Novel Coumarin Derivatives with Expected Biological Activity

Abstract
:Introduction
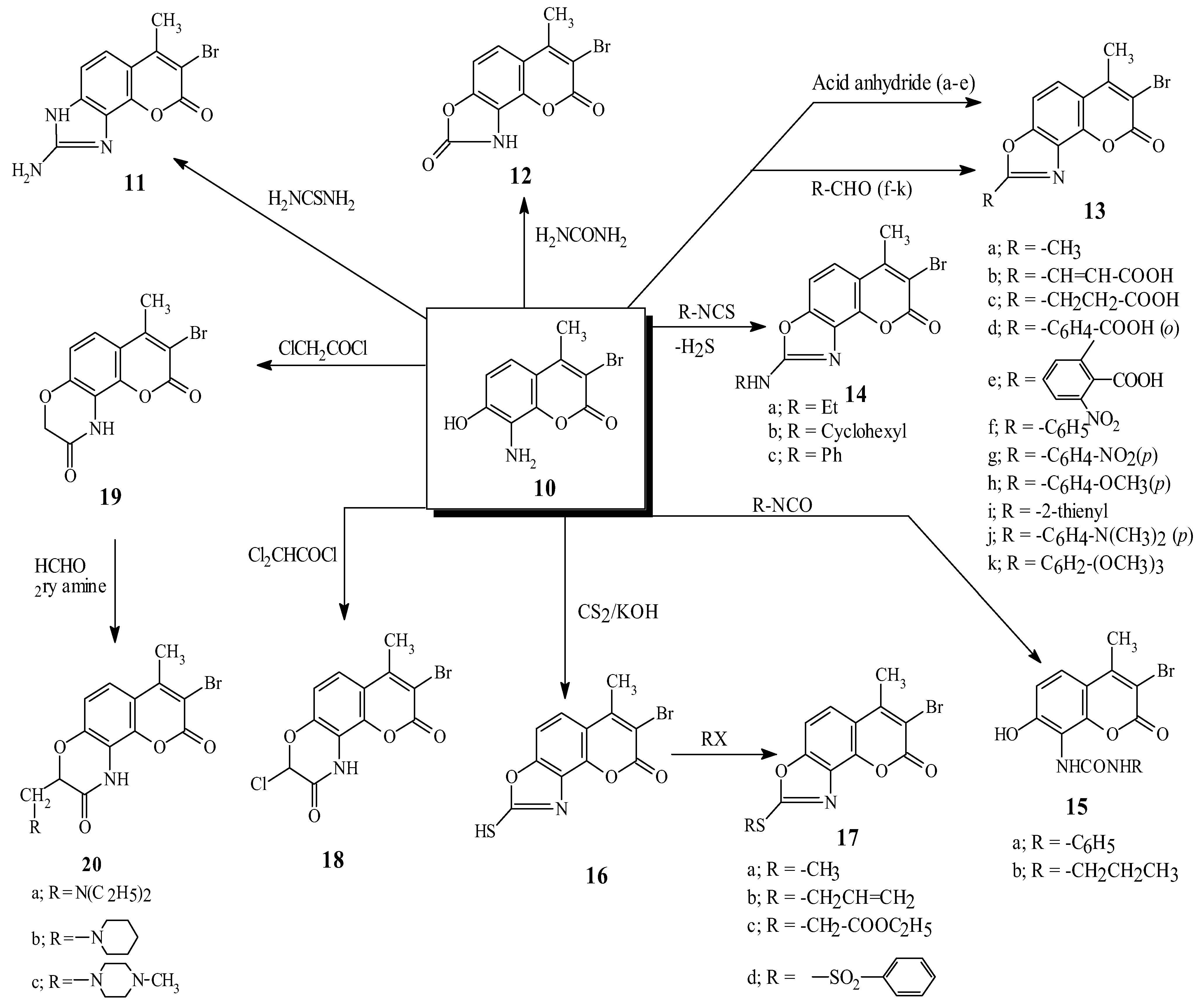
Results and Discussion
Biological Screening
Experimental
3-Bromo-7-hydroxy-4-methyl-8-nitrocoumarin (1a)
3-Bromo-7-methoxy-4-methyl-8-nitrocoumarin (2)
8-Amino-3-bromo-7-methoxy-4-methylcoumarin (3)
3-Bromo-8-chloroacetamido-7-methoxy-4-methylcoumarin (4)
3-Bromo-7-methoxy-4-methyl-8[substituted thiocarbonylmercaptoacetyl amino]coumarins (5a-d)
8-Acetylamino-3-bromo-7-methoxy-4-methylcoumarin (6)
8-Bromo-8-diacetylamino-7-methoxy-4-methylcoumarin (7)
3-(Benzylidinyl)amino-3-bromo-7-methoxy-4-methyl-coumarin (8)
N-Ethyl-N‵-[3-bromo-7-methoxy-4-methylcoumarin-8-yl]thiourea (9)
8-Amino-3-bromo-7-hydroxy-4-methylcoumarin (10)
2-Amino-7-bromo-6-methyl-8H-pyrano[2,3-e]benzimidazol- 8-one (11)
4-Bromo-6-methyl-8H-pyrano[2,3-e]benzoxazole-2,8-dione (12)
7-Bromo-6-methyl-2-substituted-8H-pyrano[2,3-e]benzoxazol-8-ones (13a-e)
7-Bromo-6-methyl-2-substituted-8H-pyrano[2,3-e]benzoxazol-8-one (13f-k)
2-Substituted amino-7-bromo-6-methyl-8H-pyrano[2,3-e]-benzoxazol-8-one (14a-c)
N-Substituted-N‵-[3-bromo-7-hydroxy-4-methylcoumarin-8-yl]urea (15a,b)
7-Bromo-6-methyl-8H-pyrano[2,3-e]benzoxazol-8-one-2-thione (16)
7-Bromo-6-methyl-2-thio substituted-8H-pyrano[2,3-e]-benzoxazol-8-ones (17a-d)
