Experimental
General
Melting points were determined in open capillary tubes in a Buchi 510 circulating oil apparatus and are uncorrected. Infrared spectra were recorded on a Perkin Elmer 781 spectrophotometer.1H-NMR spectra were recorded at room temperature for CDCl3 solutions, (unless otherwise stated) on a Jeol JNM-EX 90A FT NMR and Bruker Avance DPX 250 MHz spectrometers using tetramethylsilane as an internal standard. All chemical shifts are reported as δ values (ppm). Mass spectra were obtained on a GCMS-QP 1000 EX mass spectrometer (70 eV). The microorganism strains used in this study were β-lactam sensitive ones.
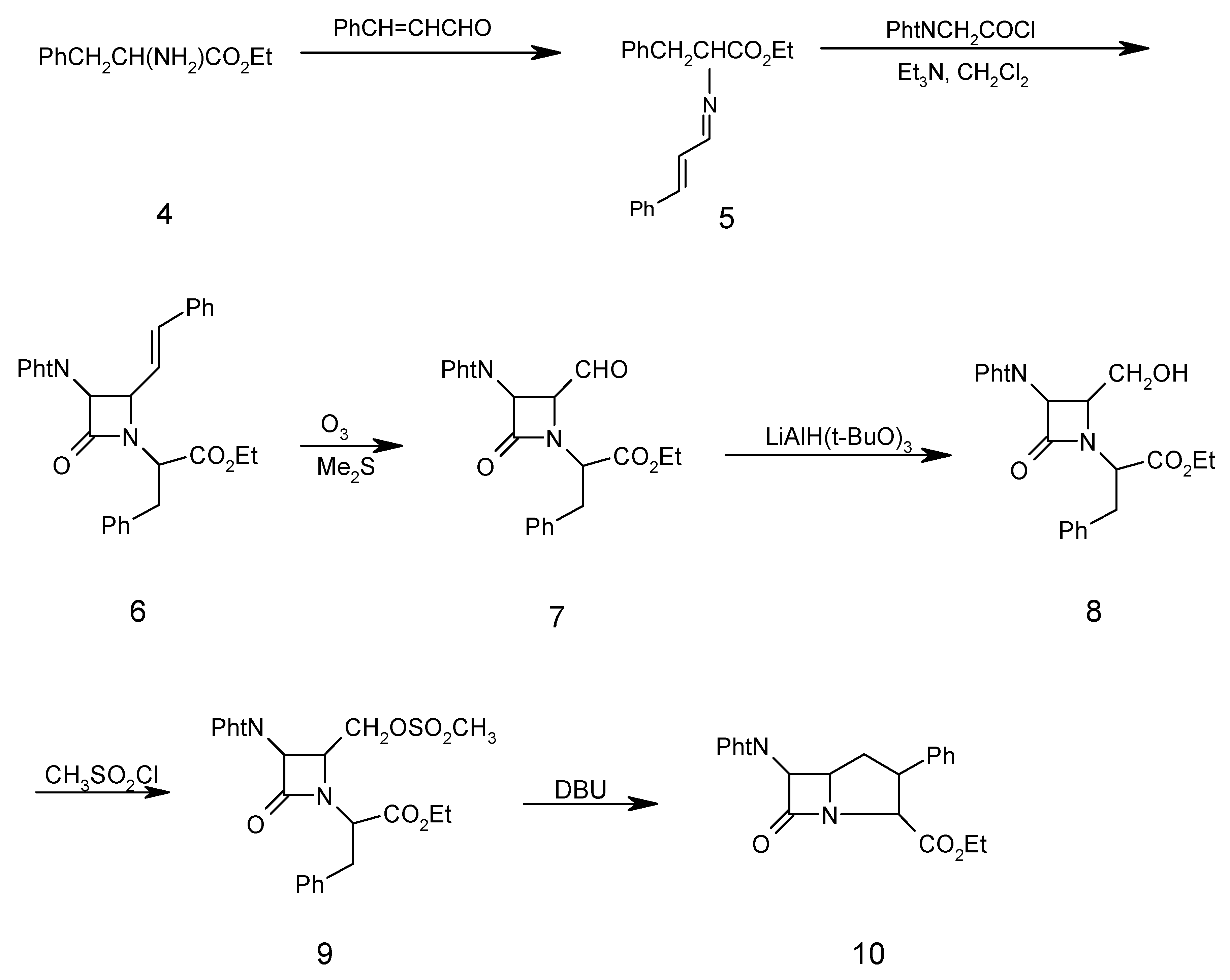
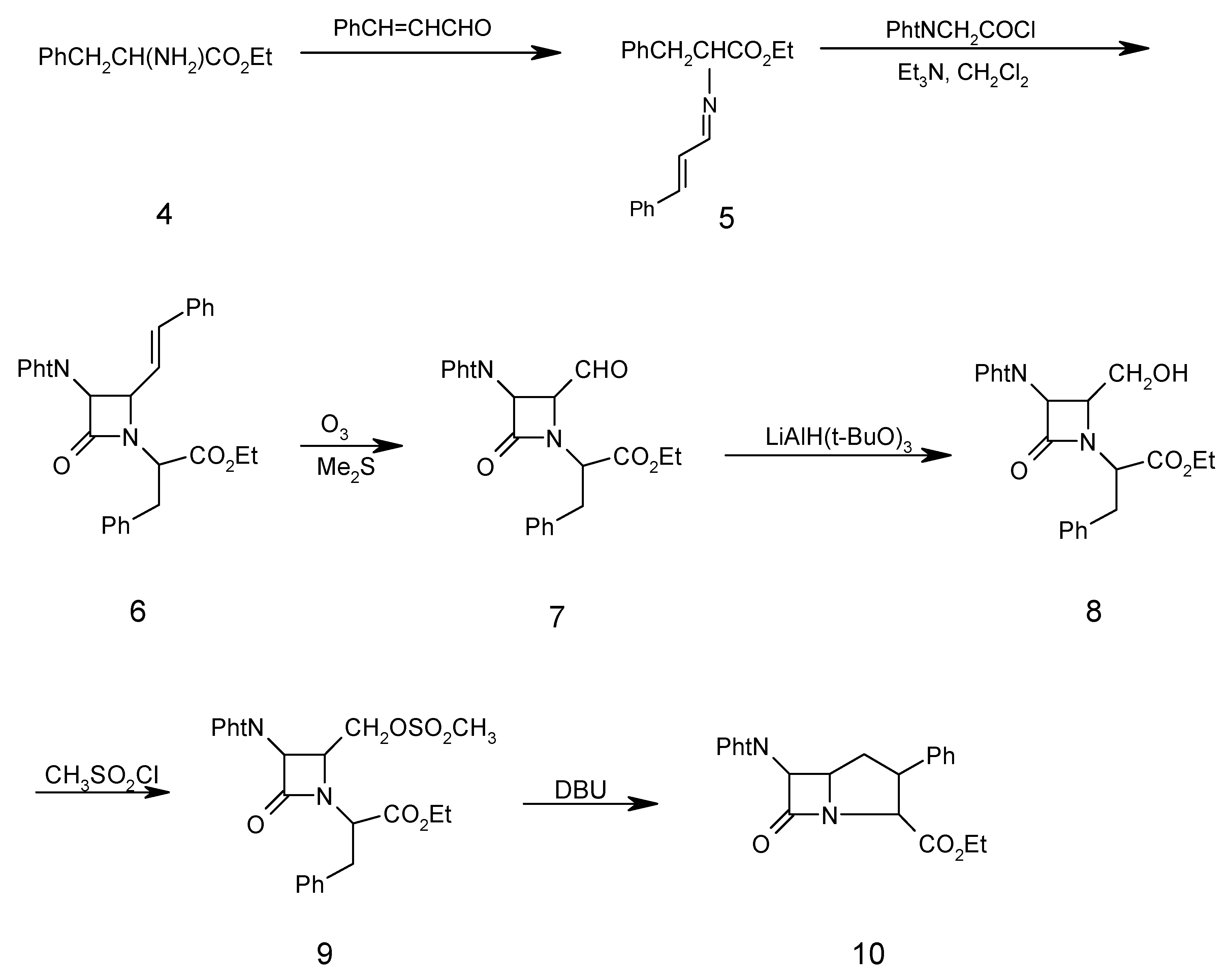
Ethyl α-amino-(N-cinnamylidene)-ß-phenyl propionate (5)
A mixture of cinnamaldehyde (1.50 ml, 10.00 mmol), D-phenylalanine ethyl ester (4, 2.00 g, 10.00 mmol), triethylamine (1.01 g, 10.00 mmol), and Na2SO4 (10.00g, 72.40 mmol) in dry methylene chloride (40.00 mL) was refluxed for 5 hours. The resulting mixture was filtered and washed with H2O (3 x 30 mL). The organic layer was separated and dried (Na2SO4). Evaporation of the solvent under reduced pressure gave the chiral Schiff base 5 as a solid which was recrystallized from ethanol (2.50 g, 81 %); m.p.130-132°C; 1H-NMR: 1.30 (3H, t, CH3), 3.45 (2H, d, PhCH2), 4.34 (2H, q, OCH2CH3), 4.40 (1H, t, CHCO2Et), 5.85 (1H, d, J=15Hz, PhCH=CH), 6.45 (1H, dd, J=15Hz, PHCH=CHCH), 7.20-7.40 (10H, m, 2Ph), 8.30 (CH=N); IR (KBr) (cm-1): 1640 (C=N), 1740 (COOEt); MS (m/z): 307 (M+), 235 (M- COOEt), 193 (M- PhCH=CHCH=N), 104 (PhCH2CH), 91 (PhCH2), 77 (Ph); Anal. Calc. for C20H21NO2: C, 78.15; H, 6.98; N, 4.56%; found: C, 78.20; H, 6.80; N, 4.60%
