Abstract
The long-held “shielding cone” model of the through-space NMR shielding effect of a carbon-carbon double bond predicts only the effect of the magnetic anisotropy of the double bond; it ignores other important contributors to the overall shielding. GIAO-SCF and GIAO-MP2 calculations have been performed on a simple model system, methane moved sequentially above ethene or 2-methylpropene. These calculations permit the net NMR shielding surface to be mapped. Based on those results, a new and very different graphical model for predicting the effect of a proton’s position relative to a carbon-carbon double bond on its chemical shift is presented.
Introduction
The long-held “shielding cone” model used for predicting the through-space magnetic influence of the magnetic anisotropy of a carbon-carbon double bond on the chemical shift of a nearby proton, found in most textbooks on NMR spectroscopy [1], is based on the McConnell equation [2]. It was designed to predict the long-range shielding effect of the magnetic anisotropy of the carbon-carbon double bond. This model has been used improperly in many textbooks to predict the effect on a chemical shift of a proton due to its proximity and position relative to the double bond. Recently this model has been called into question as the result of ab initio SCF calculations [3,4,5,6,7,8] and experimental observation [9] of protons over carbon-carbon double bonds which report deshielding rather than shielding for protons above and within 3 Å of the plane of a carbon-carbon double bond. Substantial(as much as 2 ppm) deshielding is observed in some systems for protons within this region.
Methods and Calculations
We have used the GIAO (gauge-including atomic orbital)-SCF method [10,11,12] within Gaussian 98 [13] to map the proton NMR shielding surface of a simple model system: methane moved sequentially over ethene. From the methane proton shielding data we have derived an empirical mathematical function to predict the NMR shielding increment (Δσ) of a proton as a function of Cartesian coordinates relative to the center of a carbon-carbon double bond [8]. The computed shielding increment is defined as the difference in isotropic shielding value of a proton in methane oriented over a double bond compared to that of a proton of methane by itself. This ab initio approach includes a variety of effects influencing the chemical shift, including the magnetic anisotropy of nearby bonds, electric field effects, orbital interactions, and dispersion, to the extent these are modeled at the HF/6-31G(d,p) level. In this paper we report the results of a series of calculations on this simple system performed at the MP2/6-31G(d,p) level. Because the MP2 method includes electron correlation effects and models dispersion effects better than HF methods, MP2 calculations should provide a more accurate description of the net NMR shielding [14]. The observed chemical shift difference (Δδ) is the difference in observed chemical shift between a proton oriented over a double bond (e.g., protons labeled a, Figure 1) and a proton not over a double bond, but otherwise similarly situated (e.g., protons labeled b, Figure 1) [15]. For convenience of comparison, the differences are defined such that a proton deshielded (negative Δσ) by a double bond also has a negative value of Δδ. The most recent shielding model [8] provides predictions of (de)shielding increments which agree quite well with experimental chemical shift differences (Table 1) in several diverse structures.
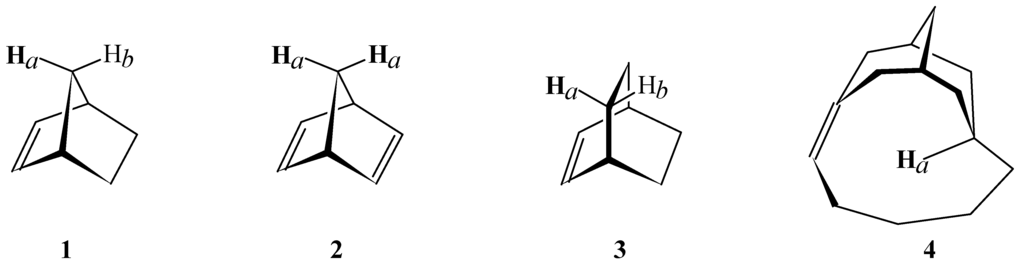
Figure 1.
Structures having a proton over a carbon-carbon double bond.
Figure 1.
Structures having a proton over a carbon-carbon double bond.
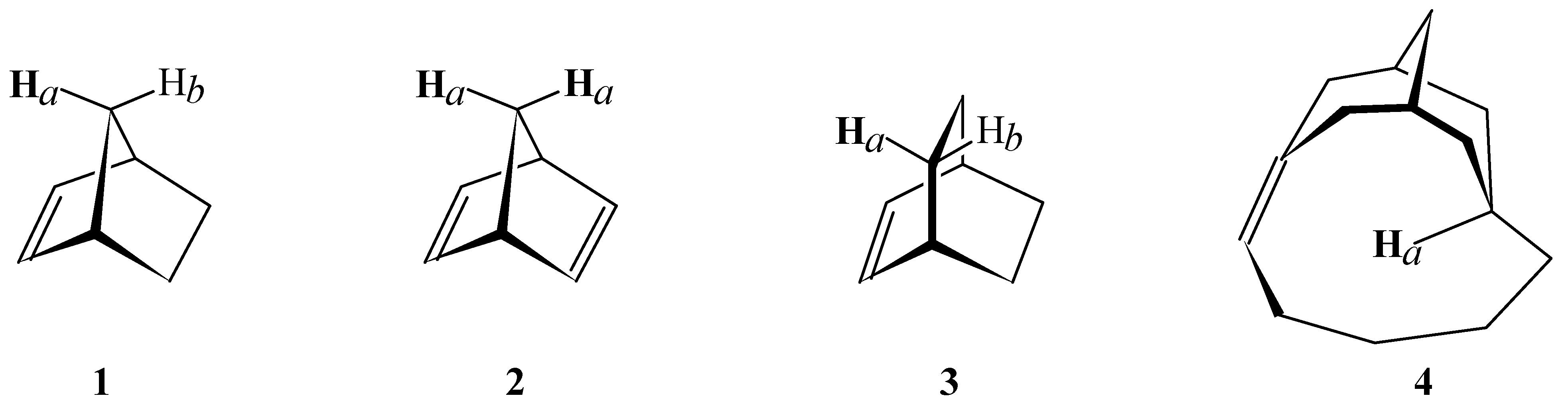
Figure 2.
