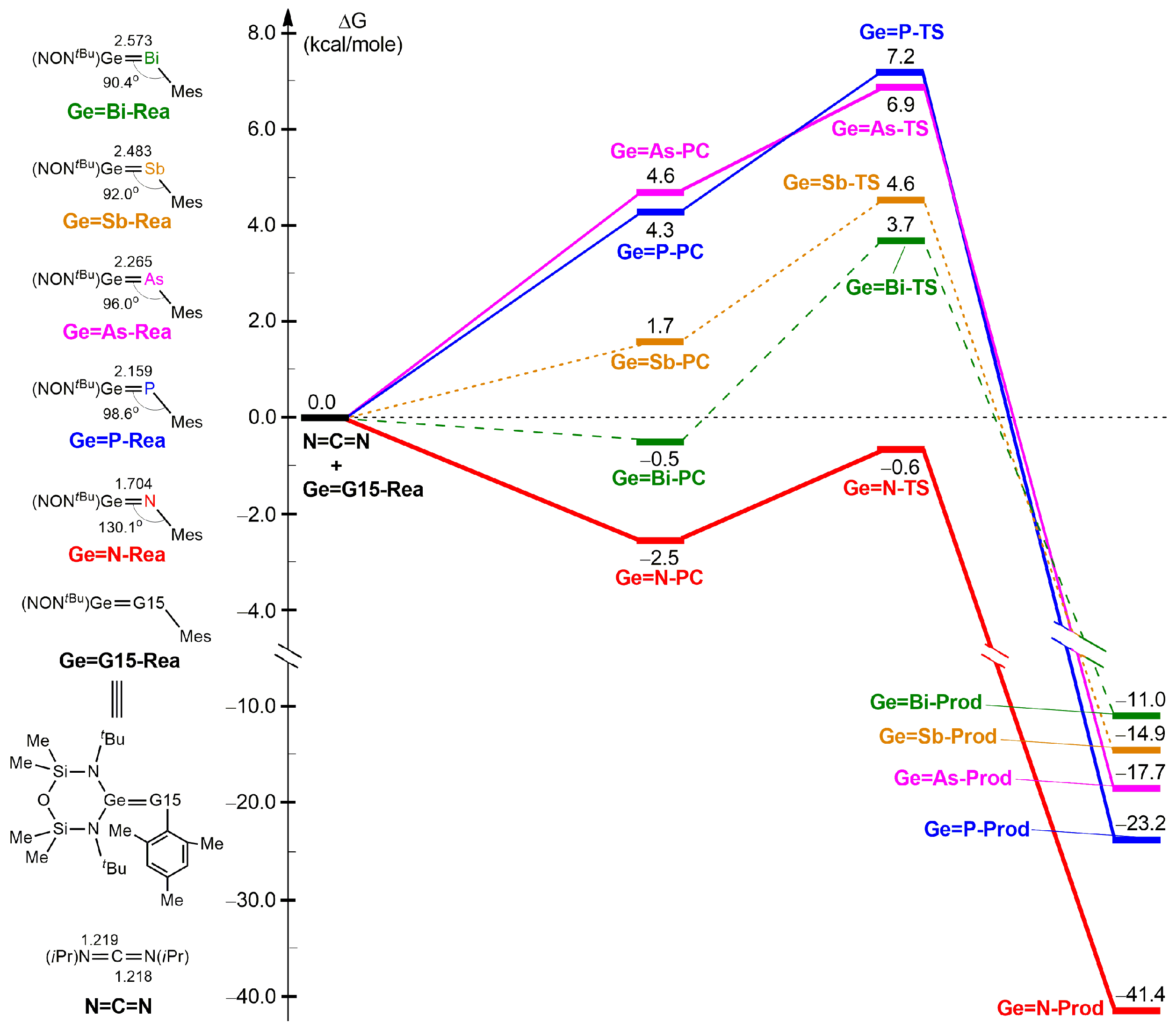
2.1. The Nature of the Ge=G15 Bonding in Ge=G15-Rea
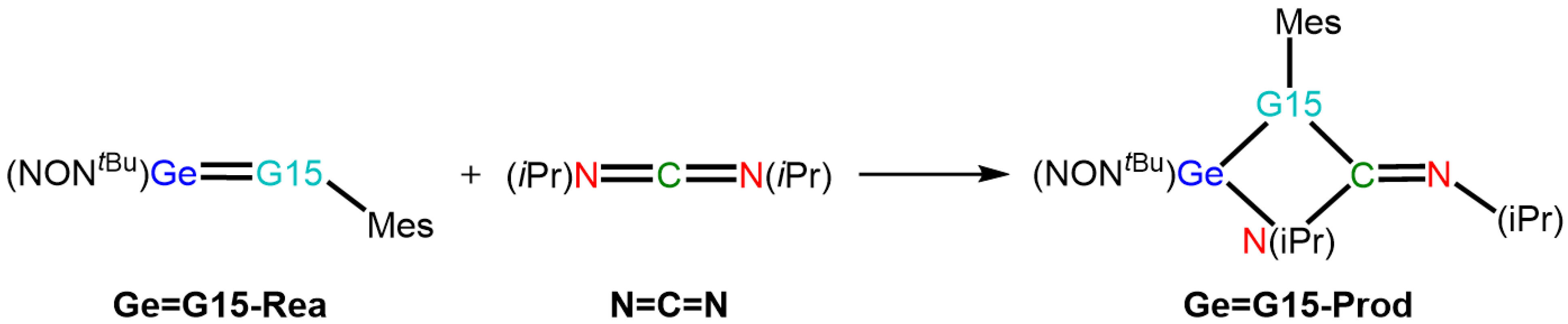
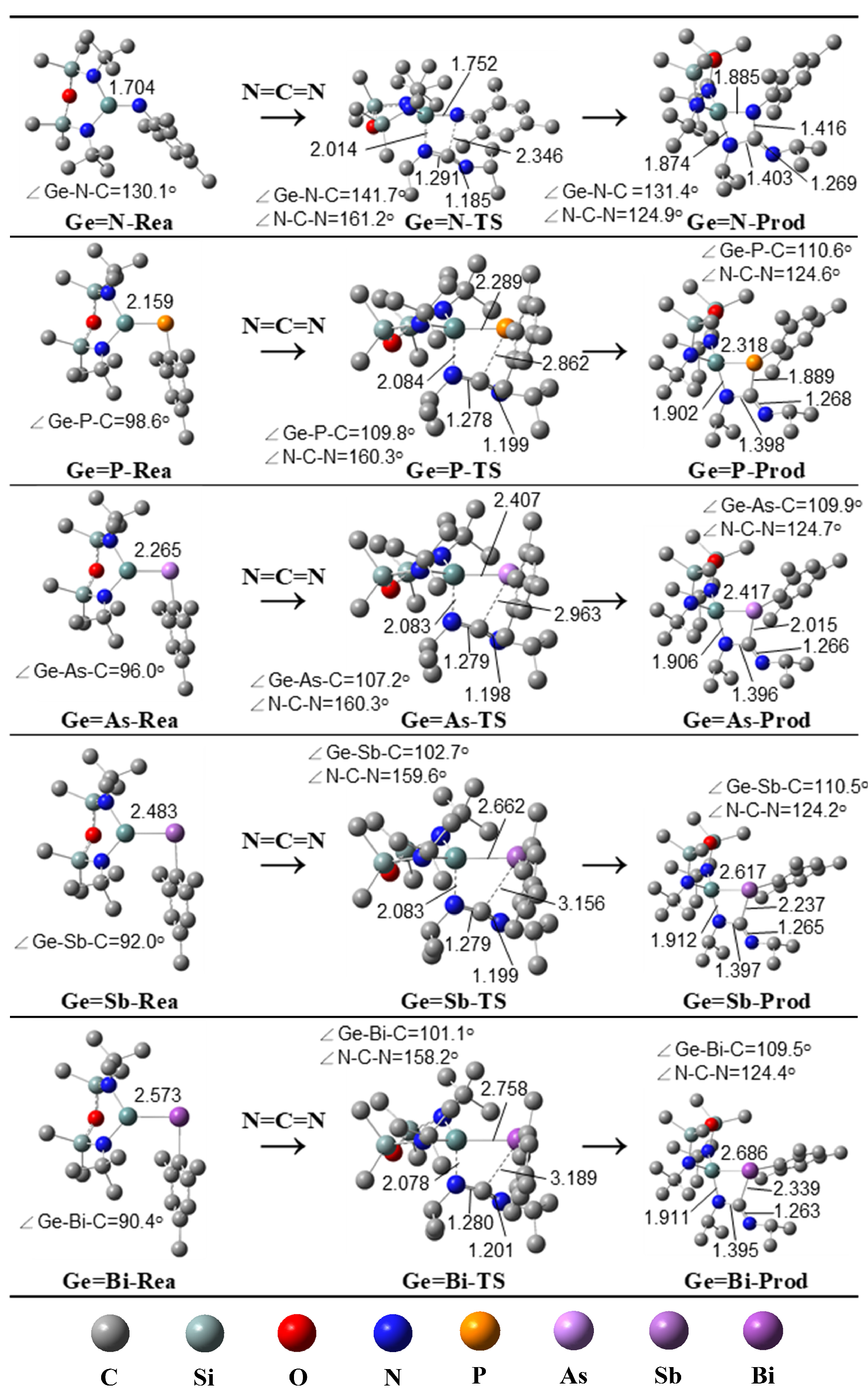
2.2. The [2 + 2] Cycloaddition Reactions of Ge=G15-Rea with N=C=N Outlined in Equation (1)
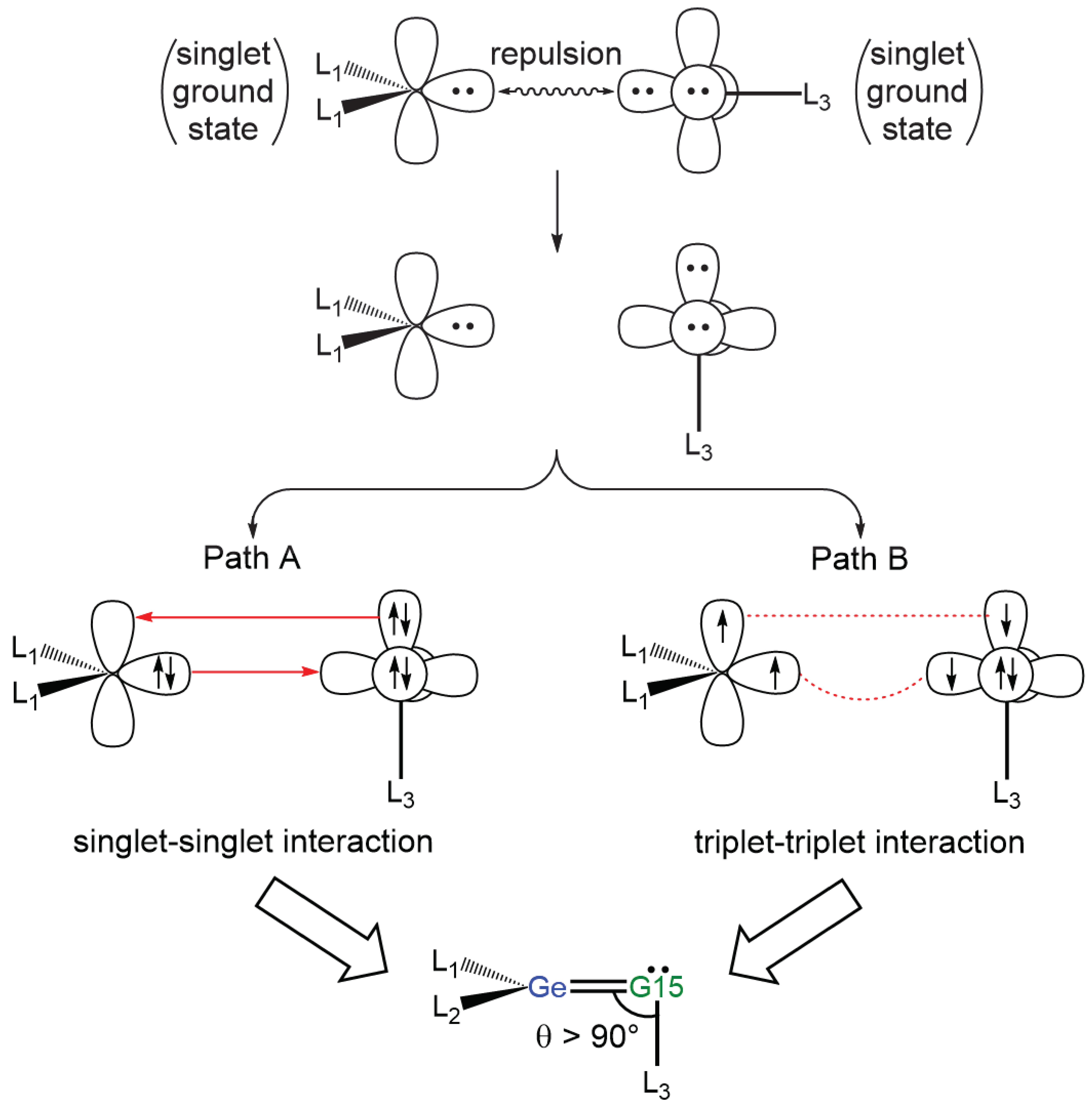
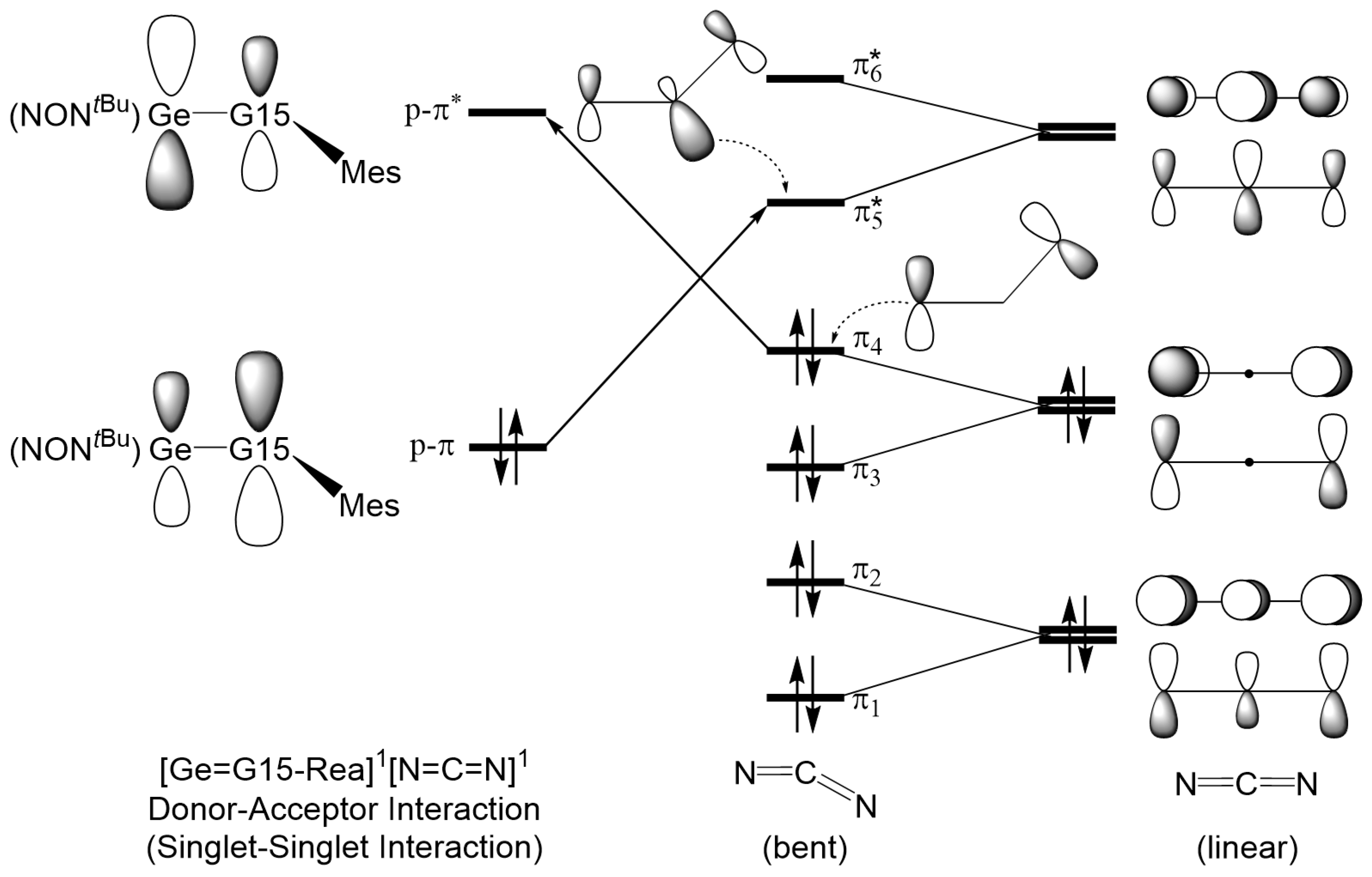
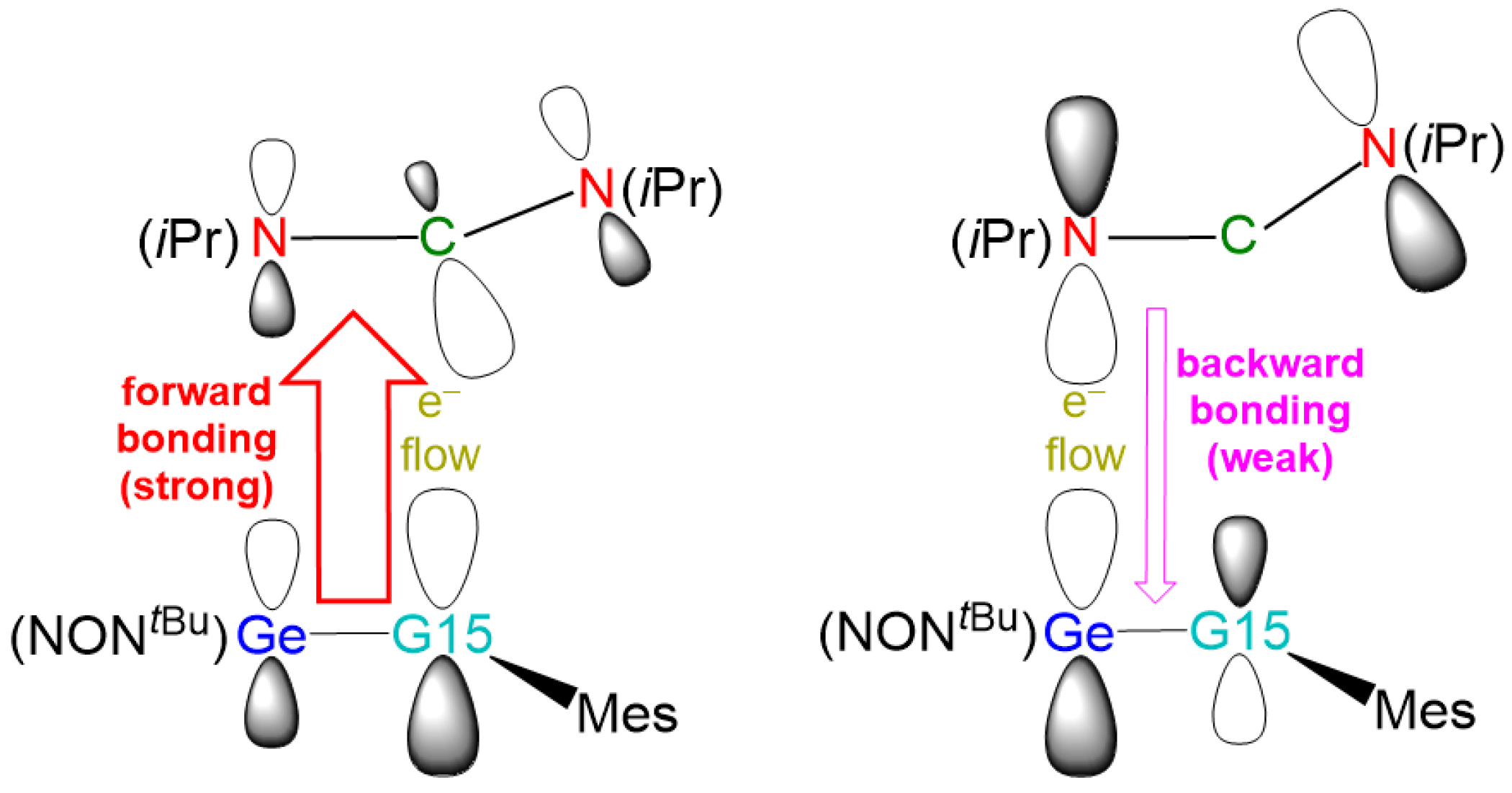
2.3. The Interaction Models
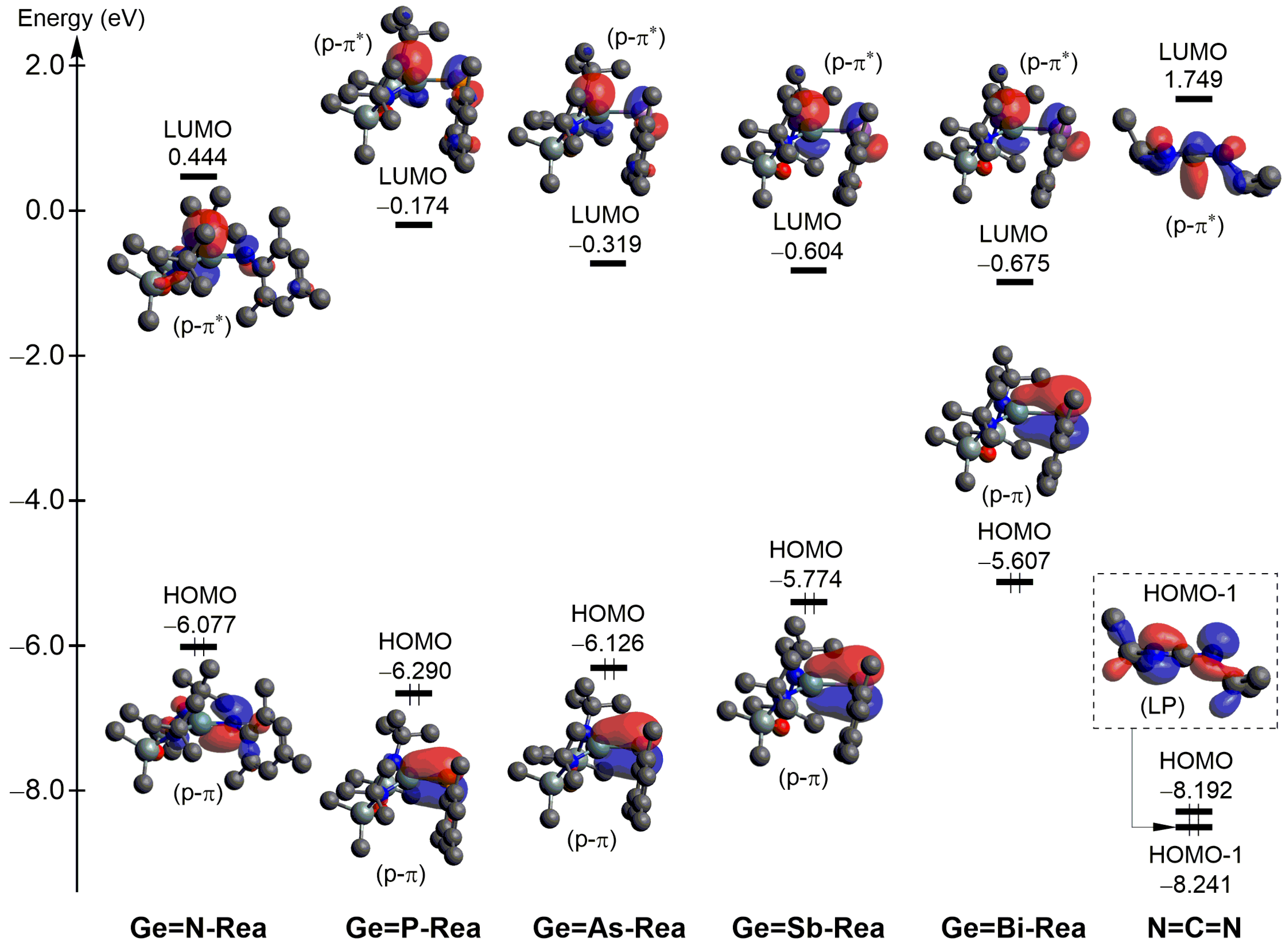
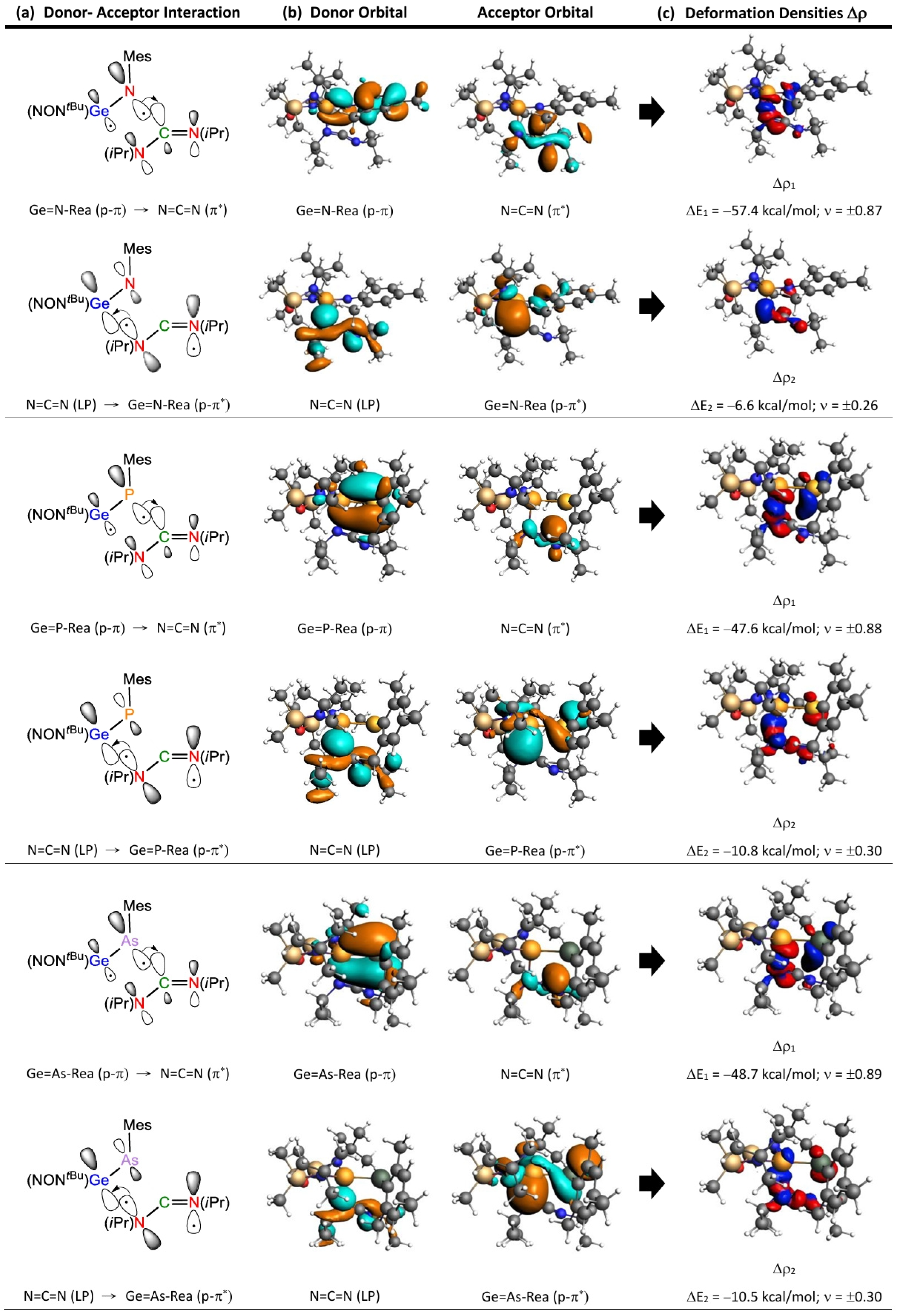
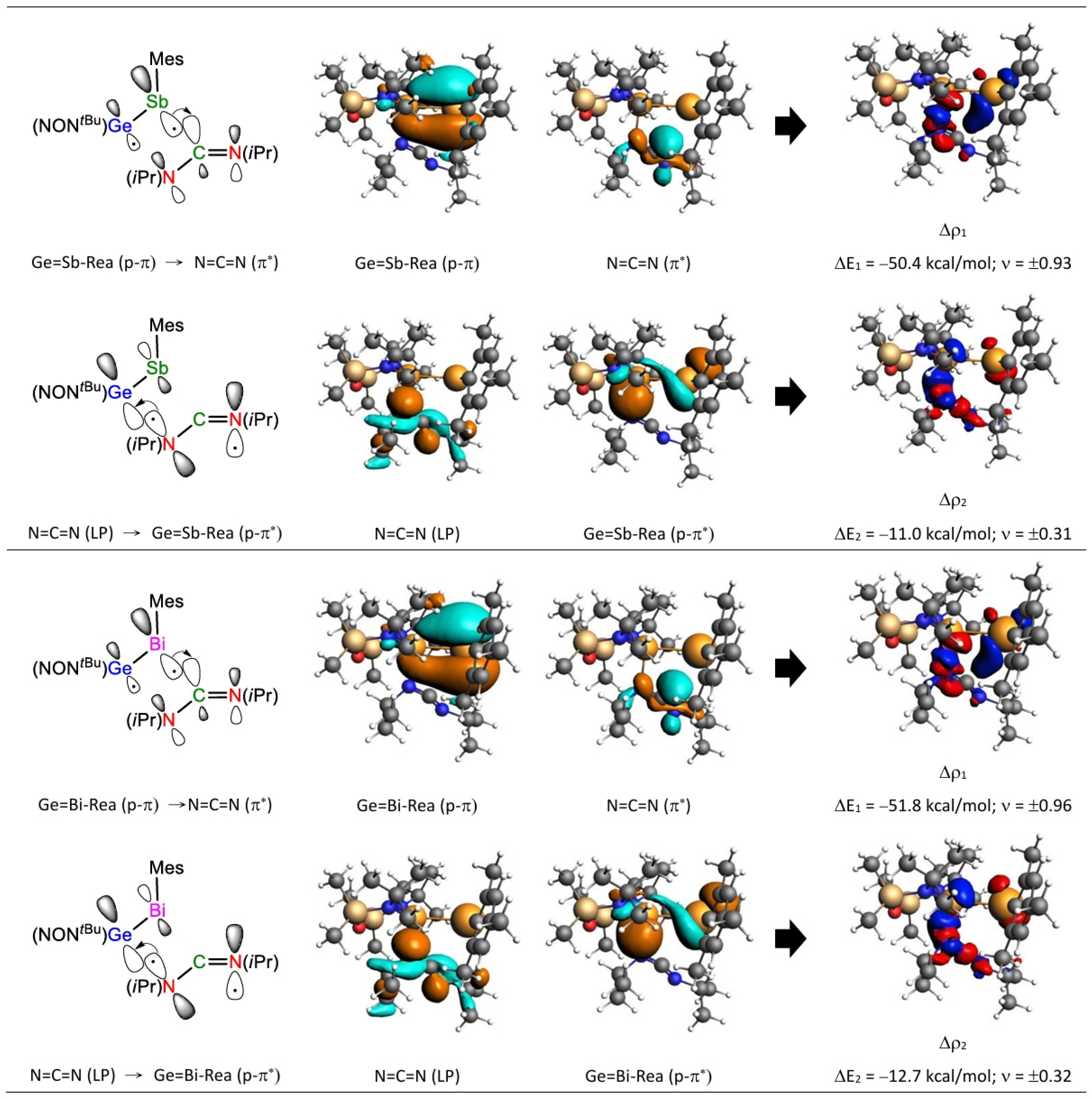
2.4. The FMO and EDA–NOCV Analyses
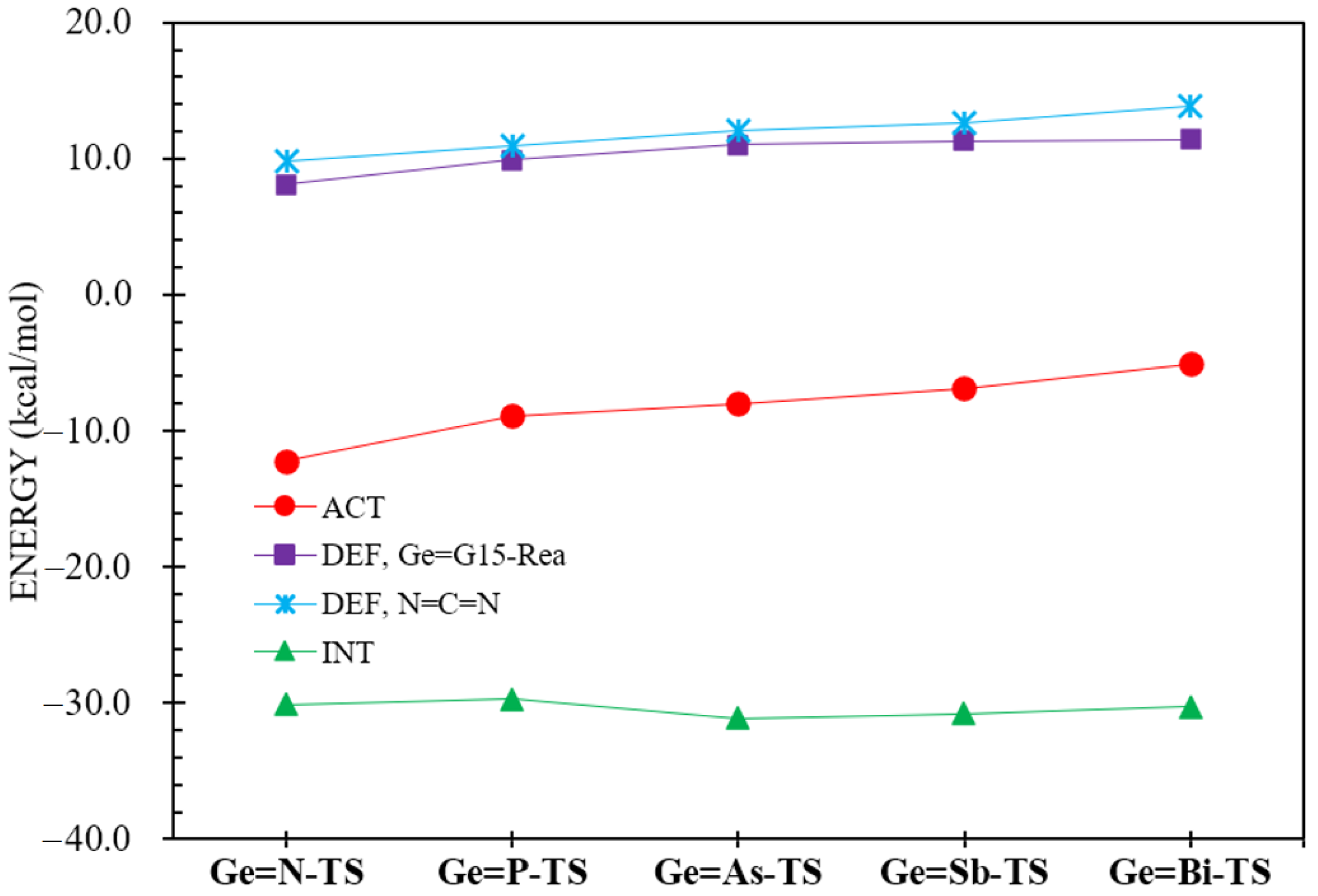
2.5. The ASM Analyses
3. Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kutzelnigg, W. Chemical Bonding in Higher Main Group Elements. Angew. Chem. Int. Ed. Engl. 1984, 23, 272–295. [Google Scholar] [CrossRef]
- Jacobsen, H.; Ziegler, T. Nonclassical double bonds in ethylene analogs: Influence of Pauli repulsion on trans bending and pi-bond strength. A density functional study. J. Am. Chem. Soc. 1994, 116, 3667–3679. [Google Scholar] [CrossRef]
- West, R.; Fink, M.J.; Michl, J. Tetramesityldisilene, a Stable Compound Containing a Silicon-Silicon Double Bond. Science 1981, 214, 1343–1344. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene: Isolation of a true phosphobenzene. J. Am. Chem. Soc. 1981, 103, 4587–4589. [Google Scholar] [CrossRef]
- Barrau, J.; Escudié, J.; Satgé, J. Multiply bonded germanium species. Recent developments. Chem. Rev. 1990, 90, 283–319. [Google Scholar] [CrossRef]
- Power, P.P. π-Bonding and the Lone Pair Effect in Multiple Bonds between Heavier Main Group Elements. Chem. Rev. 1999, 99, 3463–3504. [Google Scholar] [CrossRef] [PubMed]
- Sasamori, T.; Tokitoh, N. Doubly bonded systems between heavier Group 15 elements. Dalton Trans. 2008, 11, 1395–1408. [Google Scholar] [CrossRef]
- Fischer, R.C.; Power, P.P. π-Bonding and the Lone Pair Effect in Multiple Bonds Involving Heavier Main Group Elements: Developments in the New Millennium. Chem. Rev. 2010, 110, 3877–3923. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.Y.; Sekiguchi, A. Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Power, P.P. Main-Group Elements as Transition Metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef]
- Präsang, C.; Scheschkewitz, D. Reactivity in the periphery of functionalised multiple bonds of heavier group 14 elements. Chem. Soc. Rev. 2016, 45, 900–921. [Google Scholar] [CrossRef]
- Nesterov, V.; Breit, N.C.; Inoue, S. Advances in Phosphasilene Chemistry. Chem.-Eur. J. 2017, 23, 12014–12039. [Google Scholar] [CrossRef] [PubMed]
