
Hydrogenation-Facilitated Spontaneous N-O Cleavage Mechanism for Effectively Boosting Nitrate Reduction Reaction on Fe2B2 MBene


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
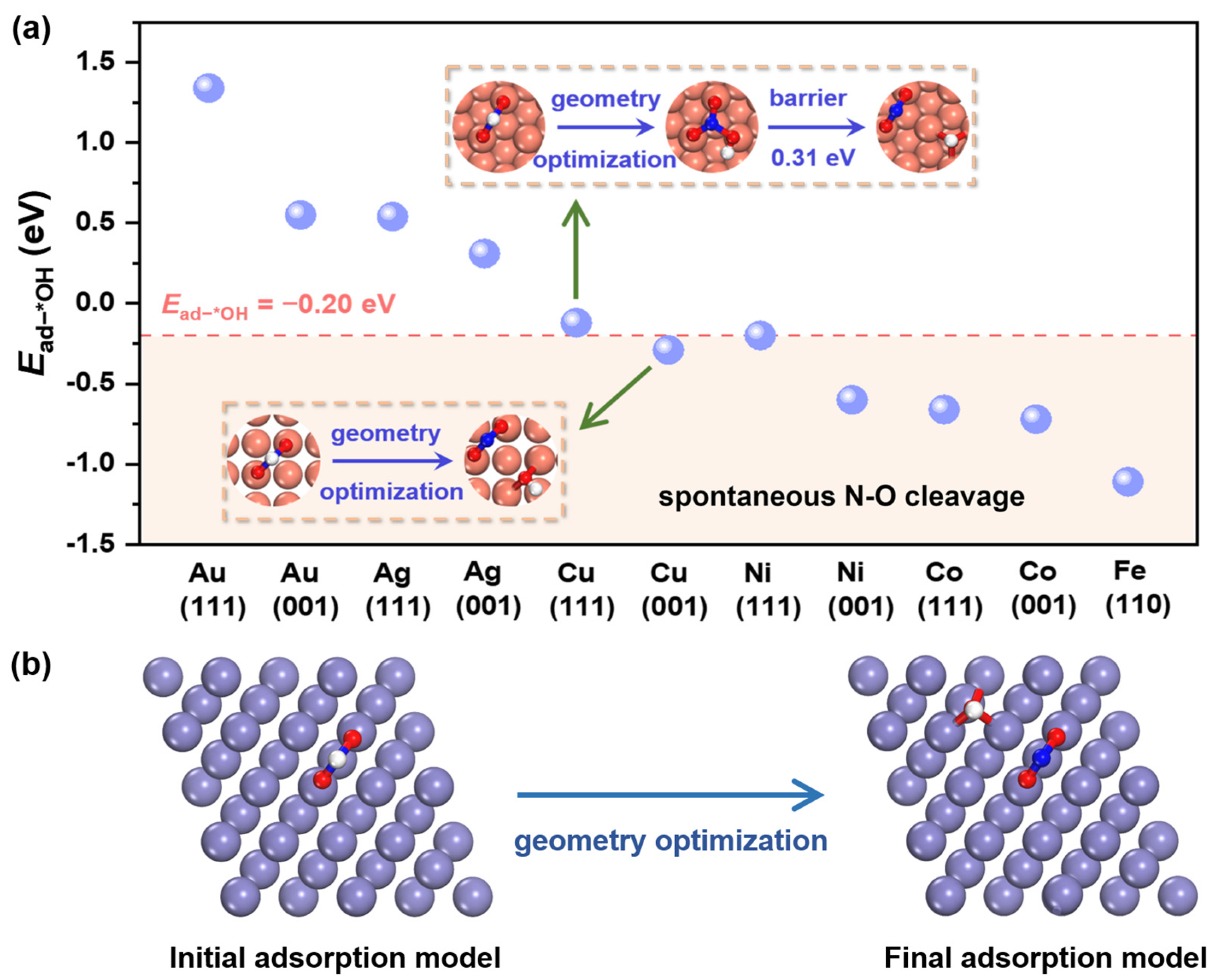
2.1. Hydrogenation-Facilitated Spontaneous N-O Cleavage Mechanism
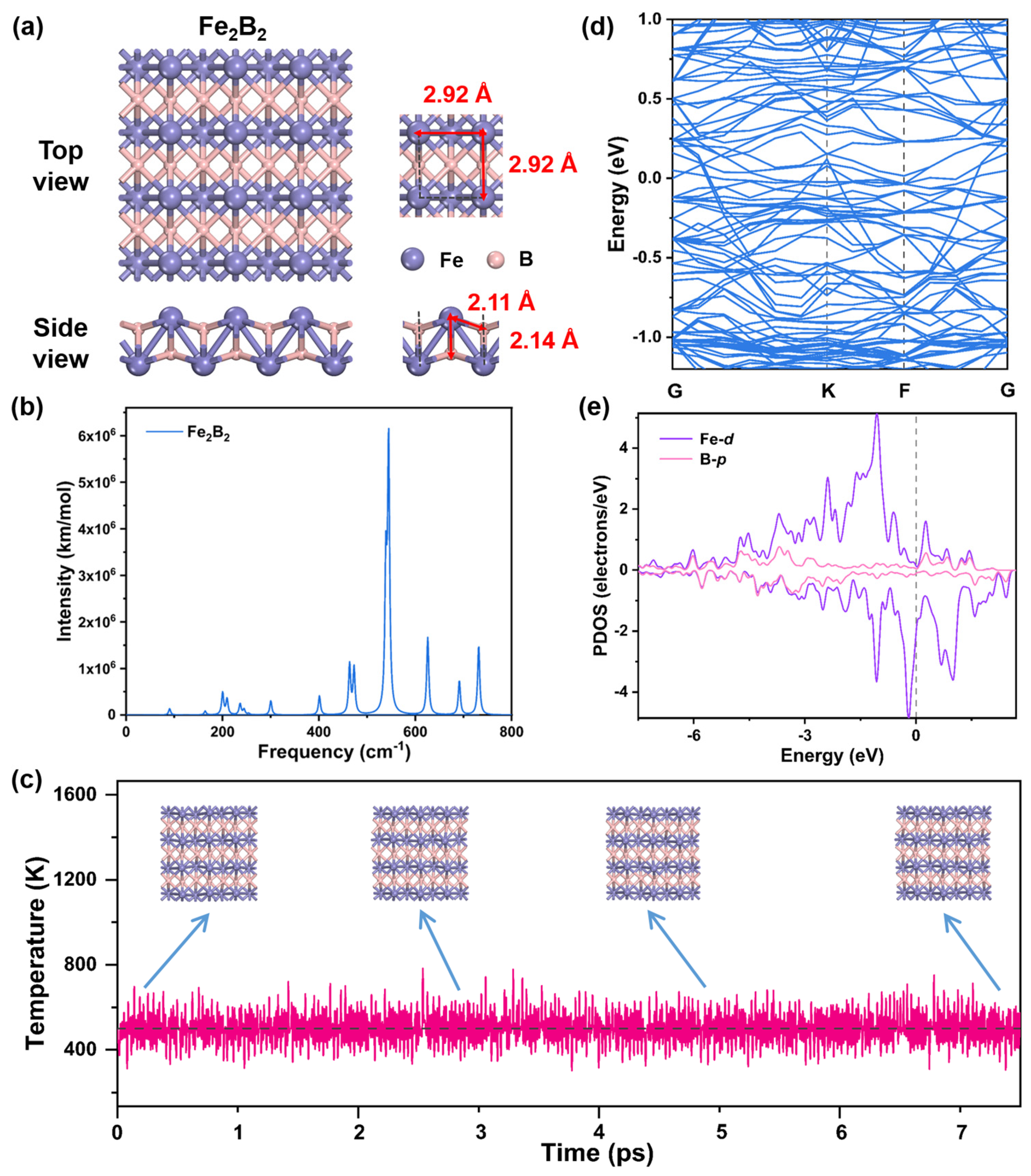
2.2. Structures and Stability of Fe2B2
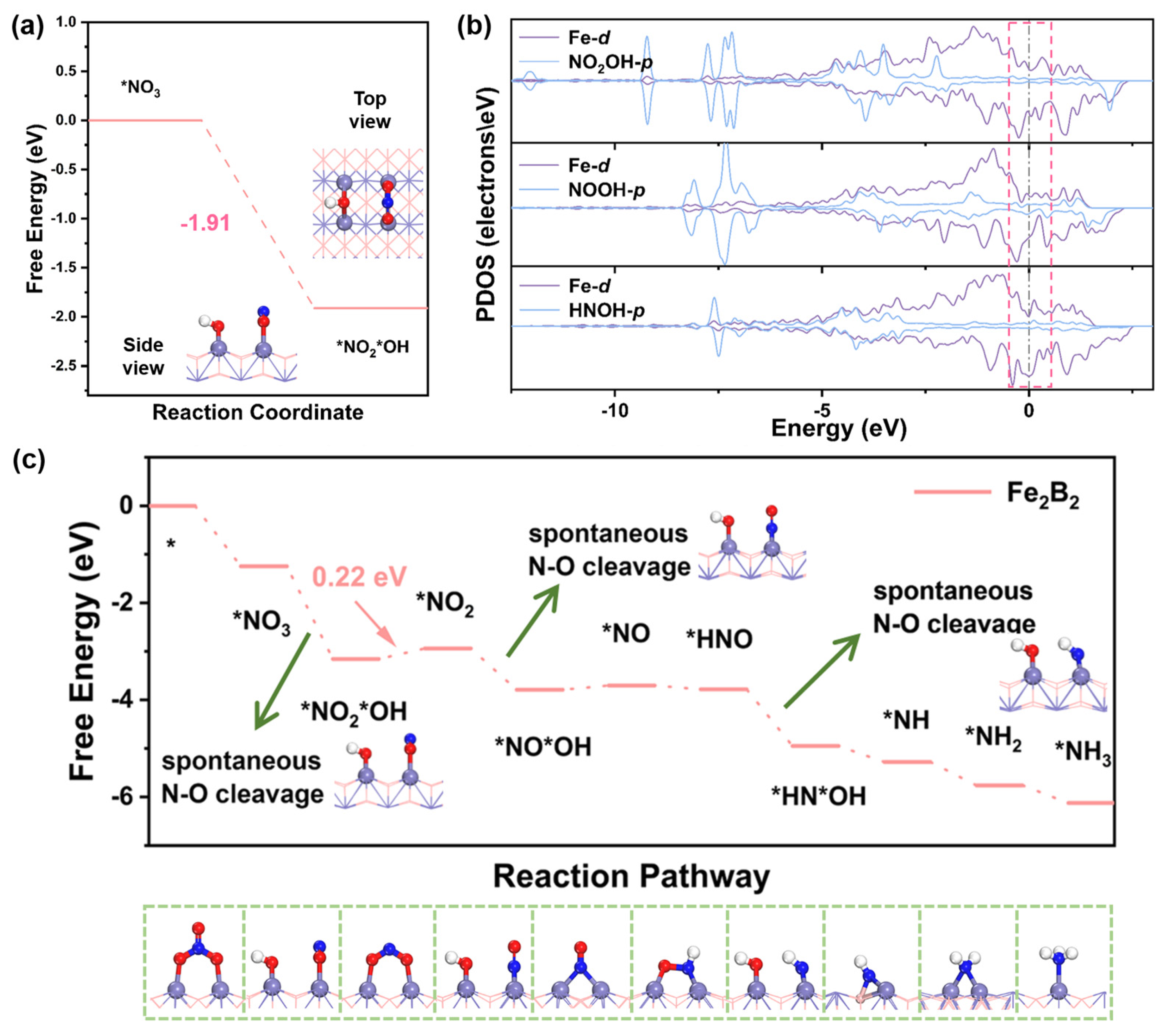
2.3. NO3− Adsorption and Activation
2.4. NO3RR Performance of Fe2B2
2.5. Selectivity Toward NH3 Synthesis
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Langevelde, P.H.; Katsounaros, I.; Koper, M.T.M. Electrocatalytic Nitrate Reduction for Sustainable Ammonia Production. Joule 2021, 5, 290–294. [Google Scholar] [CrossRef]
- Singh, S.; Anil, A.G.; Kumar, V.; Kapoor, D.; Subramanian, S.; Singh, J.; Ramamurthy, P.C. Nitrates in the environment: A critical review of their distribution, sensing techniques, ecological effects and remediation. Chemosphere 2022, 287, 131996. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, B.; Chatterjee, R.P.; Ruj, B.; Chakrabortty, S.; Nayak, J. Strategic management of nitrate pollution from contaminated water using viable adsorbents: An economic assessment-based review with possible policy suggestions. J. Environ. Manag. 2022, 303, 114081. [Google Scholar] [CrossRef] [PubMed]
- Picetti, R.; Deeney, M.; Pastorino, S.; Miller, M.R.; Shah, A.; Leon, D.A.; Dangour, A.D.; Green, R. Nitrate and nitrite contamination in drinking water and cancer risk: A systematic review with meta-analysis. Environ. Res. 2022, 210, 112988. [Google Scholar] [CrossRef]
- Neef, H.J. International overview of hydrogen and fuel cell research. Energy 2009, 34, 327–333. [Google Scholar] [CrossRef]