Reference structure for structure 4.
Figure 2.
Reference structure for structure 4.
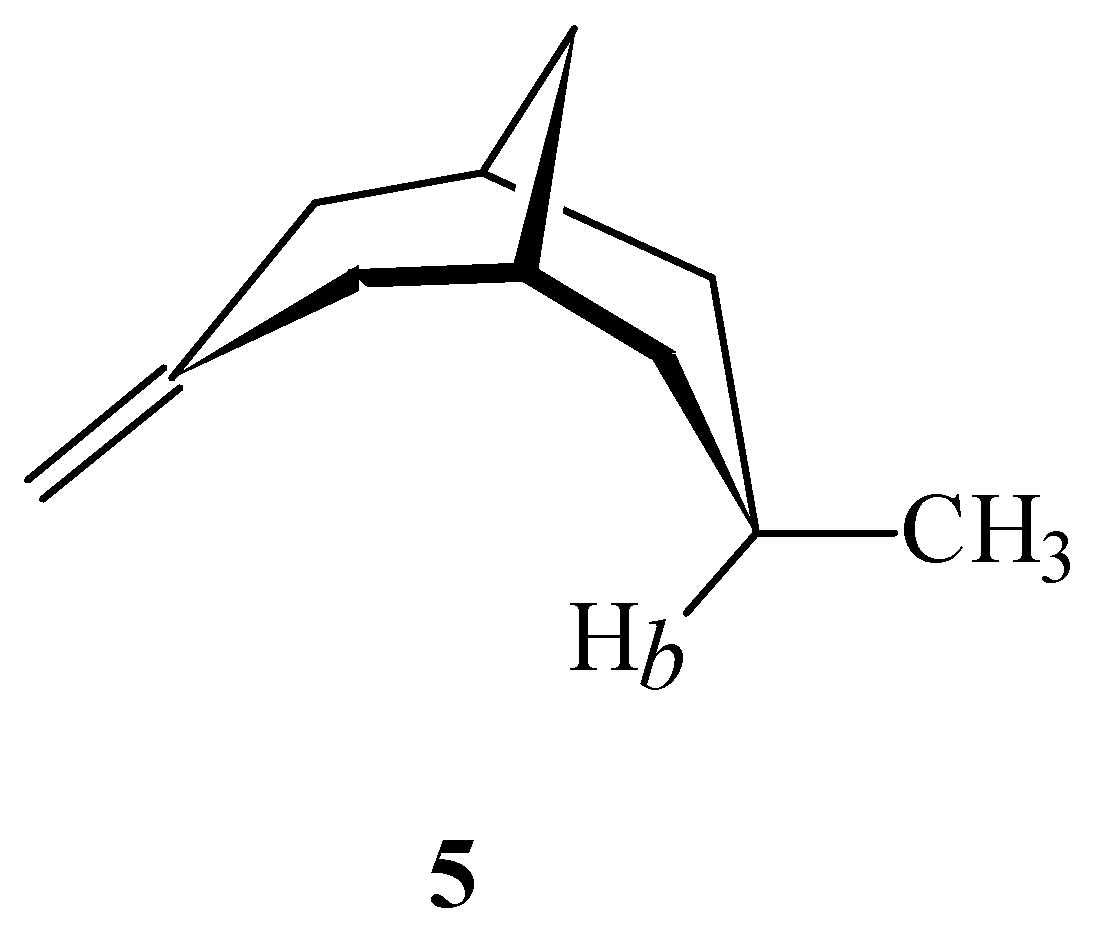

Table 1.
Shielding increments (Δσ) in ppm of protons over a carbon-carbon double bond in the structures in Figure 1 as predicted by McConnell's equation, predicted by the equation derived from ab initio GIAO-SCF calculations, and experimentally observed chemical shift differences (Δδ).
| Structure | McConnell Predicted Δδ | Ab Initio GIAO-SCF Predicted Δδ | Experimentally Observed Δδ |
|---|---|---|---|
| 1 | -0.10 | -0.28 | -0.24 |
| 215 | -0.10 | -0.46 | -0.80 |
| 3 | -0.06 | 0.30 | 0.27 |
| 4 | 0.12 | -1.99 | -2.12 |
Results and Discussion
We have prepared color graphs of the two-dimensional isosurface of the shielding increments predicted by the GIAO-SCF shielding-derived function (Figure 3) and the shielding increment derived from the McConnell equation (Figure 4). These graphs present  or
or  in a plane along the carbon-carbon double bond axis normal to the molecular plane of ethene from 2.0 Å to 3.5 Å above the molecular plane. The McConnell equation is the basis of the “shielding cone” model in common (but improper) use for predicting NMR shielding in the vicinity of a carbon-carbon double bond. The dramatic difference between the McConnell shielding model and the GIAO-SCF model is quite evident from a comparison of the two plots. When shielding increments calculated using the two methods are compared to experimental observations of chemical shift differences (Table 1) it is clear that the (mostly deshielding) values derived from the ab initio GIAO-SCF computations agree with experimental data much more closely.