- Bag, P.; Weetman, C.; Inoue, S. Experimental Realisation of Elusive Multiple-Bonded Aluminium Compounds: A New Horizon in Aluminium Chemistry. Angew. Chem. Int. Ed. 2018, 57, 14394–14413. [Google Scholar] [CrossRef]
- Chu, T.; Nikonov, G.I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. [Google Scholar] [CrossRef]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018, 118, 9678–9842. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-D.; Sasamori, T. Activation of Small Molecules by Compounds that Contain Triple Bonds Between Heavier Group-14 Elements. Chem. Asia. J. 2018, 13, 3800–3817. [Google Scholar] [CrossRef]
- Melen, R.L. Frontiers in molecular p-block chemistry: From structure to reactivity. Science 2019, 363, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Pan, S.; Holzmann, N.; Schwerdtfeger, P.; Frenking, G. Chemical Bonding and Bonding Models of Main-Group Compounds. Chem. Rev. 2019, 119, 8781–8845. [Google Scholar] [CrossRef]
- Power, P.P. An Update on Multiple Bonding Between Heavier Main Group Elements: The Importance of Pauli Repulsion, Charge- Shift Character, and London Dispersion Force Effects. Organometallics 2020, 39, 4127–4138. [Google Scholar] [CrossRef]
- Hanusch, F.; Groll, L.; Inoue, S. Recent advances of group 14 dimetallenes and dimetallynes in bond activation and catalysis. Chem. Sci. 2021, 12, 2001–2015. [Google Scholar] [CrossRef]
- Weetman, C. Main Group Multiple Bonds for Bond Activations and Catalysis. Chem.-Eur. J. 2021, 27, 1941–1954. [Google Scholar] [CrossRef]
- Dankert, F.; Hering-Junghans, C. Heavier group 13/15 multiple bond systems: Synthesis, structure and chemical bond activation. Chem. Commun. 2022, 58, 1242–1262. [Google Scholar] [CrossRef] [PubMed]
- Niesmann, J.; Klingebiel, U.; Rudolph, S.; Herbst-Inner, R.; Noltemeyer, M. [2 + 2]-Cycloadditionen Eines Iminosilans-Synthese Viergliedriger Heterocyclen. J. Organomet. Chem. 1996, 15, 43–49. [Google Scholar] [CrossRef]
- Xiong, Y.; Yao, S.; Driess, M. Chemical Tricks to Stabilize Silanones and Their Heavier Homologues with E=O Bonds (E=Si–Pb): From Elusive Species to Isolable Building Blocks. Angew Chem. Int. Ed. 2013, 52, 4302–4311. [Google Scholar] [CrossRef] [PubMed]
- Muck, F.M.; Baus, J.A.; Ulmer, A.; Burschka, C.; Tacke, R. Reactivity of the Donor-Stabilized Guanidinatosilylene [ArNC(NMe2)NAr]Si[N(SiMe3)2] (Ar = 2,6-Diisopropylphenyl). Eur. J. Inorg. Chem. 2016, 2016, 1660–1670. [Google Scholar] [CrossRef]