- Panwar, N.L.; Kothari, R.; Tyagi, V.V. Thermo chemical conversion of biomass—Eco friendly energy routes. Renew. Sust. Energ. Rev. 2012, 16, 1801–1816. [Google Scholar] [CrossRef]
- Duca, M.; Koper, M.T.M. Powering denitrification: The perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 2012, 5, 9726–9742. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Q.; Xie, L.; Ji, Y.; Li, T.; Zhang, B.; Li, N.; Tang, B.; Liu, Y.; Gao, S.; et al. High-Performance Electrochemical Nitrate Reduction to Ammonia under Ambient Conditions Using a FeOOH Nanorod Catalyst. ACS Appl. Mater. Interfaces 2022, 14, 17312–17318. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Karamad, M.; Yong, X.; Huang, Q.; Cullen, D.A.; Zhu, P.; Xia, C.; Xiao, Q.; Shakouri, M.; Chen, F.-Y.; et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 2021, 12, 2870. [Google Scholar] [CrossRef]
- Lu, X.; Song, H.; Cai, J.; Lu, S. Recent development of electrochemical nitrate reduction to ammonia: A mini review. Electrochem. Commun. 2021, 129, 107094. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Zhang, Z.; Mo, Z.; Wang, C.; Gao, S. Flower-like open-structured polycrystalline copper with synergistic multi-crystal plane for efficient electrocatalytic reduction of nitrate to ammonia. Nano Energy 2022, 97, 107124. [Google Scholar] [CrossRef]
- Yang, X.; Ling, F.; Zi, X.; Wang, Y.; Zhang, H.; Zhang, H.; Zhou, M.; Guo, Z.; Wang, Y. Low-Coordinate Step Atoms via Plasma-Assisted Calcinations to Enhance Electrochemical Reduction of Nitrogen to Ammonia. Small 2020, 16, 2000421. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Cui, P.; Wang, F.; Ding, L.