in a plane along the carbon-carbon double bond axis normal to the molecular plane of ethene from 2.0 Å to 3.5 Å above the molecular plane. The McConnell equation is the basis of the “shielding cone” model in common (but improper) use for predicting NMR shielding in the vicinity of a carbon-carbon double bond. The dramatic difference between the McConnell shielding model and the GIAO-SCF model is quite evident from a comparison of the two plots. When shielding increments calculated using the two methods are compared to experimental observations of chemical shift differences (Table 1) it is clear that the (mostly deshielding) values derived from the ab initio GIAO-SCF computations agree with experimental data much more closely.
or in a plane along the carbon-carbon double bond axis normal to the molecular plane of ethene from 2.0 Å to 3.5 Å above the molecular plane. The McConnell equation is the basis of the “shielding cone” model in common (but improper) use for predicting NMR shielding in the vicinity of a carbon-carbon double bond. The dramatic difference between the McConnell shielding model and the GIAO-SCF model is quite evident from a comparison of the two plots. When shielding increments calculated using the two methods are compared to experimental observations of chemical shift differences (Table 1) it is clear that the (mostly deshielding) values derived from the ab initio GIAO-SCF computations agree with experimental data much more closely.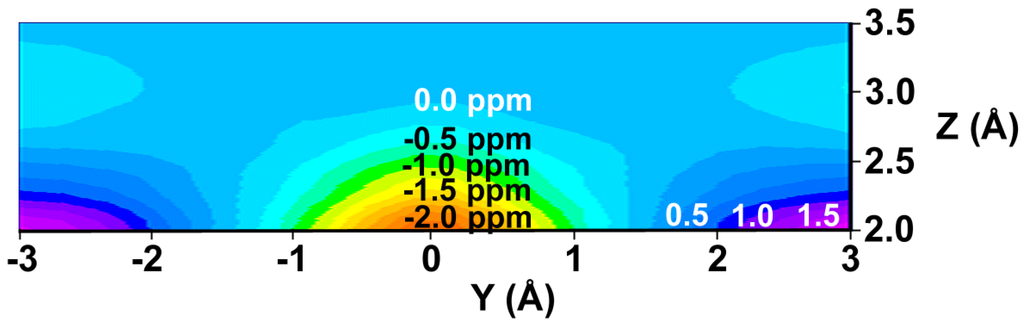
Figure 3.
(De)shielding isosurface above the plane of ethene (Z direction) along the C=C bond (Y) axis predicted by GIAO-SCF calculations (predicted shielding in ppm indicated on the graph; positive (white) numbers represent shielding, negative (black) numbers represent deshielding).
Figure 3.
(De)shielding isosurface above the plane of ethene (Z direction) along the C=C bond (Y) axis predicted by GIAO-SCF calculations (predicted shielding in ppm indicated on the graph; positive (white) numbers represent shielding, negative (black) numbers represent deshielding).
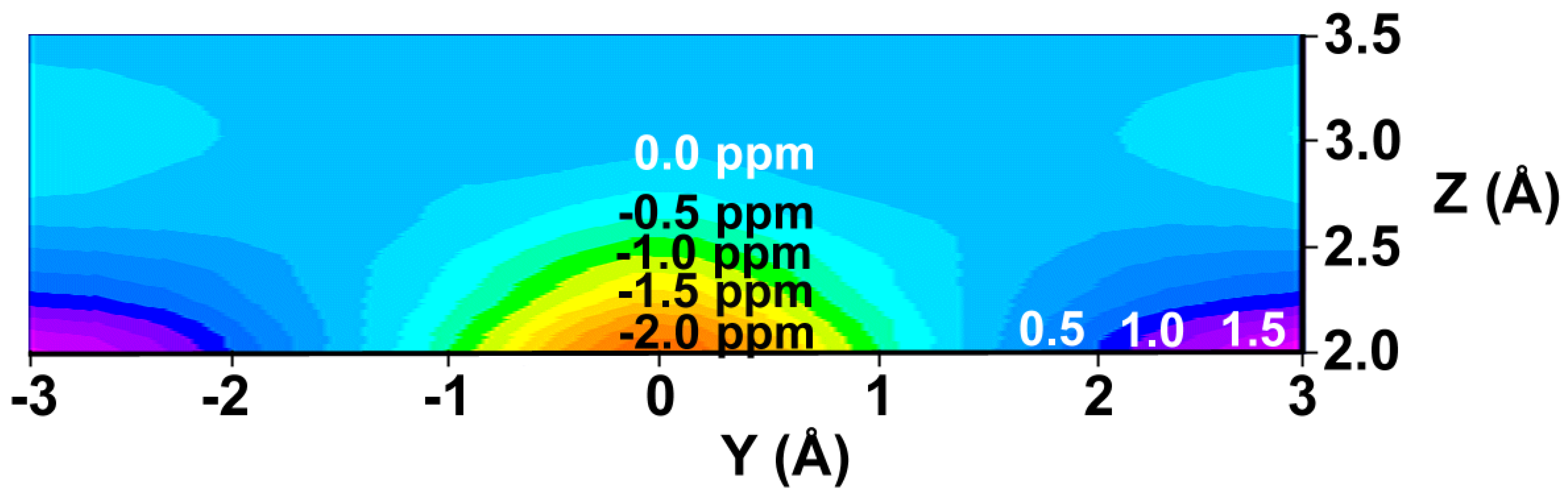
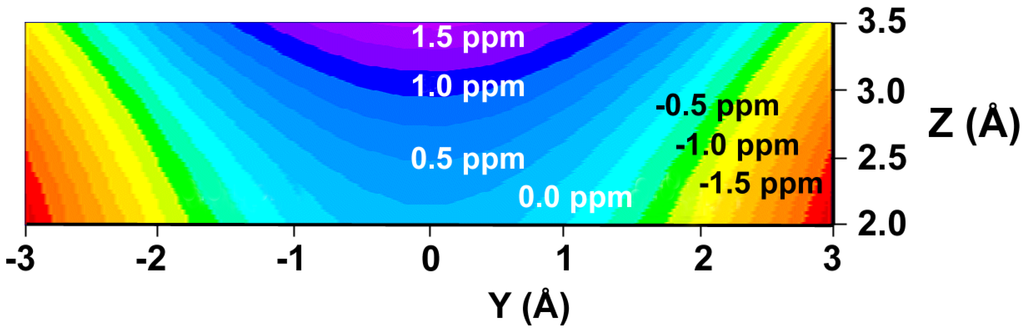
Figure 4.