- Hadlington, T.J.; Szilvásib, T.; Driess, M. Metal Nitrene-Like Reactivity of A SiQN Bond Towards CO2. Chem. Commun. 2018, 54, 9352–9355. [Google Scholar] [CrossRef] [PubMed]
- Yuvaraj, K.; Jones, C. Synthesis and Reactivity of Boryl Substituted Silaimines. Dalton Trans. 2019, 48, 11961–11965. [Google Scholar] [CrossRef]
- Wright, R.J.; Phillips, A.D.; Hardman, N.J.; Power, P.P. The “Diindene” ArInInAr (Ar = C6H3-2,6-Dipp2, Dipp = C6H3-2,6-Pri2). Dimeric Versus Monomeric In(I) Aryls: para-Substituent Effects in Terphenyl Ligands. J. Am. Chem. Soc. 2002, 124, 8538–8539. [Google Scholar] [CrossRef]
- Wright, R.J.; Phillips, A.D.; Hino, S.; Power, P.P. Synthesis and Reactivity of Dimeric Ar′TlTlAr′ and Trimeric (Ar′′Tl)3(Ar′, Ar′′ = Bulky Terphenyl Group) Thallium(I) Derivatives: Tl(I)−Tl(I) Bonding in Species Ligated by Monodentate Ligands. J. Am. Chem. Soc. 2005, 127, 4794–4799. [Google Scholar] [CrossRef]
- Rivieṙe-Baudet, M.; Rivieṙe, P.; Satgé, J. Germa-2azetidines: Precurseurs Des Premieres Germaimines. J. Organomet. Chem. 1978, 154, C23–C27. [Google Scholar] [CrossRef]
- Lai, C.-H.; Su, M.-D.; Chu, S.-Y. Effects of First-Row Substituents on Silicon−Phosphorus Triple Bonds. Inorg. Chem. 2002, 41, 1320–1322. [Google Scholar] [CrossRef]
- Lai, C.-H.; Su, M.-D.; Chu, S.-Y. Relative stability of multiple bonds between germanium and arsenic. A theoretical study. Polyhedron 2002, 21, 579. [Google Scholar] [CrossRef]
- Hu, Y.-H.; Su, M.-D. Substituent Effect on Relative Stabilities of the Phosphorus and Tin Multiple Bonds. Chem. Phys. Lett. 2003, 378, 289–298. [Google Scholar] [CrossRef]
- Chen, C.H.; Su, M.D. Theoretical Design of Silicon−Phosphorus Triple Bonds: A Density Functional Study. Eur. J. Inorg. Chem. 2008, 2008, 1241–1247. [Google Scholar] [CrossRef]
- Wen, X.-T.; Li, Y.-C.; Su, M.-D. Substituent Effects on the Geometries and Energies of the Antimony–Silicon Multiple Bond. Bull. Chem. Soc. Jpn. 2014, 87, 816–818. [Google Scholar] [CrossRef]
- Meller, A.; Ossig, G.; Maringgele, W.; Stalke, D.; Herbst-Irmer, R.; Freitag, S.; Sheldrick, G.M. Synthesis and X-ray crystal structure of germaimines. J. Chem. Soc. Chem. Commun. 1991, 16, 1123–1124. [Google Scholar] [CrossRef]
- Ando, W.; Ohtaki, T.; Kabe, Y. Synthesis, Structure, and Photoreaction of Bis(germanimines). Organometallics 1994, 13, 434–435. [Google Scholar] [CrossRef]
- Veith, M.; Rammo, A. Synthese Von Cyclischen, Chiralen Verbindungen Mit Ge-El-Doppelbindungen (El = N, S, (O)) Und Deren Dynamisches Verhalten in Lösung. Z. Anorg. Allg. Chem. 1997, 623, 861–872. [Google Scholar] [CrossRef]