8-Bromo-3-chloro-7-methyl-9H-pyrano[2,3-e]benzo-1,4-oxazine-2,9-dione (18)
8-Bromo-7-methyl-9H-pyrano[2,3-e]benzo-1,4-oxazine-2,9-dione (19)
8-Bromo-7-methyl-2-substituted-9H-pyrano[2,3-e]benzo-1,4-oxazine-2,9-dione (20a-c)
References and Notes
- Soine, T.O. J. Pharm. Sci 1964, 53, 231.
- Badran, M.M.; Ismail, M.M.; El-Hakeem, M.A. Egypt. J. Pharm. Sci 1992, 33, 1081.
- El-Farargy, A.F. Egypt. J. Pharm. Sci 1991, 32, 625.
- Tunek, H. Monatsh. 1962, 93, 684. [Google Scholar]
- Nofal, Z.M.; El-Masry, A.H.; Fahmy, H.H.; Sarhan, A.I. Egypt. J. Pharm. Sci. 1997, 38, 1.
- Kitagawa, H.; Iwaki, R. Yakugaku Zasshi 1958, 78, 491, (Chem. Abstr. 1958, 52, 18874c-d.).
- Vogel, A.I. Practical Organic Chemistry; Longmans, Green and Co: London, 1976; p. 736. [Google Scholar]
- Nabih, I.; Michael, J.; Zoorob, H.; El-Zahar, M. Egypt. J. Chem. 1986, 26, 3.
- Propp, F. J. Med. Chem 1964, 7, 210.
- Schouberg, A.; Latif, N. J. Am. Chem. Soc. 1954, 76, 6208.
- Mannich, C.; Honig, P. Arch. Pharm. 1927, 265, 298.
- El-Merzabani, M.M.; El-Aaser, A.A.; Attia, M.A.; El-Duweini, A.; Ghazal, A.M. J. Planta Medica 1979, 36, 150.
- Lazarus, H.; Tegeler, M.; Mazzone, H.; Le Roy, J.; Boone, B.; Foley, G. Cancer Chemother-Rep. 1966, 50, 543. [PubMed]
- Samples Availability: available from MDPI.