Shielding isosurface above the plane of ethene (Z direction) along the C=C bond (Y) axis predicted by the McConnell equation (predicted shielding in ppm indicated on the graph).
Figure 4.
Shielding isosurface above the plane of ethene (Z direction) along the C=C bond (Y) axis predicted by the McConnell equation (predicted shielding in ppm indicated on the graph).
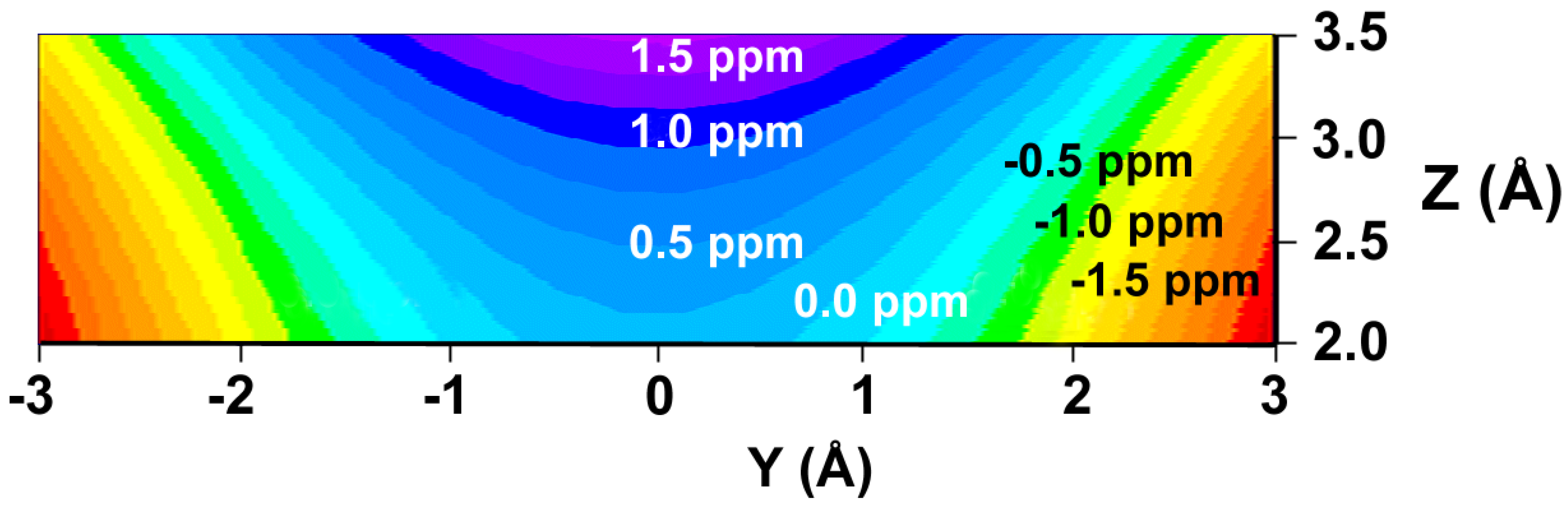
The major reason for the dramatic difference between the two sets of predicted shielding increments is the simplicity of the McConnell model. It computes the magnetic shielding at a point in space relative to the center of a carbon-carbon double bond arising only from the magnetic anisotropy of the double bond. Our ab initio GIAO-SCF approach, while utilizing a simple model system, also includes the important contributions of electric field effects, orbital interactions, and dispersion between the C-H bond and the alkene double bond to the extent that these are calculated by Hartree-Fock theory using the 6-31G(d,p) basis set. The data are not corrected for basis set superposition error (BSSE). Previous estimations of BSSE using the counterpoise method [16] have shown BSSE to be negligible (<0.01 ppm) in this series of computations [6,7].
The ab initio model was developed using the simplest alkene (ethene) yet all applications of the shielding function involve more highly substituted alkenes. To determine the effect of the degree of substitution of the double bond on the calculated shielding increment over a double bond, a series of calculations of methane with one proton oriented perpendicular to the plane of the double bond of 2-methylpropene (Figure 5) was performed. In this series of calculations methane was moved incrementally along the C=C axis with the proximal hydrogen of methane at a fixed distance 2.0 Å above the plane of the double bond. These calculations were performed at the MP2/6-31G(d,p) level.
