Results and Discussion
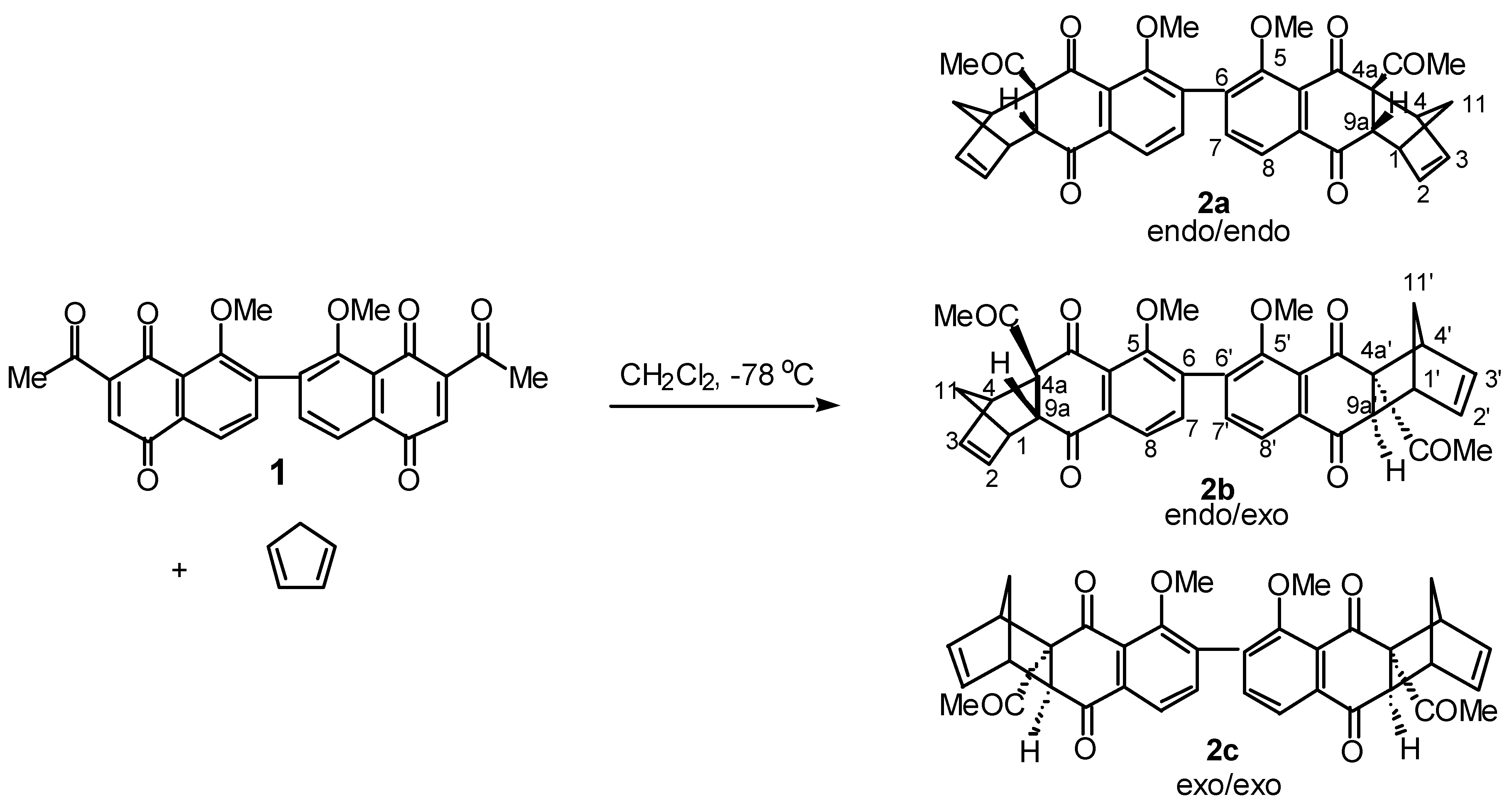
A study of the Diels-Alder addition of cyclopentadiene to bis-naphthoquinone was initiated. An excess of freshly distilled cyclopentadiene was added under nitrogen −78 °C to a dichloromethane so- lution of bis-naphthoquinone (
1) which was prepared as reported previously [
3]. After 10 min., no starting material remained and the formation of several new products of similar polarity was evident upon TLC analysis.