{kind=link}
{kind=link}
{kind=link}
| Compound No. | % of dead tumour cells at 100μg/ml |
|---|---|
| 1 | 20% |
| 4 | All alive |
| Sa | 30% |
| 6 | All alive |
| 8 | 20% |
| 12 | 10% |
| 14a | All alive |
| 1Sa | 40% |
| 17a | All alive |
| 20b | All alive |
| Comp. No. | M.P.°C (Solvent of cryst.) | Yield % | Molecular formula (mol. mass) | Analysis % Calcd./Found | ||
|---|---|---|---|---|---|---|
| C | H | N | ||||
| 1a | 246 | 80 | C10H6BrNO5 | 40.02 | 2.01 | 4.66 |
| (Acetone) | (300.04) | 39.90 | 2.10 | 5.01 | ||
| 1b | 235 | 20 | C10H6BrNO5 | 40.02 | 2.01 | 4.66 |
| (Methanol) | (300.04) | 39.86 | 2.05 | 4.58 | ||
| 2 | 236 | 70 | C11H8BrNO5 | 42.05 | 2.56 | 4.45 |
| (Acetone) | (314.07) | 42.30 | 2.40 | 4.20 | ||
| 3 | 222-223 | 68 | C11H10BrNO3 | 46.50 | 3.54 | 4.92 |
| (Ethanol) | (284.09) | 46.85 | 3.92 | 5.21 | ||
| 4 | 276 | 85 | C13H11BrClNO4 | 43.30 | 3.07 | 3.88 |
| (Acetone) | (360.57) | 43.50 | 3.30 | 3.70 | ||
| 5a | 211 | 49 | C18H21BrN2O4S2 | 45.66 | 4.47 | 5.91 |
| (Ethanol) | (473.39) | 46.00 | 4.80 | 5.60 | ||
| 5b | 156-158 | 72 | C19H21BrN2O4S2 | 47.01 | 4.36 | 5.77 |
| (Ethanol) | (485.40) | 46.90 | 4.80 | 5.60 | ||
| 5c | 185-186 | 70 | C18H19BrN2O5S2 | 44.35 | 3.92 | 5.74 |
| (Ethanol) | (487.37) | 44.76 | 4.30 | 5.80 | ||
| 5d | 173-175 | 75 | C19H22BrN3O4S2 | 45.60 | 4.43 | 8.39 |
| (Ethanol) | (500.41) | 45.21 | 4.73 | 8.00 | ||
| 6 | 257-258 | 72 | C13H12BrNO4 | 47.87 | 3.71 | 4.29 |
| (Ethanol) | (326.13) | 48.20 | 3.90 | 4.60 | ||
| 7 | 178-179 | 92 | C15H14BrNO5 | 48.93 | 3.83 | 3.80 |
| (Ethanol) | (368.10) | 49.20 | 3.60 | 4.00 | ||
| Comp. No. | M.P.°C (Solvent of cryst.) | Yield % | Molecular formula (mol. mass) | Analysis % Calcd./Found | ||
|---|---|---|---|---|---|---|
| C | H | N | ||||
| 8 | 144-146 | 77 | C18H14BrNO3 | 58.08 | 3.79 | 3.76 |
| (Ethanol) | (372.19) | 57.73 | 3.40 | 3.30 | ||
| 9 | 101-102 | 63 | C14H15BrN2O3S | 45.29 | 4.07 | 7.54 |
| (EtOH/H2O) | (371.24) | 45.30 | 4.10 | 7.30 | ||
| 10 | 260-dec. | 60 | C10H8BrNO3 | 44.47 | 2.98 | 5.18 |
| (MeOH/H2O) | (270.06) | 44.50 | 3.10 | 5.30 | ||
| 11 | 238-dec. | 60 | C11H8BrN3O2 | 44.92 | 2.74 | 14.28 |
| (ethanol) | (294.09) | 44.80 | 2.70 | 14.20 | ||
| 12 | 290 | 55 | C11H6BrNO4 | 44.62 | 2.04 | 4.72 |
| (ethanol) | (296.05) | 44.50 | 2.30 | 4.90 | ||
| 13a | >300 | 30 | C12H8BrNO3 | 49.00 | 2.74 | 4.76 |
| (DMF/H2O) | (294.08) | 48.72 | 3.11 | 4.39 | ||
| 13b | 233 | 52 | C14H8BrNO5 | 48.02 | 2.30 | 3.99 |
| (DMF/H2O) | (350.10) | 47.70 | 2.60 | 4.38 | ||
| 13c | 230 | 45 | C14H10BrNO5 | 47.75 | 2.86 | 3.97 |
| (DMF/H2O) | (352.12) | 47.52 | 2.40 | 3.62 | ||
| 13d | 284 | 75 | C18H10BrNO5 | 54.02 | 2.51 | 3.49 |
| (DMF/H2O) | (400.16) | 54.40 | 2.50 | 3.90 | ||
| 13e | >300 | 60 | C18H9BrN2O7 | 48.56 | 2.03 | 6.29 |
| (DMF/H2O) | (445.17) | 48.84 | 2.41 | 6.70 | ||
| 13f | 204-206 | 48 | C17H10BrNO3 | 57.32 | 2.83 | 3.93 |
| (EtOAc/ | (356.15) | 57.20 | 3.02 | 3.50 | ||
| pet. ether) | ||||||
| 13g | 216 | 86 | C17H9BrN2O5 | 50.89 | 2.26 | 6.98 |