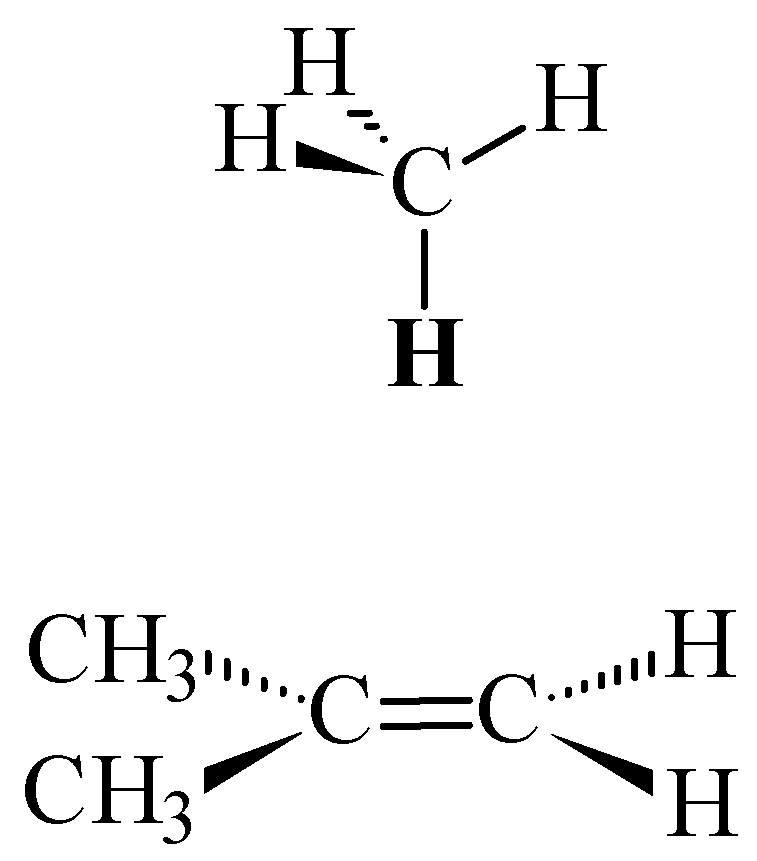
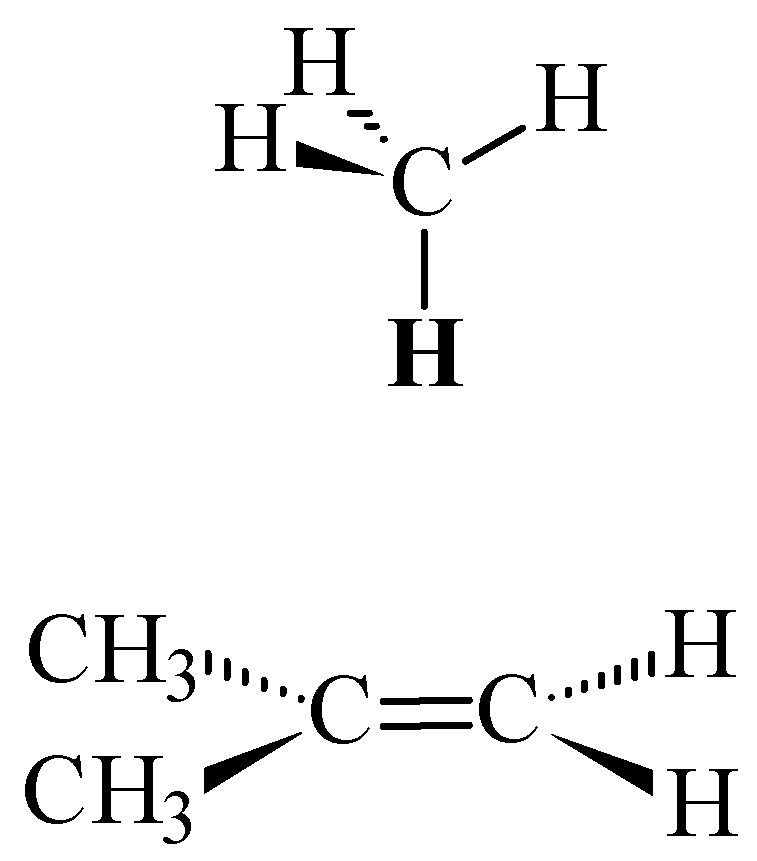
Figure 5.
Methane oriented over 2-methylpropene with one proton perpendicular to the plane of the carbon-carbon double bond.
Figure 5.
Methane oriented over 2-methylpropene with one proton perpendicular to the plane of the carbon-carbon double bond.
The output gives both HF/6-31G(d,p) and MP2/6-31G(d,p) isotropic shielding values. The results are shown in Figure 6, along with the intermolecular deshielding curve for a similar series of calculations 2.0 Å above ethene. The results indicate that the degree of substitution does indeed perturb the shielding surface slightly, shifting the region of maximum deshielding toward the less substituted end of the double bond by approximately 0.15 Å and decreasing the magnitude of the deshielding effect slightly (<12% decrease). This perturbation is consistent with the calculated natural population analysis charges [17] on the carbon atoms of the carbon-carbon double bond. The more highly substituted carbon has a natural population analysis charge of 0.00 whereas the less substituted (terminal) carbon has a natural population analysis charge of -0.45. The more electron-rich end of the double bond provides greater shielding of the proton, as expected. Given the uncertainty in predictions of shielding based on this equation, the accuracy with which our function-predicted shielding increments match the experimental chemical shift differences attests to the validity of applying the model to alkenes having different degrees of substitution of the double bond.