N-(α-Carboethoxy-β-phenylethyl)-3-phthalimido-4-styryl-2-azetidinone (6)
To a solution of chiral Schiff base 5 (3.07 g, 10.00 mmol) and triethylamine (1.01 g, 10.00 mmol) in dry methylene chloride (70 mL) at –10oC was added phthalimidoacetyl chloride (3.00 g, 15.00 mmol) in dry methylene chloride (30.00 mL) dropwise. After the addition was complete, the solution was stirred at room temperature for 24 hours, then the reaction mixture was washed with water (3 x 30 mL). The organic layer was separated and dried (Na2SO4), filtered and the solvent was evaporated under reduced pressure to give the β-lactam 6 as a solid that was recrystallized (from ethanol) in 70% yield; m.p. 152-154oC; 1H-NMR: 1.30 (3H, t, CH3), 3.45 (2H, d, PhCH2) 4.34 (2H, q, OCH2CH3), 4.40 (1H, t, CHCO2Et), 4.48 (1H, dd, J=5.5 Hz, CHCH=CH), 4.60 (1H, s, CHN(CO)2), 5.85 (1H, dd, PhCH=CH, J=16 Hz), 6.4 (1H, d, J=12 Hx, PhCH=CH), 7.20 (5H, m, PhCH2), 7.40 (5H, m, PhCH=CH), 7.76 (4H, m, C6H4(CO)2N), IR (KBr) (cm-1), 1780 (β-lactam CO), 1740 (CO2Et), 1730, 1770 (phthalimido CO); MS (m/z): 494 (M+), 421 (M+-CO2Et), 308 (M+-COCHPhtN), 234 (M+-CO2Et-COCHC6H4(CO)2N), 146 (C6H4(CO)2N), 104 (PhCH2CH), 91 (PhCH2); Anal. Calc. for C30H26N2O5: C, 72.86; H, 5.30; N, 5.66%; found: C, 72.80; H, 5.20; N, 5.70%
N-(α-Carboethoxy-β-phenylethyl)-3-phthalimido-4-formyl-2-azetidinone (7)
β-Lactam 6 (2.48 g, 5.00 mmol) in dry CH2Cl2 (50 mL) was saturated with N2 at –78oC, then a mixture of O3/N2 was bubbled in until KI-starch paper showed excess ozone (15 min), which was then removed by passing a stream of N2 for 10 min. Dimethyl sulfide (0.75 g, 12.00 mmol) was added and the temperature of the solution was allowed to rise to 25o over 1h. The