| (EtOAc/ | (401.16) | 50.62 | 2.43 | 6.72 | ||
| pet. ether) | ||||||
| 13h | 240 | 30 | C18H12BrNO4 | 55.97 | 3.13 | 3.62 |
| (EtOAc/ | (386.18) | 55.63 | 3.52 | 3.40 | ||
| pet. ether) | ||||||
| 13i | 217 | 35 | C15H8BrNO3S | 49.74 | 2.22 | 3.86 |
| (EtOAc/ | (362.17) | 49.31 | 2.27 | 3.60 | ||
| pet. ether) | ||||||
| 13j | 223-224 | 70 | C19H15BrN2O3 | 57.15 | 3.78 | 7.01 |
| (EtOAc/ | (399.23) | 56.82 | 3.60 | 6.76 | ||
| pet. ether) | ||||||
| 13k | 201 | 55 | C20H16BrNO6 | 53.82 | 3.61 | 3.13 |
| (EtOAc/ | (446.23) | 53.51 | 3.50 | 3.52 | ||
| pet. ether) | ||||||
| 14a | 273-275 | 86 | C13H11BrN2O3 | 48.31 | 3.43 | 8.67 |
| (benzene) | (323.14) | 48.12 | 3.26 | 8.40 | ||
| 14b | 267 | 87 | C17H17BrN2O3 | 54.12 | 4.54 | 7.42 |
| (benzene) | (377.23) | 54.19 | 4.96 | 7.17 | ||
| 14c | 190 | 71 | C17H11BrN2O3 | 55.00 | 2.98 | 7.54 |
| (benzene) | (371.18) | 54.63 | 2.52 | 7.82 | ||
| 15a | 206-dec | 53 | C17H13BrN2O4 | 52.46 | 3.36 | 7.19 |
| (benzene) | (389.19) | 52.10 | 3.00 | 7.49 | ||
| 15b | 196 | 60 | C14H15BrN2O4 | 47.33 | 4.25 | 7.88 |
| (benzene/pet | (355.18) | 47.50 | 4.45 | 8.13 | ||
| ether) | ||||||
| 16 | 281-2 | 85 | C11H6BrNO3S | 42.32 | 1.93 | 4.48 |
| (ethanol) | (312.11) | 41.90 | 2.00 | 4.10 | ||
| 17a | 200 | 87 | C12H8BrNO3S | 44.18 | 2.47 | 4.29 |
| (ethanol) | (326.14) | 44.50 | 2.80 | 4.30 | ||
| 17b | 134-136 | 55 | C14H10BrNO3S | 47.74 | 2.86 | 3.97 |
| (ethanol-ether) | (352.18) | 47.60 | 3.10 | 4.00 | ||
| 17c | 175-177 | 40 | C15H12BrNO5S | 45.23 | 3.03 | 3.51 |
| (ethanol-ether) | (398.21) | 45.60 | 3.00 | 3.90 | ||
| 17d | 204-dec | 57 | C17H10BrNO5S2 | 45.14 | 2.22 | 3.09 |
| (ethanol) | (452.27) | 45.50 | 2.00 | 3.50 | ||
| 18 | 254 | 95 | C12H7BrClNO4 | 41.83 | 2.04 | 4.06 |
| (ethanol) | (344.52) | 41.80 | 2.40 | 4.00 | ||
| 19 | 275 | 88 | C12H8BrNO4 | 46.47 | 2.59 | 4.51 |
| (ethanol) | (310.08) | 46.10 | 3.00 | 4.88 | ||
| 20a | 193 | 44 | C17H19BrN2O4 | 51.65 | 4.84 | 7.08 |
| (EtOH/pet. | (395.24) | 51.91 | 4.61 | 7.30 | ||
| ether) | ||||||
| 20b | 182 | 46 | C18H19BrN2O4 | 53.08 | 4.70 | 6.88 |
| (ethanol) | (407.25) | 53.27 | 4.54 | 7.20 | ||
| 20c | 225-226 | 49 | C18H20BrN3O4 | 51.19 | 4.77 | 9.95 |
| (ethanol) | (422.26) | 50.83 | 4.52 | 9.80 | ||
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Nofal, Z.M.; El-Zahar, M.I.; Abd El-Karim, S.S. Novel Coumarin Derivatives with Expected Biological Activity. Molecules 2000, 5, 99-113. https://doi.org/10.3390/50200099
Nofal ZM, El-Zahar MI, Abd El-Karim SS. Novel Coumarin Derivatives with Expected Biological Activity. Molecules. 2000; 5(2):99-113. https://doi.org/10.3390/50200099
Chicago/Turabian StyleNofal, Z. M., M. I. El-Zahar, and S. S. Abd El-Karim. 2000. "Novel Coumarin Derivatives with Expected Biological Activity" Molecules 5, no. 2: 99-113. https://doi.org/10.3390/50200099
APA StyleNofal, Z. M., El-Zahar, M. I., & Abd El-Karim, S. S. (2000). Novel Coumarin Derivatives with Expected Biological Activity. Molecules, 5(2), 99-113. https://doi.org/10.3390/50200099