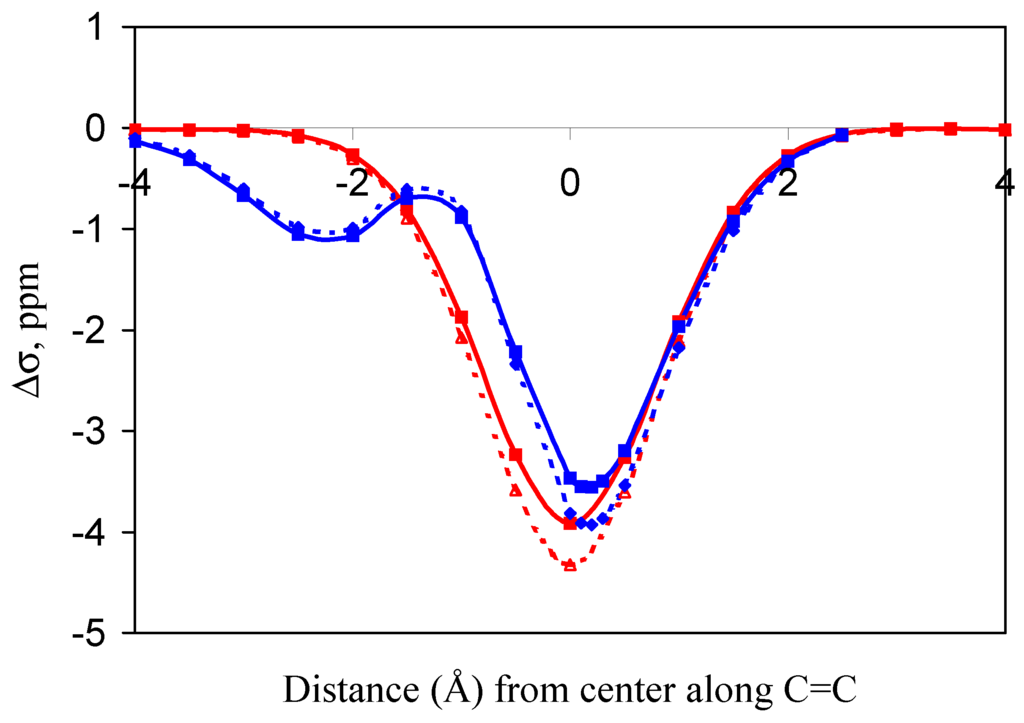
Figure 6.
(De)shielding curve for methane oriented as in Figure 5 with one proton 2.0 Å above 2-methylpropene 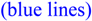 and moved along the double bond axis and methane similarly moved over ethene
and moved along the double bond axis and methane similarly moved over ethene 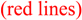 . Dotted lines are HF results; solid lines are MP2 results.
. Dotted lines are HF results; solid lines are MP2 results.
and moved along the double bond axis and methane similarly moved over ethene . Dotted lines are HF results; solid lines are MP2 results.
Figure 6.
(De)shielding curve for methane oriented as in Figure 5 with one proton 2.0 Å above 2-methylpropene and moved along the double bond axis and methane similarly moved over ethene . Dotted lines are HF results; solid lines are MP2 results.
and moved along the double bond axis and methane similarly moved over ethene . Dotted lines are HF results; solid lines are MP2 results.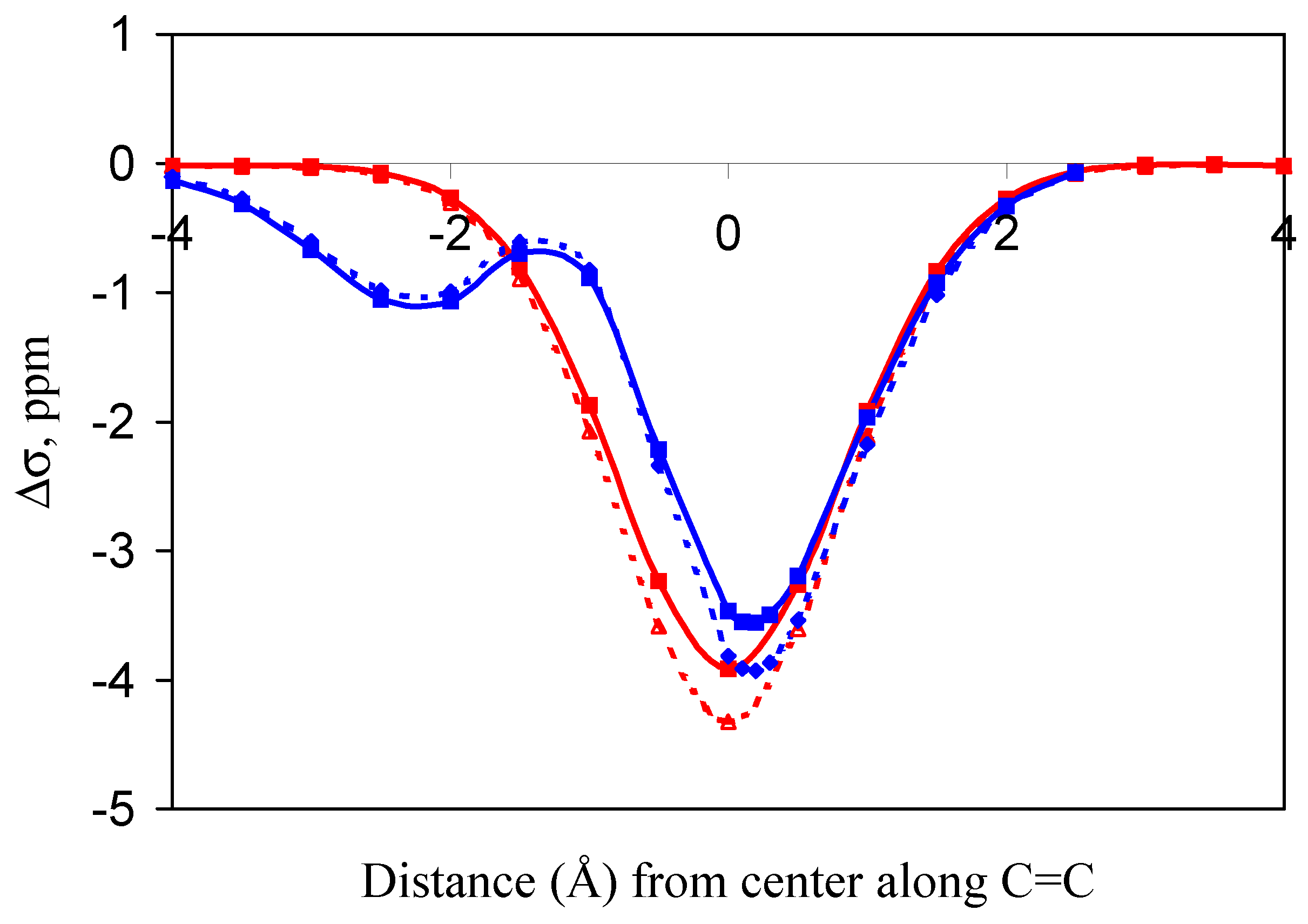
The shielding values calculated at the MP2 level are quite similar to those calculated at the SCF (HF) level. Above the center of the carbon-carbon double bond, where deshielding is at a maximum, the MP2 shielding values (solid lines) are approximately 9% less than the SCF (HF) values (dotted lines) for both the ethene series and the 2-methylpropene series . This is within the uncertainty of the predictions based on the SCF (HF) shielding function previously described [8].