reaction mixture was washed with H2O (3 x 40 mL) and with saturated brine (2 x 30 mL). The organic layer was separated and dried (Na2SO4). Evaporation of the solvent gave a syrup. The oily residue was heated under high vacuum for 20 hours at 50oC to remove benzaldehyde. The product 7 was obtained as a brownish oil (1.89 g, 90%); 1H-NMR: 1.30 (3H, t, CH3), 3.45 (2H, d, PhCH2), 4.34 (2H, q, OCH2CH3), 4.5 (1H, t, CHCO2Et), 4.75 (1H, dd, J=5.20, CHCHO), 5.30 (1H, d, J=5.30, CHN(CO)2), 7.25 (5H, m, PhCH2), 7.80 (4H, m, C6H4(CO)2N), 9.62 (1H, d, CHO); IR (CDCl3) (cm-1): 1790 (β-lactam CO), 1730, 1770 (phthalimido CO), 1760 (CO2Et), 1740 (CHO); MS (m/z): 420 (M+), 39 (M+-CHO), 105 (PhCH2CH2), 77 (Ph). Anal. Calc. for C23H20N2O6: C, 65.71; H, 4.79; N, 6.66%; found: C, 65.80; H, 4.69; N, 6.56%
N-(α-Carboethoxy-β-phenylethyl)-3-phthalimido-4-hydroxymethyl-2-azetidinone (8)
To aldehyde 7 (1.50 g, 3.50 mmol) in tetrahydrofuran (80 mL) at 0oC was added lithium tri(tert-butoxy)aluminium hydride (1.78 g, 7.00 mmol). After stirring under nitrogen for 3h at 0oC, the mixture was acidified with dilute hydrochloric acid (2%) to pH 5 and silica gel (1.00 g) was added. The suspension was stirred for 10 minutes, filtered and the solvent was evaporated. Ethyl acetate (60 mL) was added to the residue and the organic phase was washed with brine (3 x 30 mL), dried over Na2SO4 and the solvent was evaporated. The crude alcohol 8 was chromatographed on silica gel. Elution with chloroform-ethyl acetate (9:1) afforded 8 as an oil in 60% yield; 1H-NMR: 1.30 (3H, t, CH3), 3.45 (2H, d, PhCH2), 3.75 (2H, d, CH2OH), 4.34 (2H, q, CH2CH3), 4.40 (1H, t, CHCO2Et), 4.65 (1H, br, OH), 4.75 (1H, m, CHCH2OH), 5.30 (1H, d, J=5.20, CHCHCH2OH), 7.25 (5H, m, PhCH2), 7.80 (4H, m, C6H4(CO)2N); IR (CH2Cl2) (cm-1): 3300-3490 (OH), 1780 (β-lactam CO), 1765, 1720 (phthalimido CO), 1740 (CO2Et); MS (m/z) 422(M+), 421 (M+-1), 407 (M+-CH3), 236 (M+-COCHC6H4(CO)2N), 187 (COCHC6H4(CO)2N), 104 (PhCH2CH), 91(PhCH2), 77 (Ph); Anal. Calc. for C23H22N2O6: C, 65.40; H, 5.25; N, 6.63%; found: C, 65.38; H, 5.30; N, 6.70%.
N-(α-Carboethoxy-β-phenylethyl)-3-phthalimido-4-methansulfonyloxymethyl-2-azetidinone (9)
Methansulfonyl chloride (0.27 g, 2.35 mmol) in CH2Cl2 (3 mL) was added dropwise at –78oC to alcohol 8 (1.00 g, 2.35 mmol) and triethylamine (0.47 g, 4.7 mmol) in dry CH2Cl2 (25 mL). After the addition was complete, the mixture was washed with pH 4.40 buffer (0.20 M, NaH2PO4 solution) and water (2 x 30 mL), dried over Na2SO4 and the solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel. Elution with chloroform gave 9 as an oil in 60% yield; 1H-NMR: 1.30 (3H, t, CH3), 2.85 (3H, s, OSO2CH3), 3.45 (2H, d, PhCH2), 3.96 (1H, m , CHCH2OSO2CH3), 4.34 (2H, q, CH2CH3), 4.40 (1H, t, CHCO2Et), 4.80 (1H, d, J=5.2 Hz, CHCHN(CO)2), 7.25 (5H, m, PhCH2), 7.80 (4H, m, C6H4(CO)2N), IR (CH2Cl2)(cm-1): 1780 (β-lactam CO), 1765, 1725 (phthalimido CO), 1740 (CO2Et), 1380, 1180 (SO2); MS (m/z): 500 (M+), 485 (M+-CH3), 314 (M+-COCHC6H4(CO)2N), 187 (COCHC6H4(CO)2N), 95 (SO3CH3); Anal. Calc. for C24H24N2O8S: C, 57.59; H, 4.83; N, 5.60; S, 6.41%; found: C, 57.65; H, 4.90; N, 5.50; S, 6.50%
6-Phthalimido-7-oxo-3-phenyl-1-azabicyclo [3.2.0]heptane-2-carboxylic acid ethyl ester (10)