- Rit, A.; Tirfoin, R.; Aldridge, S. Exploiting Electrostatics To Generate Unsaturation: Oxidative Ge=E Bond Formation Using a Non π-Donor Stabilized [R(L)Ge:]+ Cation. Angew. Chem. Int. Ed. 2016, 55, 378–382. [Google Scholar] [CrossRef]
- Schneider, J.; Krebs, K.M.; Freitag, S.; Eichele, K.; Schubert, H.; Wesemann, L. Intramolecular Tetrylene Lewis Adducts: Synthesis and Reactivity. Chem.-Eur. J. 2016, 22, 9812–9826. [Google Scholar] [CrossRef]
- Sharma, M.K.; Sinhababu, S.; Mahawar, P.; Mukherjee, G.; Pandey, B.; Rajaraman, G.; Nagendran, S. Donor-Acceptor-Stabilised Germanium Analogues of Acid Chloride, Ester, and Acyl Pyrrole Compounds: Synthesis and Reactivity. Chem. Sci. 2019, 10, 4402–4411. [Google Scholar] [CrossRef]
- Evans, M.J.; Anker, M.D.; Mouchfiq, A.; Lein, M.; Fulton, J.R. The “Metallo”-Diels-Alder Reactions: Examining the Metalloid Behavior of Germani- mines. Chem.-Eur. J. 2020, 26, 2606–2609. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Burke, F.M.; Chapple, P.M.; Fulton, J.R. Synthesis and Reactivity of Acyclic Germanimines: Silyl Rearrangement and Cycloadditions. Inorg. Chem. 2021, 60, 8293–8303. [Google Scholar] [CrossRef]
- Guthardt, R.; Oetzel, L.; Lang, T.; Bruhn, C.; Siemeling, U. Reactions of Mesityl Azide with Ferrocene-Based N-Heterocyclic Germylenes, Stannylenes and Plumbylenes, Including PPh2-Functionalised Congeners. Chem.-Eur. J. 2022, 28, e202200996. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, L.; He, Y.; Chen, Y.; Wang, D.; Liu, J.; Tan, G. A Germanimidoyl Chloride: Synthesis, Characterization and Reactivity. Chem. Commun. 2023, 59, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, Y.; Li, T.; Wang, D.; Xu, L.; Tan, G. Synthesis and Reactivity of N-Heterocyclic Carbene Coordinated Formal Germanimidoyl-Phosphinidenes. Inorg. Chem. 2023, 62, 20906–20912. [Google Scholar] [CrossRef]
- Escudié, J.; Couret, C.; Satgé, J.; Andrianarison, M.; Andriamizaka, J.D. 2,2-Dimesityl-1-(2,4,6-tri-tert-butylphenyl)-germaphosphene: The first stable compound with a germanium- phosphorus double bond. J. Am. Chem. Soc. 1985, 107, 3378–3379. [Google Scholar] [CrossRef]
- Escudlé, J.; Couret, C.; Andrianarison, M.; Raharinirina, A.; Satgé, J. New Aspects of the Reactivity of the First Stable Germa-and Stannaphosphenes. Phosphorus Sulfur Rel. Elem. 1987, 30, 377–380. [Google Scholar] [CrossRef]
- Draeger, M.; Escudié, J.; Couret, C.; Ranaivonjatovo, H.; Satgé, J. Structure of the stable germaphosphene Mes2Ge=Par: A Ge=P connection with the geometry of a true double bond. Organometallics 1988, 7, 1010–1013. [Google Scholar] [CrossRef]