and the 2-methylpropene series . This is within the uncertainty of the predictions based on the SCF (HF) shielding function previously described [8].Conclusions
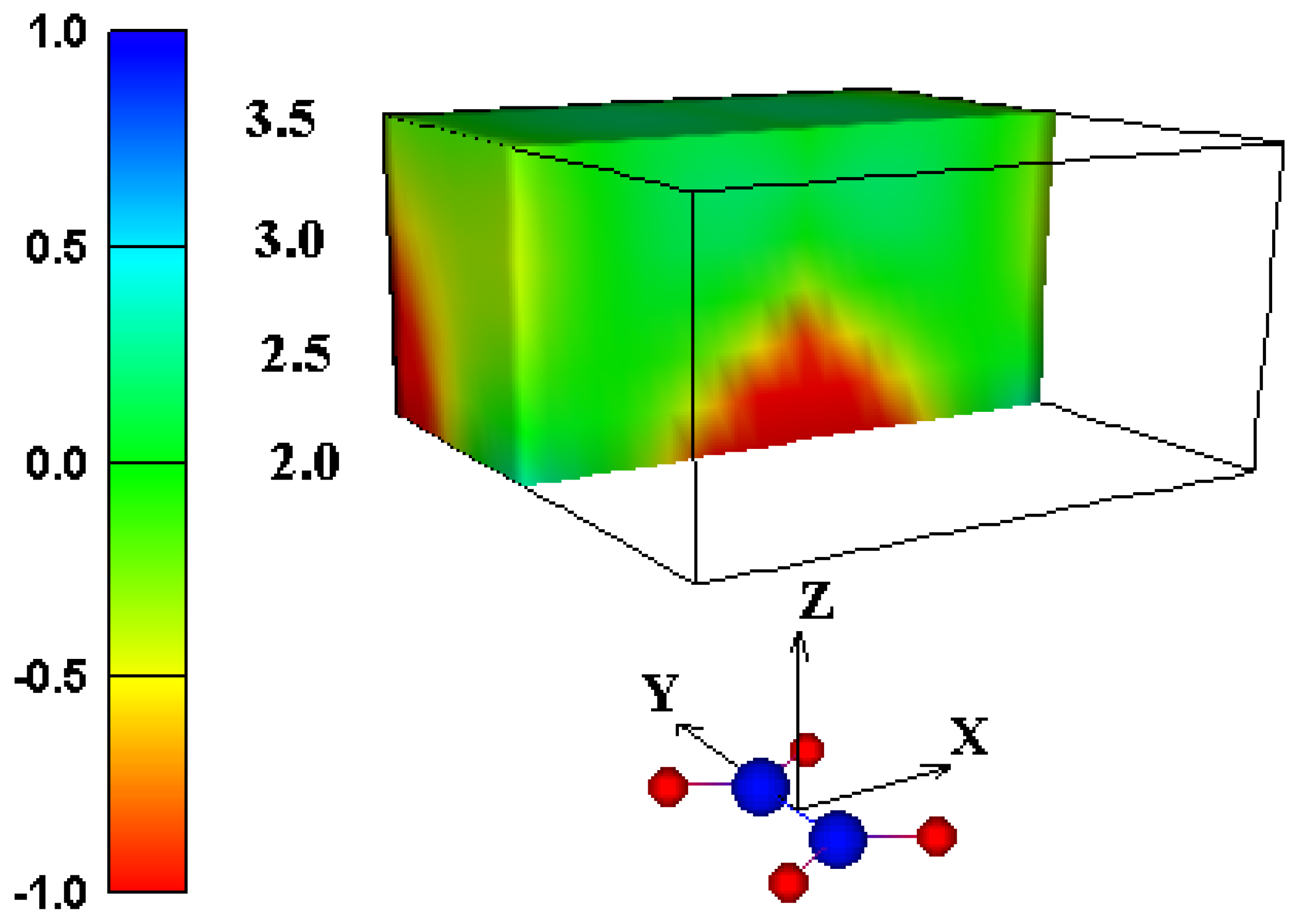
In conclusion, the experimental and computational evidence strongly suggests that the traditional shielding cone model should not be used for predicting effects on chemical shifts of protons over acarbon-carbon double bond. We propose a new graphical model (Figure 3 and Figure 7) for predicting the net through-space intermolecular (de)shielding effect of a carbon-carbon double bond on the NMR chemical shift of a nearby proton. The shape of the deshielding region resembles a dome, with greater deshielding closer to the region over the center of the carbon-carbon double bond. Predictions using this model stand in sharp contrast to predictions based on the traditional shielding cone model, but are in close agreement with reported chemical shifts of protons in such an environment.