-X.; Wang, H. High Efficiency Electrochemical Nitrogen Fixation Achieved with a Lower Pressure Reaction System by Changing the Chemical Equilibrium. Angew. Chem.-Int. Edit. 2019, 58, 15541–15547. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.; Yang, D.; Qian, X.; Wang, Z.; Xu, Y.; Li, X.; Xue, H.; Wang, L. Direct fabrication of bi-metallic PdRu nanorod assemblies for electrochemical ammonia synthesis. Nanoscale 2019, 11, 5499–5505. [Google Scholar] [CrossRef]
- Yang, X.; Nash, J.; Anibal, J.; Dunwell, M.; Kattel, S.; Stavitski, E.; Attenkofer, K.; Chen, J.G.; Yan, Y.; Xu, B. Mechanistic Insights into Electrochemical Nitrogen Reduction Reaction on Vanadium Nitride Nanoparticles. J. Am. Chem. Soc. 2018, 140, 13387–13391. [Google Scholar] [CrossRef]
- Chen, J.G.; Crooks, R.M.; Seefeldt, L.C.; Bren, K.L.; Bullock, R.M.; Darensbourg, M.Y.; Holland, P.L.; Hoffman, B.; Janik, M.J.; Jones, A.K.; et al. Beyond fossil fuel–driven nitrogen transformations. Science 2018, 360, eaar6611. [Google Scholar] [CrossRef]
- Foster, S.L.; Bakovic, S.I.P.; Duda, R.D.; Maheshwari, S.; Milton, R.D.; Minteer, S.D.; Janik, M.J.; Renner, J.N.; Greenlee, L.F. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 2018, 1, 490–500. [Google Scholar] [CrossRef]
- Soloveichik, G. Electrochemical synthesis of ammonia as a potential alternative to the Haber–Bosch process. Nat. Catal. 2019, 2, 377–380. [Google Scholar] [CrossRef]
- Zhang, G.; Li, X.; Chen, K.; Guo, Y.; Ma, D.; Chu, K. Tandem Electrocatalytic Nitrate Reduction to Ammonia on MBenes. Angew. Chem.-Int. Edit. 2023, 62, e202300054. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Xue, Z.; Tan, R.; Tian, J.; Hou, H.; Zhang, X.; Zhao, Y. Unraveling the activity trends of T-C2N based Single-Atom catalysts for electrocatalytic nitrate reduction via high-throughput screening. J. Colloid Interface Sci. 2024, 674, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Qiao, L.; Peng, S.; Bai, H.; Liu, C.; Ip, W.F.; Lo, K.H.; Liu, H.; Ng, K.W.; Wang, S.; et al. Recent Advances in Electrocatalysts for Efficient Nitrate Reduction to Ammonia. Adv. Funct. Mater. 2023, 33, 2303480. [Google Scholar] [CrossRef]
- Sathishkumar, N.; Chen, H.-T. Mechanistic Understanding of the Electrocatalytic Nitrate Reduction Activity of Double-Atom Catalysts. J. Phys. Chem. C 2023, 127, 994–1005. [Google Scholar] [CrossRef]
- Hu, T.; Wang, C.; Wang, M.; Li, C.M.; Guo, C. Theoretical Insights into Superior Nitrate Reduction to Ammonia Performance of Copper Catalysts. ACS Catal. 2021, 11, 14417–14427. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Liu, C.; Zheng, L.; Petit, E.; Qi, K.; Zhang, Y.; Wu, H.; Wang, W.; Tiberj, A.; et al. 3.4% Solar-to-Ammonia Efficiency from Nitrate Using Fe Single Atomic Catalyst Supported on MoS2 Nanosheets. Adv. Funct. Mater. 2022, 32, 2108316. [Google Scholar] [CrossRef]
- Niu, H.; Zhang, Z.; Wang, X.; Wan, X.; Shao, C.; Guo, Y. Theoretical Insights into the Mechanism of Selective Nitrate-to-Ammonia Electroreduction on Single-Atom Catalysts. Adv. Funct. Mater. 2021, 31, 2008533. [Google Scholar] [CrossRef]
- Chen, F.-Y.; Wu, Z.-Y.; Gupta, S.; Rivera, D.J.; Lambeets, S.V.; Pecaut, S.; Kim, J.Y.T.; Zhu, P.; Finfrock, Y.Z.; Meira, D.M.; et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 2022, 17, 759–767. [Google Scholar] [CrossRef]
- Xiang, T.; Liang, Y.; Zeng, Y.; Deng, J.; Yuan, J.; Xiong, W.; Song, B.; Zhou, C.; Yang, Y. Transition Metal Single-Atom Catalysts for the Electrocatalytic Nitrate Reduction: Mechanism, Synthesis, Characterization, Application, and Prospects. Small 2023, 19, 2303732. [Google Scholar] [CrossRef]
