Toward the Understanding of the Metabolism of Levodopa I. DFT Investigation of the Equilibrium Geometries, Acid-Base Properties and Levodopa-Water Complexes



Abstract
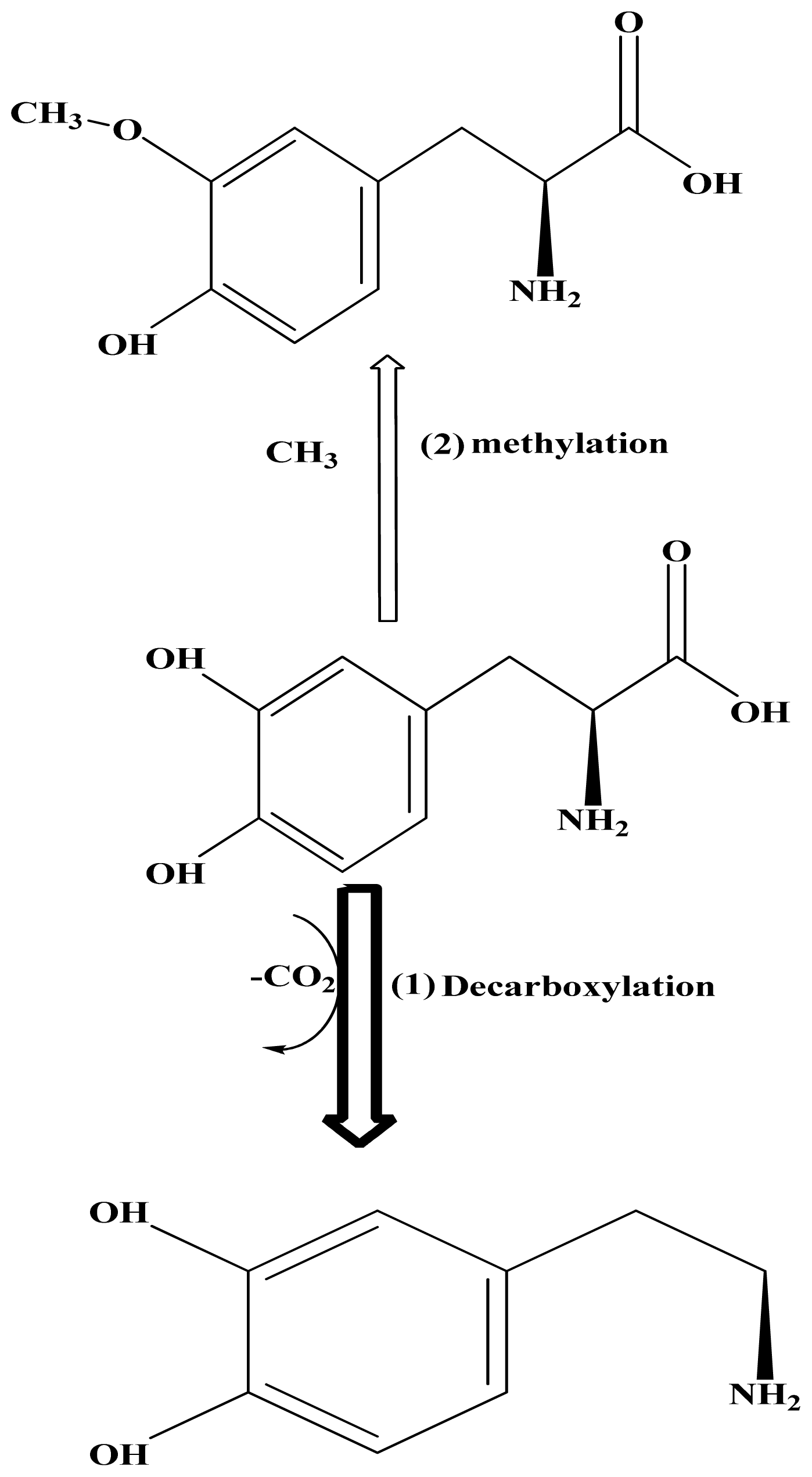
:1. Introduction
2. Results and Discussion
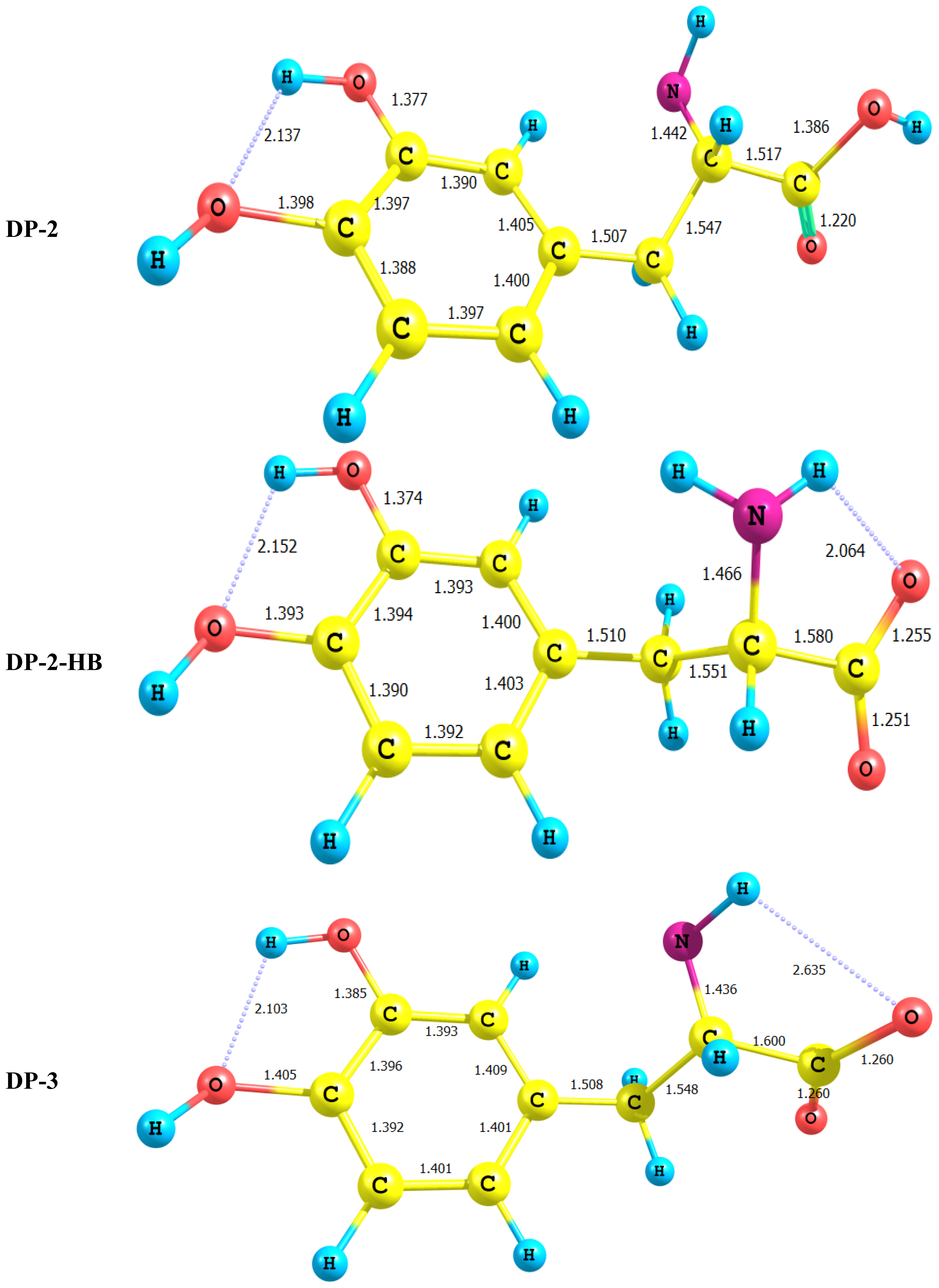