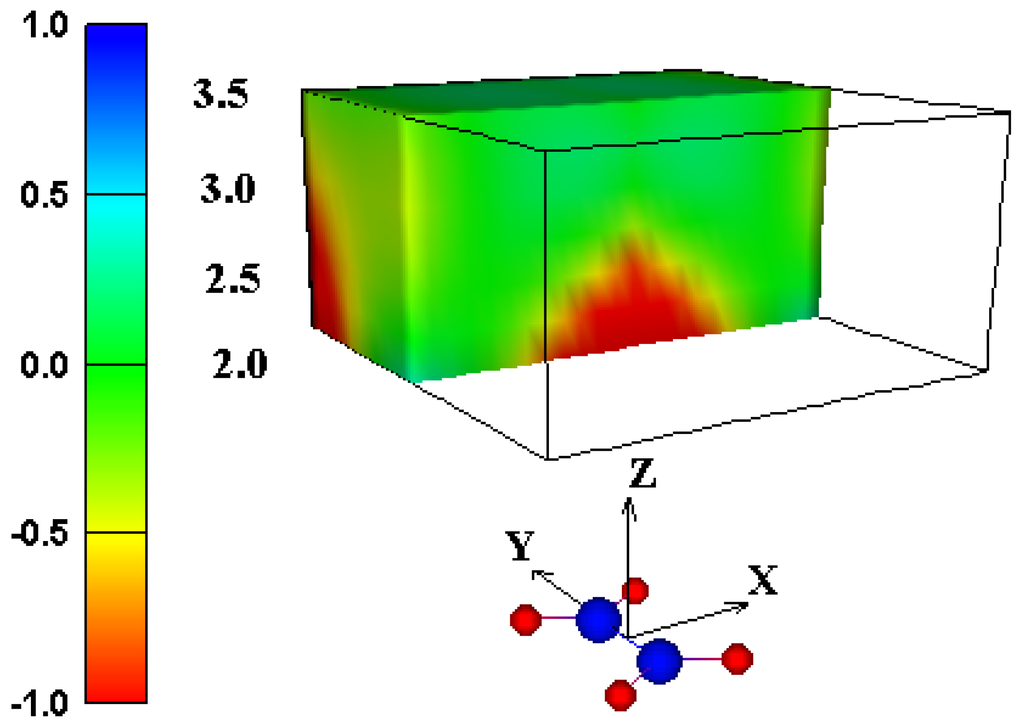
Figure 7.
Shielding isosurface above ethene (shown as red and blue spheres) predicted by GIAO-SCF calculations (animated in WWW version).
Figure 7.
Shielding isosurface above ethene (shown as red and blue spheres) predicted by GIAO-SCF calculations (animated in WWW version).
Acknowledgements
The authors gratefully acknowledge support of this work by the donors of The Petroleum Research Fund, administered by the American Chemical Society, the North Carolina Supercomputing Center, the UNCW Information Technology Services Division of and the UNCW College of Arts and Sciences.
References and Notes
- For example, Gunther, H. NMR Spectroscopy: Basic Principles, Concepts, and Applications in Chemistry, 2nd ed.; Wiley: Chichester, 1995; pp. 78–84. [Google Scholar]
- McConnell, H. M. Theory of Nuclear Magnetic Shielding in Molecules. I. Long-Range Dipolar Shielding of Protons. J. Chem. Phys. 1957, 27, 226–229. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. Computational Evidence of NMR Deshielding of Protons over a Carbon-Carbon Double Bond. J. Am. Chem. Soc. 1998, 120, 11510–11511. [Google Scholar]
- Elguero, J.; Alkorta, I. Ab Initio hybrid DFT-GIAO calculations of the shielding produced by carbon-carbon bonds and aromatic rings in 1H NMR. New J. Chem. 1998, 22, 381–385. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. An Empirical Proton NMR Shielding Equation for Alkenes Based on Ab Initio Calculations. Struct. Chem. 1998, 9(6), 403–410. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. Proceedings of ACS Symposium "Modeling NMR Chemical Shifts: Gaining Insights into Structure and Environment," American Chemical Society; Washington, D.C., 1999; pp. 207–219.
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. An Improved Model for Predicting Proton NMR Shielding by Alkenes Based on Ab Initio GIAO Calculations. Struct. Chem. 1999, 10(5), 375–380. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Brown, J. D.; Ingrassia, S. T.; Minga, E. K. An Algorithm for Predicting Proton NMR Deshielding over a Carbon-Carbon Double Bond. J. Mol. Graphics Mod. 2000, 18(1), 1–6. [Google Scholar]
- Sorensen, T. S.; Whitworth, S. M. □-Hydrido Bridging in Tricycloalkyl Carbocations. J. Am. Chem. Soc. 1990, 112, 8135–8144. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-Consistent Perturbation Theory of Diamagnetism. I. A Gauge-Invariant LCAO Method for N.M.R. Chemical Shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Chesnut, D. B.; Foley, C. K. Some Simple Basis Sets for Accurate 13C Chemical Shift Calculations. Chem. Phys. Lett. 1985, 118, 316–321. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J. F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98; Revision A.7; Gaussian, Inc.: Pittsburgh PA, 1998. [Google Scholar]
- Gauss, J. Effects of Electron Correlation on the Calculation of Nuclear Magnetic Resonance Chemical Shifts. J. Phys. Chem. 1993, 99(5), 3629–3643. [Google Scholar] [CrossRef]
- For structure 2, the shielding and chemical shift values for Hb were taken from structure 1. For structure 4, the value for Hb in structure 5 (Figure 2) was used as reference.
- Boys, S. F.; Bernardi, F. The Calculations of Small Molecular Interaction by the Difference of Separate Total Energies. Some Procedures with Reduced Error. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Reed, A. E.; Weinstock, R. B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83(2), 735–746. [Google Scholar] [CrossRef]
© 2000 by MDPI (http://www.mdpi.org).