1H nmr analysis of the reaction mixture displayed features consistent with forma- tion of adduct (
2). The presence of signals resonating at δ 6.0-6.5 were assigned to the vinylic protons whilst a resonance at δ 4.13 was characteristic of the bridgehead proton assigned to H-9a.
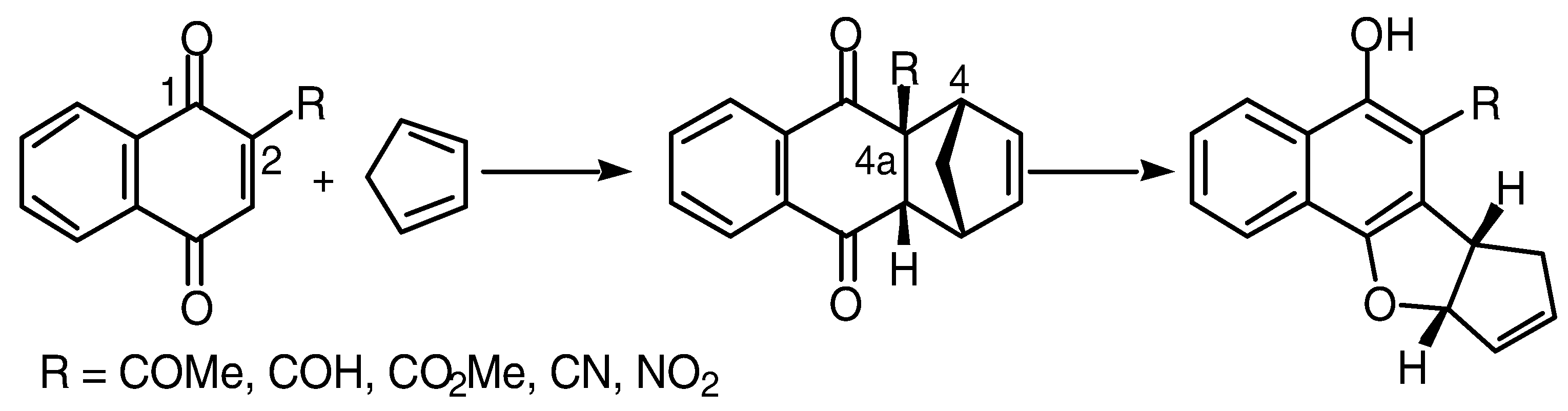
When using monomeric quinones as the dienophile exclusive formation of the
endo cycloaddition adducts was observed [
2]. However, the possibility of forming several diastereomeric adducts arises when bis-dienophile (
1) is used. In bis-naphthoquinone (
1) three modes of addition can potentially result,
endo-endo, endo-exo and
exo-exo, thus the possibility of diastereomeric adducts is not unex- pected. The presence of multiple vinylic, bridgehead and methoxy signals in the
1H nmr spectrum pro- vided evidence for a mixture of products.
Direct fragmentation of the product mixture using SnCl
4 in dichloromethane was then examined in order to simplify product analysis (
Scheme 3). After stirring for 10 min. at 0 °C several new products of lower polarity were evident upon TLC analysis, however, rapid decomposition of the crude reaction mixture in CDCl
3 occurred. The addition of the Lewis acid catalyst (SnCl
4) to bis-naphthoquinone (
1) prior to addition of cyclopentadiene afforded baseline material due to decomposition of bis- naphthoquinone (
1) by the Lewis acid.