- Chen, G.F.; Yuan, Y.F.; Jiang, H.F.; Ren, S.Y.; Ding, L.X.; Ma, L.; Wu, T.P.; Lu, J.; Wang, H.H. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper-molecular solid catalyst. Nat. Energy 2020, 5, 605–613. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, S.; Wang, Y.; Wei, P.; Shao, J.; Liu, T.; Wang, G.; Bao, X. Enhancing Electrochemical Nitrate Reduction to Ammonia over Cu Nanosheets via Facet Tandem Catalysis. Angew. Chem.-Int. Edit. 2023, 62, e202303327. [Google Scholar] [CrossRef]
- Zhu, D.; Chen, M.; Huang, Y.; Li, R.; Huang, T.; Cao, J.-j.; Shen, Z.; Lee, S.C. FeCo alloy encased in nitrogen-doped carbon for efficient formaldehyde removal: Preparation, electronic structure, and d-band center tailoring. J. Hazard. Mater. 2022, 424, 127593. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, S.; Wu, Z.; Jiang, M.; Kuang, M.; Liu, Y.; Luo, W.; Zhang, D.; Yang, J. Relay Catalysis of Fe and Co with Multi-Active Sites for Specialized Division of Labor in Electrocatalytic Nitrate Reduction Reaction. Adv. Funct. Mater. 2024, 34, 2403838. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Hayat, A.; Bashir, T.; Ahmed, A.M.; Ajmal, Z.; Alghamdi, M.M.; El-Zahhar, A.A.; Sohail, M.; Amin, M.A.; Al-Hadeethi, Y.; Ghasali, E.; et al. Novel 2D MBenes-synthesis, structure, properties with excellent performance in energy conversion and storage: A review. Mater. Sci. Eng. R-Rep. 2024, 159, 100796. [Google Scholar] [CrossRef]