- Ranaivonjatovo, H.; Escudié, J.; Couret, C.; Satgé, J. A new stable germaphosphene and some of its chemical properties. J. Organomet. Chem. 1991, 415, 327–333. [Google Scholar] [CrossRef]
- Lee, V.Y.; Kawai, M.; Sekiguchi, A.; Ranaivonjatovo, H.; Escudié, J. A “Push-Pull” Phosphasilene and Phosphagermene and Their Anion-Radicals. Organometallics 2009, 28, 4262–4265. [Google Scholar] [CrossRef]
- Ballestero-Martınez, E.; Hadlington, T.J.; Tibor Szilvasi, T.; Yao, S.; Driess, M. From zinco(II) arsaketenes to silylene-stabilised zinco arsinidene complexes. Chem. Commun. 2018, 54, 6124–6127. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.Y.; Manami Kawai, M.; Gapurenko, O.A.; Minkin, V.I.; Gornitzka, H.; Sekiguchi, A. Arsagermene, a compound with an –As=Ge< double bond. Chem. Commun. 2018, 54, 10947–10949. [Google Scholar]
- Lee, V.Y.; Sekiguchi, A. Heavy Analogs of Alkenes, 1,3-Dienes, Allenes and Alkynes: Multiply Bonded Derivatives of Si, Ge, Sn and Pb. In Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds; Wiley: Chichester, UK, 2010; Chapter 5. [Google Scholar]
- Baceiredo, A.; Kato, T. Organosilicon Compounds (Volume 1: Theory and Experiment Synthesis); Lee, V.Y., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 9. [Google Scholar]
- Chen, J.-Y.; Su, M.-D. Diboration of the E=E Double Bond by [2]Metallocenophanes (E = N, P, As, Sb, and Bi): A Theoretical Study. Organometallics 2010, 29, 5812–5820. [Google Scholar] [CrossRef]
- Morokuma, K. Molecular Orbital Studies of Hydrogen Bonds. III. C=O···H–O Hydrogen Bond in H2CO···H2O and H2CO···2H2O. J. Chem. Phys. 1971, 55, 1236–1244. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the Calculation of Bonding Energies by the Hartree Fock Slater Method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. A Theoretical Study of the Ethylene-Metal Bond in Complexes Between Copper(1+), Silver(1+), Gold(1+), Platinum(0) or Platinum(2+) and Ethylene, Based on the Hartree-Fock-Slater Transition-State Method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Ziegler, T. Oxidative insertion as front-side SN2 substitution: A theoretical study of the model reaction system Pd + CH3Cl. Organometallics 1995, 14, 2288–2296. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M. Understanding reactivity with Kohn-Sham molecular orbital theory: E2-SN2 mechanistic spectrum and other concepts. J. Comput. Chem. 1999, 20, 114–128. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. Rev. Comput. Chem. 2000, 15, 1–86. [Google Scholar]