Partial characterisation of the original Diels-Alder product mixture (5 mg) was accomplished using preparative HPLC affording four separate products in low yield (0.6-2 mg). Formation of the
endo- endo adduct (
2a) as the major product was confirmed upon spectroscopic analysis. High resolution mass spectrometry established the molecular formula C
36H
30O
8 whilst two bands in the IR spectrum at 1725 cm
−1 and 1678 cm
−1 were attributed to the carbonyls of the ketone and ene-dione respectively. The
1H nmr spectrum displayed two singlets at δ 2.42 and δ 3.50 assigned to the methyl groups of the acetyl and methoxy, whilst multiplets at δ 3.58-3.60 and δ 3.80-3.82 were assigned to H-1 and H-4 re- spectively. A two proton multiplet at δ 6.24-6.26 was assigned to the vinylic protons H-2 and H-3, whilst a doublet at δ 4.13 with coupling constant
J9a,1 3.9 Hz was assigned to the bridgehead proton H- 9a. The coupling constant
J9a,1 3.9 Hz is consistent with formation of the bis-
endo adduct (
2a) [
2].
A second product (1 mg) displayed features consistent with endo-exo adduct (2b) formation, most noticeably three vinylic multiplets were observed which integrated for one, two and one proton(s) re- spectively. The two proton multiplet at δ 6.25-6.27 was assigned to the endo ring vinylic protons H-2 and H-3. These protons resonated at a similar chemical shift to those reported for the analogous pro-tons in the endo-endo adduct (2a). The remaining one proton multiplets at δ 6.41-6.44 and δ 6.11-6.13 were thus assigned to the vinylic protons of the exo adduct portion of the molecule, H-2’ and H-3’. Two singlets at δ 2.30 and δ 2.42 were assigned to the acetyl group of the exo and endo rings respec- tively, whilst multiple aromatic signals provided further evidence for the unsymmetrical diastereomer (2b).
The two remaining products (<1.5 mg) exhibited complex 1H nmr spectra and the structures to date have not been elucidated. It is not unreasonable to assume that the formation of unsymmetrical dimers arising from reaction of only one half of bis-naphthoquinone (1) may well result in a complicated mixture. A poor mass recovery was obtained from the Diels-Alder addition of bis-naphthoquinone (1) to cyclopentadiene thereby precluding a synthetically useful approach to the bis-cyclopentannulated products (3).
Experimental
General
1H and 13C NMR spectra were obtained using a Bruker AM 400 NMR and were recorded at 400 and 10 MHz respectively. Liquid secondary ion mass spectrometry (LSIMS) high resolution mass spectra were recorded at a nominal resolution of 5000 using 4-nitrobenzyl alcohol (NBA) and a 5:1 mix (v/v) of dithiothreitol : dithioerythritol as matrix. High performance liquid chromatography (HPLC) was car- ried out using a Waters Associates system consisting of, a mdel M-6000A pump, a millipore model U6K injector, a model 440 ultra-violet detector at 256 nm and a R401 differential refractometer. Sepa- ration was carried out using the indicated solvents on a Partisil 10 M9 semipreparative column of the following dimensions: outer diameter 12.80 mm, inner diameter 9.40 mm, length 500.0 mm, and parti- cle size 10.0 μm. Cyclopentadiene was obtained from Aldrich Chemical Company (USA) and was cracked immediately prior to use.