1,8-Diazabicyclo [5.4.0]undec-7-ene (1.53 g, 10.00 mmol) was added to mesylate β-lactam 9 (2.02 g, 5.00 mmol) in THF-DMF 9:1 (20 mL). The mixture was refluxed for four hours and then the solvent was evaporated. Ethyl acetate (30 mL) was added to the residue and washed with water (2 x 30 mL), dried over Na2SO4 and the sovlent was evaporated under reduced pressured to give the crude product which was chromatographed on silica gel. Elution with 1:1 n-hexane-dichloromethane afforded yellow crystals of 10 in 20% yield, m.p. 97oC; 1H-NMR: 1.30 (3H, t, CH3), 2.20 (2H, m, CHCH2CH), 3.85-3.90 (2H, m, CHCH2CH), 4.30 (2H, q, CH2CH3), 4.60 (1H, d, J=16 Hz, NCHCO2Et), 4.95 (1H, d, J=5.2 Hz, CH(CO)2N), 7.10 (5H, m, Ph), 7.50-7.80 (4H, m, C6H4(CO)2N); IR (KBr) (cm-1): 1780 (β-lactam CO), 1755, 1735 (phthalimido CO), 1720 (CO2Et); MS(m/z): 404 (M+), 389 (M+-CH3), 217 (M+-COCHC6H4(CO)2N), 187 (COCHC6H4(CO)2N), 77 (Ph); Anal. Calc. for C23H20N2O5: C, 68.31; H, 4.98; N, 6.93;%; found; C, 68.40; H, 5.05; N, 6.90%
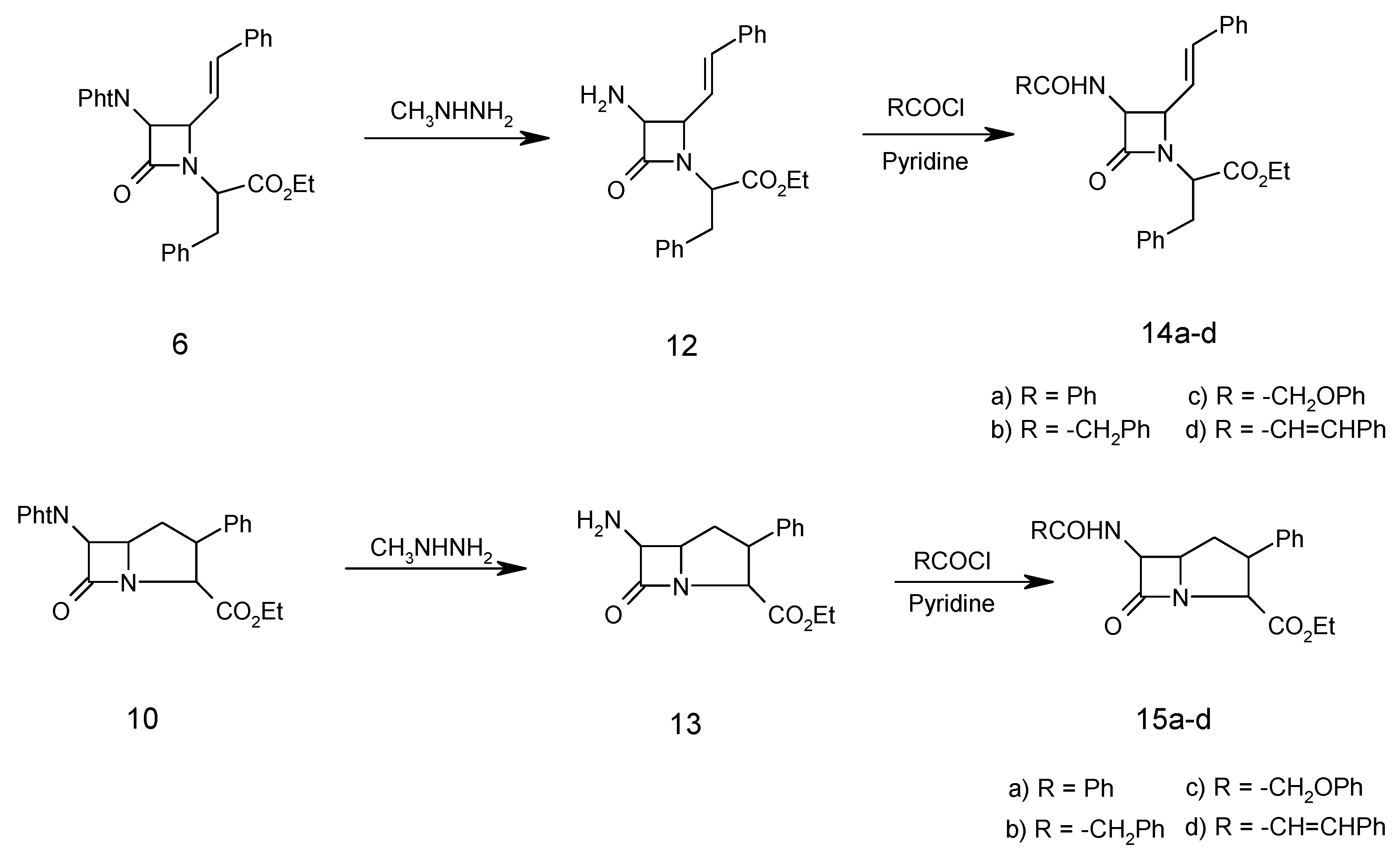
General procedure for dephthaloylation of protected β-lactam 6
The monocyclic β-lactam 6 (4.94 g, 10.00 mmol) was dissolved in ethanol (25 mL) and methylhydrazine (0.046 g, 10.00 mmol) was added. After refluxing for 2 hours, the reaction mixture was stored overnight and then concentrated to dryness under vacuum. The residue (methylphthalhydrazide) was stirred for 2 hours with 5N HCl (25 mL) and filtered. The aqueous and acid extracts were combined and concentrated hydrochloric acid (3 mL) was added. After 2 hours, the aqueous solution was evaporated to give the β-lactam N-(α-carboethoxy-β-phenylethyl)-3-amino-4-styryl-2-azetidinone (12) as a hydrochloride salt (3.09g, 85%); m.p. of the free amino β-lactam 110°C; 1H-NMR: 1.30 (3H, t, CH3), 3.45 (2H, d, PhCH2) 4.34 (2H, q, OCH2CH3), 4.40 (1H, t, CHCO2Et), 4.48 (1H, dd, J=5.5 Hz, CHCH=CH), 4.60 (1H, m, CHNH2), 5.85 (1H, dd, PhCH=CH, J=16 Hz), 6.4 (1H, d, J=12 Hx, PhCH=CH), 7.20(5H, m, PhCH2), 7.40(5H, m, PhCH=CH); IR (KBr) (cm-1): 3300-3400 (H2N), 1780 (β-lactam CO), 1740 (ester CO); MS (m/z): 364 (M+), 348 (M+-NH2), 307 (M+-COCHNH2), 291 (M+-CO2Et), 104 (PhCH2CH), 91 (PhCH2);Anal. Calc. for C22H24N2O3: C, 72.51; H, 6.64; N, 7.69%; found: C, 72.45; H, 6.74; N, 7.75%.
General procedure for acylation of free amino β-lactam 12.
Pyridine (1.50 g, 18.96 mmol) was added to a solution of 12 (1.09 g, 3.00 mmol) followed by dropwise addition of phenylacetyl chloride (0.93 g, 6.00 mmol) in CH2Cl2 (20 mL). The solution was stirred for 2h. at 25°C, then it was washed successively with 10% HCl, 10% NaHCO3 and water, dried (MgSO4), filtered and the solvent was then evaporated to give the impure amide which was recrystallized from ethanol to give N-(α-carboethoxy-β-phenylethyl)-3-phenylacetylamino-4-styryl-2-azetidinone (14b, 3.86g, 80%); m.p. 95°C; 1H-NMR: 1.30 (3H, t, CH3), 3.45 (2H, d, PhCH2), 3.50 (2H, s, PhCH2CO), 4.34 (2H, q, OCH2CH3), 4.40 (1H, t, CHCO2Et), 4.48 (1H, dd, J=5.5 Hz, CHCH=CH), 4.60 (1H, s, CHNHCO), 5.85 (1H, dd, PhCH=CH, J=16 Hz), 6.4 (1H, d, J=12 Hx, PhCH=CH), 7.20-7.40 ( 15H, m, 3Ph); IR (cm –1): 3400-3450 (NH), 1775 (β-lactam CO), 1740 (ester CO), 1685 (amide CO); MS (m/z): 482 (M+), 409 (M+-CO2Et), 348 (M+-PhCH2CONH), 307 (M+-PhCH2CONHCHCO), 91 (PhCH2); Anal. Calc. for C30H30N2O4: C, 74.67; H, 6.27; N, 5.80%; found: C, 74.75; H, 6.20; N, 5.90%.
Compounds 14a , 14c, 14d and 15a-d were treated identically and their spectra were similar, except for the expected variations due to the amide side chains.





{kind=link}
{kind=link}