- Mitoraj, M.; Michalak, A. Natural Orbitals for Chemical Valence as Descriptors of Chemical Bonding in Transition Metal Complexes. J. Mol. Model. 2007, 13, 347–355. [Google Scholar] [CrossRef]
- Mitoraj, M.; Michalak, A. Donor-acceptor properties of ligands from the natural orbitals for chemical valence. Organometallics 2007, 26, 6576–6580. [Google Scholar] [CrossRef]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond orbitals from chemical valence theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Tonner, R.; Frenking, G. Divalent Carbon(0) Chemistry, Part 1: Parent compounds. Chem. Eur. J. 2008, 14, 3260–3272. [Google Scholar] [CrossRef]
- Gimferrer, M.; Danés, S.; Vos, E.; Corral, I.; Jana, A.; Salvador, P.; Andrada, D.M. Reply to the Comment on “The oxidation state in low-valent beryllium and magnesium compounds” by S. Pan and G. Frenking, Chem. Sci. 2022, 13, 6583. Chem. Sci. 2023, 14, 384–392. [Google Scholar] [CrossRef]
- Fukui, K. Recognition of Stereochemical Paths by Orbital Interaction. Acc. Chem. Res. 1971, 2, 57–64. [Google Scholar] [CrossRef]
- Hammond, G.S. A Correlation of Reaction Rates. J. Am. Chem. Soc. 1955, 77, 334–338. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154122. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Computer Code ADF2023, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 17 April 2025).
- van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Chang, C.; Pelissier, M.; Durand, P. Regular Two-Component Pauli-Like Effective Hamiltonians in Dirac Theory. Phys. Scr. 1986, 34, 394–404. [Google Scholar] [CrossRef]
- Heullyt, J.-L.; Lindgrent, I.; Lindrothi, E.; Lundqvist, S.; Pendrill, A.-M. Diagonalisation of the Dirac Hamiltonian as a basis for a relativistic many-body procedure. J. Phys. B 1986, 19, 2799–2815. [Google Scholar] [CrossRef]
- van Zeist, W.-J.; Bickelhaupt, F.M. The Activation Strain Model of Chemical Reactivity. Org. Biomol. Chem. 2010, 8, 3118–3127. [Google Scholar] [CrossRef] [PubMed]
- Fernańdez, I.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory: Understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef]
- Fernańdez, I. Combined activation strain model and energy decomposition analysis methods: A new way to understand pericyclic reactions. Phys. Chem. Chem. Phys. 2014, 16, 7662–7671. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory. WIREs Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar] [CrossRef]
- Fernańdez, I. Discoveringthe Future of Molecular Sciences; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp. 165–187. [Google Scholar]