- Guo, S.; Ng, C.; Lu, J.; Liu, C.T. Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J. Appl. Phys. 2011, 109, 103505. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies. J. Chem. Phys. 2004, 23, 1841–1846. [Google Scholar] [CrossRef]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Nørskov, J.K.; Taylor, C.D.; Janik, M.J.; Neurock, M. Calculated Phase Diagrams for the Electrochemical Oxidation and Reduction of Water over Pt(111). J. Phys. Chem. B 2006, 110, 21833–21839. [Google Scholar] [CrossRef]
- Alberty, R.A. Standard Gibbs Free Energy, Enthalpy, and Entropy Changes as a Function of pH and pMg for Several Reactions Involving Adenosine Phosphates. J. Biol. Chem. 1969, 244, 3290–3302. [Google Scholar] [CrossRef]
- Montoya, J.H.; Tsai, C.; Vojvodic, A.; Nørskov, J.K. The Challenge of Electrochemical Ammonia Synthesis: A New Perspective on the Role of Nitrogen Scaling Relations. ChemSusChem 2015, 8, 2180–2186. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Chen, Z.; Jiang, Q. Hydrogenation-Facilitated Spontaneous N-O Cleavage Mechanism for Effectively Boosting Nitrate Reduction Reaction on Fe2B2 MBene. Molecules 2025, 30, 1778. https://doi.org/10.3390/molecules30081778
He Y, Chen Z, Jiang Q. Hydrogenation-Facilitated Spontaneous N-O Cleavage Mechanism for Effectively Boosting Nitrate Reduction Reaction on Fe2B2 MBene. Molecules. 2025; 30(8):1778. https://doi.org/10.3390/molecules30081778
Chicago/Turabian StyleHe, Yuexuan, Zhiwen Chen, and Qing Jiang. 2025. "Hydrogenation-Facilitated Spontaneous N-O Cleavage Mechanism for Effectively Boosting Nitrate Reduction Reaction on Fe2B2 MBene" Molecules 30, no. 8: 1778. https://doi.org/10.3390/molecules30081778
APA StyleHe, Y., Chen, Z., & Jiang, Q. (2025). Hydrogenation-Facilitated Spontaneous N-O Cleavage Mechanism for Effectively Boosting Nitrate Reduction Reaction on Fe2B2 MBene. Molecules, 30(8), 1778. https://doi.org/10.3390/molecules30081778