Method
To a solution of 7-(2’-acetyl-8’-methoxy-1’,4’-dioxonaphthalen-7’-yl)-2-acetyl-8-methoxy-1,4- naphthoquinone (1, 25 mg, 0.055 mmol) in dichloromethane (2 ml) cooled to 0 °C under nitrogen was added freshly distilled cyclopentadiene (0.018 ml, 0.22 mmol). After stirring for 10 min., the reaction mixture was poured into aqueous sodium hydrogen carbonate solution (5 ml) and extracted with di- chloromethane (4 x 5 ml). The combined organic extracts were dried over magnesium sulfate and the solvent removed under reduced pressure to give a dark oil. Purification by flash chromatography using hexane-ethyl acetate (6:4) as eluent afforded a yellow oil which was further purified by HPLC on a Partisil 10 M9 semipreparative column using hexane-ethyl acetate (75:15) as eluent to afford: (1S,4R,4aR,9aR,1’S,4’R,4’aR,9’aR)-4a-Acetyl-6-[[4a’-acetyl-1’,4’,4a’,9a’-tetrahydro-1’,4’-methano- 5’-methoxy-9’,10’-dioxoanthracen-6-yl]-1,4,4a,9a-tetrahydro-1,4-methano-5-methoxy-9,10-anthra-cenedione 2a (1.6 mg, 5%) as a yellow oil and (1S,4R,4aR,9aR,1’R,4’S,4a’S,9a’S)-4a-Acetyl-6-[[4a’- acetyl-1’,4’,4a’,9a’-tetrahydro-1’,4’-methano-5’-methoxy-9’,10’-dioxoanthracen-6-yl]-1,4,4a,9a-tetra- hydro-1,4-methano-5-methoxy-9,10-anthracenedione 2b (1 mg, 3%) as a yellow oil.
Spectral Data for Adduct 2a
IR (CH2Cl2 solution) cm−1: 1725 (C=O, acetyl), 1678 (C=O, ene-dione), 1190, 1025.
1H NMR (400 MHz, CDCl3) δ: 1.36-1.39 (1H, m, 11-HB), 1.47 (obscured, 11-HA), 2.42 (3H, s, COMe), 3.50 (3H, s, OMe), 3.58-3.60 (1H, m, 1-H), 3.80-3.82 (1H, m, 4-H), 4.13 (1H, d, J9a,1 3.9 Hz, 9a-H), 6.24-6.26 (2H, m, 2-H and 3-H), 7.62 (1H, d, J7,8 8.0 Hz, 7-H), 7.72 (1H, d, J8,7 8.0 Hz, 8-H).
MS (LSIMS): 591 (MH+, 13%), 525 (M-C5H6, 20), 207 (45) and 176 (100).
HRMS (LSIMS) for C36H31O8 (MH+): Calcd 591.2019; Found 591.2044.
Spectral Data for Adduct 2b
IR (CH2Cl2 solution) cm−1: 1725 (C=O, acetyl), 1679 (C=O, ene-dione), 1193, 1024.
1H NMR (400 MHz, CDCl3) δ: 1.31-1.48 (4H, m, 11-HA, 11-HB, 11’-HA and 11’-HB), 2.30 (3H, s, 4a’-COCH3), 2.42 (3H, s, 4a-COCH3), 3.45 (1H, m, 1’-H), 3.51 (3H, s, OMe), 3.52 (3H, s, OMe), 3.59-3.61 (1H, m, 1-H), 3.67-3.69 (1H, m, 4’-H), 3.80-3.83 (1H, m, 4-H), 4.12-4.15 (2H, m, 9a-H and 9a’-H), 6.11-6.13 (1H, m, 2’-H or 3’-H), 6.25-6.27 (2H, m, 2-H and 3-H), 6.41-6.44 (1H, m, 3’-H or 2’-H), 7.62-7.66 (2H, m, 7-H and 7’-H), 7.72 (1H, d, Jortho 8.1 Hz, 8-H or 8’-H), 7.75 (1H, d, Jortho 8.0 Hz, 8’-H or 8-H).
MS (LSIMS): 591 (MH+, 13%), 525 (M-C5H6, 16), 207 (55) and 176 (100).
HRMS (LSIMS) for C36H31O8 (MH+): Calcd 591.2019; Found 591.2041.

{kind=link}
{kind=link}
{kind=link}