- Ess, D.H.; Houk, K.N. Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc. 2007, 129, 10646–10647. [Google Scholar] [CrossRef]
- Ess, D.H.; Houk, K.N. Theory of 1,3-Dipolar Cycloadditions: Distortion/Interaction and Frontier Molecular Orbital Models. J. Am. Chem. Soc. 2008, 130, 10187–10198. [Google Scholar] [CrossRef] [PubMed]
- Ess, D.H.; Jones, G.O.; Houk, K.N. Transition States of Strain-Promoted Metal-Free Click Chemistry: 1,3-Dipolar Cycloadditions of Phenyl Azide and Cyclooctynes. Org. Lett. 2008, 10, 1633–1636. [Google Scholar] [CrossRef] [PubMed]
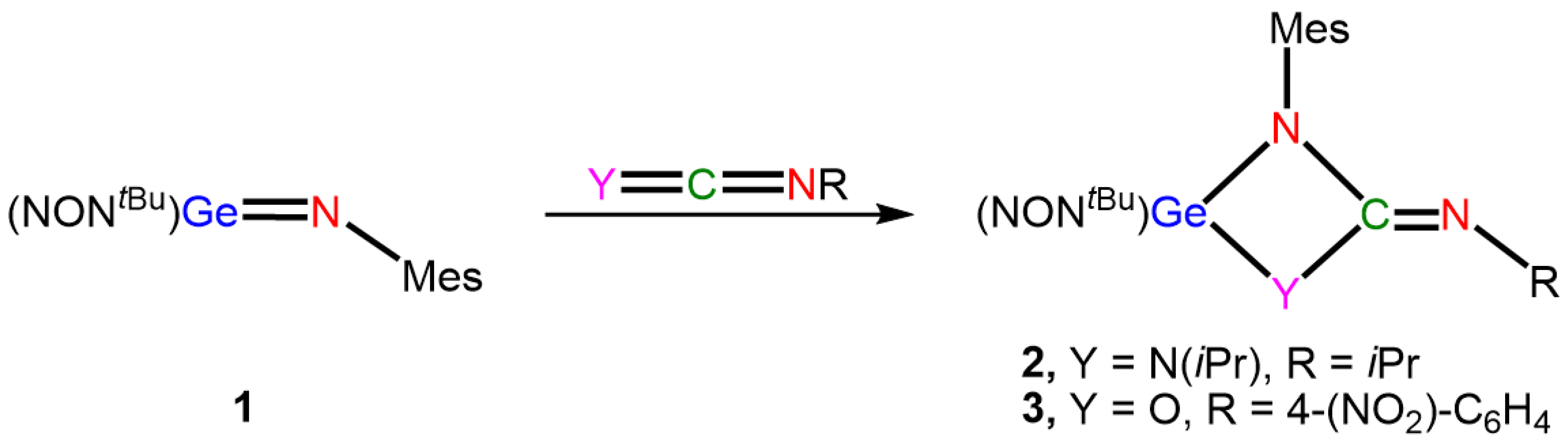



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragments | Ge=N-Rea | Ge=P-Rea | Ge=As-Rea | Ge=Sb-Rea | Ge=Bi-Rea | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| (NONtBu)Ge: (S) + :N–Mes (S) | (NONtBu)Ge: (T) + :N–Mes (T) | (NONtBu)Ge: (S) + :P–Mes (S) | (NONtBu)Ge: (T) + :P–Mes (T) | (NONtBu)Ge: (S) + :As–Mes (S) | (NONtBu)Ge: (T) + :As–Mes (T) | (NONtBu)Ge: (S) + :Sb–Mes (S) | (NONtBu)Ge: (T) + :Sb–Mes (T) | (NONtBu)Ge: (S) + :Bi–Mes (S) | (NONtBu)Ge: (T) + :Bi–Mes (T) | |
| ΔEINT (a) | −96.0 | −111.9 | −83.0 | −96.7 | −72.9 | −84.6 | −58.0 | −70.0 | −50.3 | −61.9 |
| ΔEPauli | 490.1 | 346.2 | 445.9 | 195.3 | 422.8 | 176.6 | 323.6 | 145.7 | 295.7 | 130.1 |
| ΔEElstat (b) | −249.8 (42.6%) | −204.4 (44.6%) | −241.5 (45.7%) | −140.8 (48.2%) | −236.2 (47.6%) | −133.9 (51.3%) | −192.7 (50.5%) | −117.4 (54.4%) | −180.4 (52.1%) | −108.9 (56.7%) |
| ΔEOrb (b) | −334.4 (57.1%) | −251.8 (55.0%) | −285.5 (54.0%) | −149.3 (51.1%) | −257.7 (52.0%) | −125.4 (48.0%) | −187.0 (49.0%) | −96.5 (44.7%) | −163.8 (47.4%) | −81.3 (42.3%) |
| System | Energy Difference N=C=N (LUMO)—Ge=G15-Rea (HOMO) | Energy Difference Ge=G15-Rea (LUMO)—N=C=N (HOMO-1) |
|---|---|---|
| Ge=N-Rea + N=C=N | 7.826 | 8.685 |
| Ge=P-Rea + N=C=N | 8.039 | 8.067 |
| Ge=As-Rea + N=C=N | 7.875 | 7.922 |
| Ge=Sb-Rea + N=C=N | 7.523 | 7.637 |
| Ge=Bi-Rea + N=C=N | 7.356 | 7.566 |
| Fragments | Ge=N-TS | Ge=P-TS | Ge=As-TS | Ge=Sb-TS | Ge=Bi-TS | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ge=N-Rea (S) + N=C=N (S) | Ge=N-Rea (T) + N=C=N (T) | Ge=P-Rea (S) + N=C=N (S) | Ge=P-Rea (T) + N=C=N (T) | Ge=As-Rea (S) + N=C=N (S) | Ge=As-Rea (T) + N=C=N (T) | Ge=Sb-Rea (S) + N=C=N (S) | Ge=Sb-Rea (T) + N=C=N (T) | Ge=Bi-Rea (S) + N=C=N (S) | Ge=Bi-Rea (T) + N=C=N (T) | |
| ΔEINT (a) | −30.1 | −172.8 | −29.7 | −155.0 | −31.1 | −148.1 | −30.8 | −139.4 | −30.3 | −131.1 |
| ΔEPauli | 207.0 | 291.4 | 175.0 | 236.6 | 173.7 | 232.6 | 175.2 | 223.1 | 178.5 | 219.0 |
| ΔEElstat (b) | −138.7 (58.5%) | −170.2 (36.7%) | −121.4 (59.3%) | −144.0 (36.8%) | −121.1 (59.1%) | −142.6 (37.4%) | −121.2 (58.9%) | −139.1 (38.4%) | −122.8 (58.8%) | −138.6 (39.6%) |
| ΔEOrb (b) | −95.4 (40.2%) | −291.1 (62.7%) | −80.5 (39.3%) | −244.8 (62.5%) | −81.0 (39.5%) | −235.4 (61.8%) | −82.1 (39.8%) | −220.6 (60.9%) | −83.4 (39.9%) | −208.8 (59.6%) |
| ΔEOrb(1) (c) | −57.4 (60.2%) | −234.1 (80.4%) | −47.6 (59.1%) | −199.6 (81.6%) | −48.7 (60.1%) | −195.7 (83.1%) | −50.4 (61.4%) | −182.9 (82.9%) | −51.8 (62.1%) | −170.2 (81.5%) |
| ΔEOrb(2) (c) | −6.6 (6.9%) | −27.2 (9.4%) | −10.8 (13.4%) | −24.6 (10.1%) | −10.5 (12.9%) | −23.8 (10.1%) | −11.0 (13.4%) | −24.5 (11.1%) | −12.7 (15.2%) | −26.8 (12.8%) |
| ΔERest (c) | −31.4 (32.9%) | −29.7 (10.2%) | −22.1 (27.5%) | −20.5 (8.4%) | −21.8 (27.0%) | −15.9 (6.7%) | −20.7 (25.2%) | −13.3 (6.0%) | −18.9 (22.7%) | −11.8 (5.7%) |
| ΔEDisper (b) | −3.0 (1.3%) | −3.0 (0.6%) | −2.8 (1.4%) | −2.8 (0.7%) | −2.8 (1.3%) | −2.8 (0.7%) | −2.7 (1.3%) | −2.7 (0.7%) | −2.7 (1.3%) | −2.7 (0.8%) |
| Entry | Ge=N-TS | Ge=P-TS | Ge=As-TS | Ge=Sb-TS | Ge=Bi-TS |
|---|---|---|---|---|---|
| ΔEACT (a,b) | −12.2 | −8.9 | −8.0 | −6.9 | −5.1 |
| ΔEDEF,N=C=N | 9.8 | 10.9 | 12.1 | 12.6 | 13.8 |
| ΔEDEF,Ge=G15-Rea | 8.1 | 9.9 | 11.0 | 11.3 | 11.4 |
| ΔEINT | −30.1 | −29.7 | −31.1 | −30.8 | −30.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-F.; Su, M.-D. Insights into the Factors Controlling the Origin of Activation Barriers in the [2 + 2] Cycloaddition Reactions of Heavy Imine-like Molecules Featuring a Ge=Group 15 Double Bond with Heterocumulenes. Molecules 2025, 30, 1905. https://doi.org/10.3390/molecules30091905
Zhang Z-F, Su M-D. Insights into the Factors Controlling the Origin of Activation Barriers in the [2 + 2] Cycloaddition Reactions of Heavy Imine-like Molecules Featuring a Ge=Group 15 Double Bond with Heterocumulenes. Molecules. 2025; 30(9):1905. https://doi.org/10.3390/molecules30091905
Chicago/Turabian StyleZhang, Zheng-Feng, and Ming-Der Su. 2025. "Insights into the Factors Controlling the Origin of Activation Barriers in the [2 + 2] Cycloaddition Reactions of Heavy Imine-like Molecules Featuring a Ge=Group 15 Double Bond with Heterocumulenes" Molecules 30, no. 9: 1905. https://doi.org/10.3390/molecules30091905
APA StyleZhang, Z.-F., & Su, M.-D. (2025). Insights into the Factors Controlling the Origin of Activation Barriers in the [2 + 2] Cycloaddition Reactions of Heavy Imine-like Molecules Featuring a Ge=Group 15 Double Bond with Heterocumulenes. Molecules, 30(9), 1905. https://doi.org/10.3390/molecules30091905