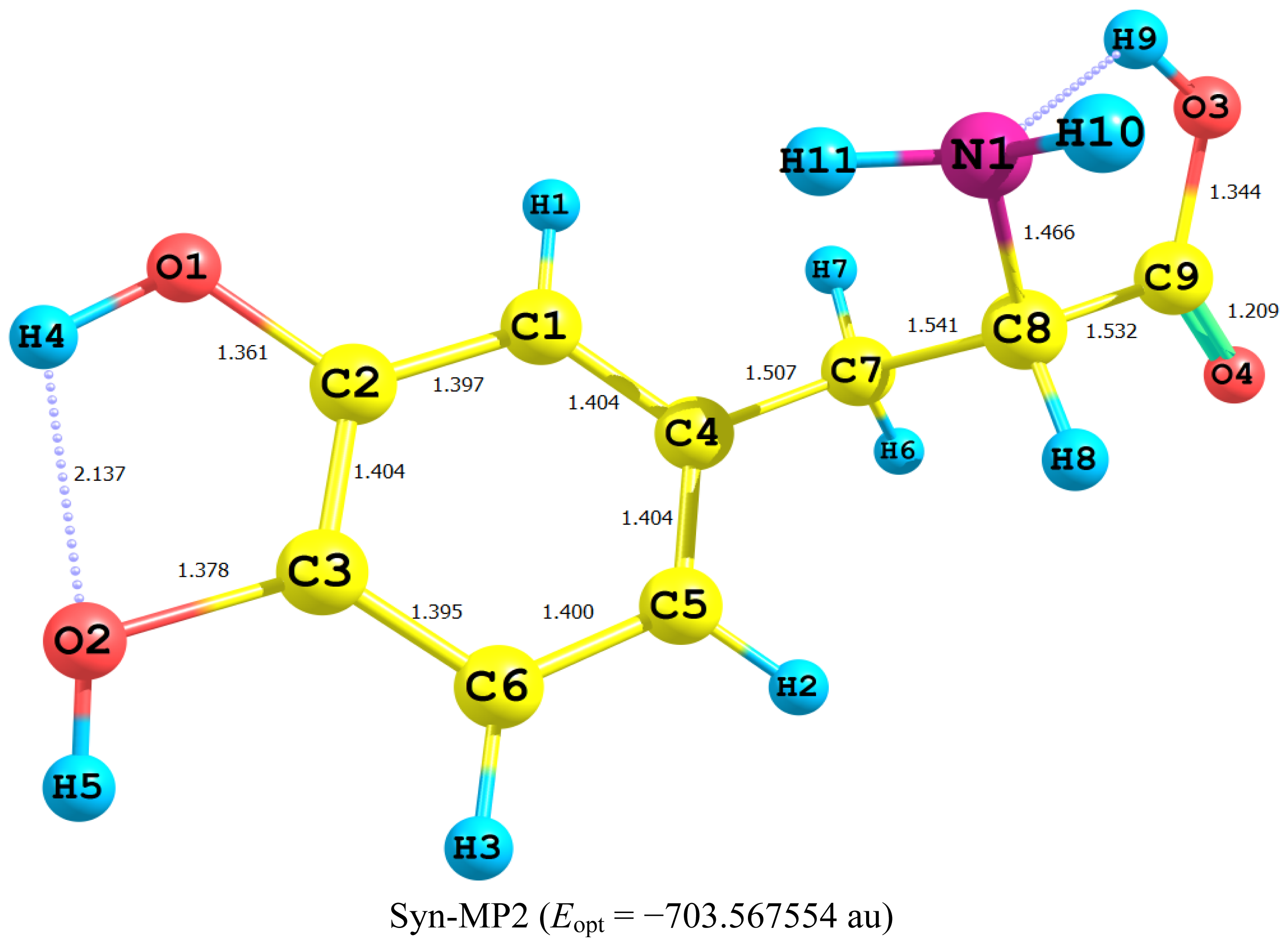
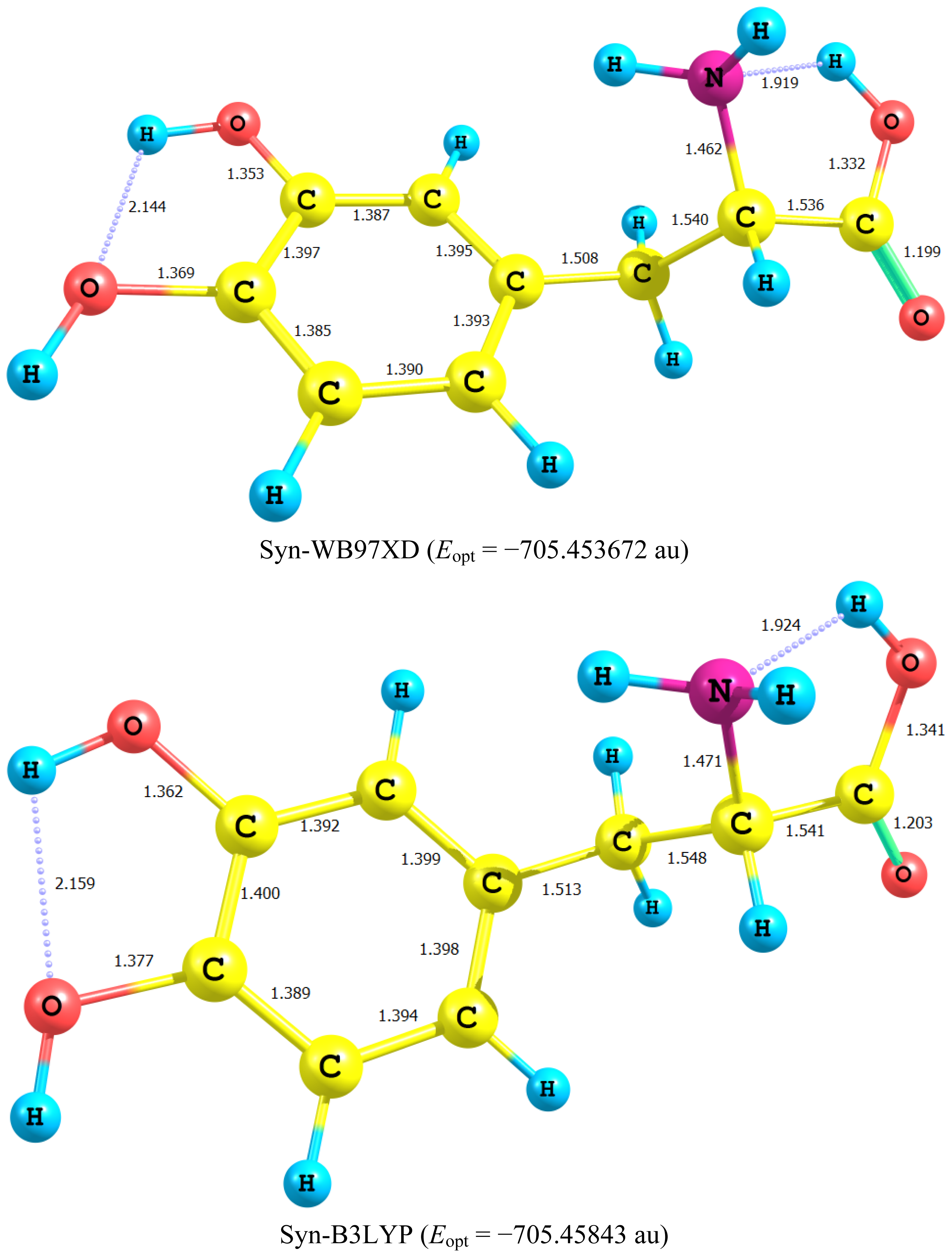
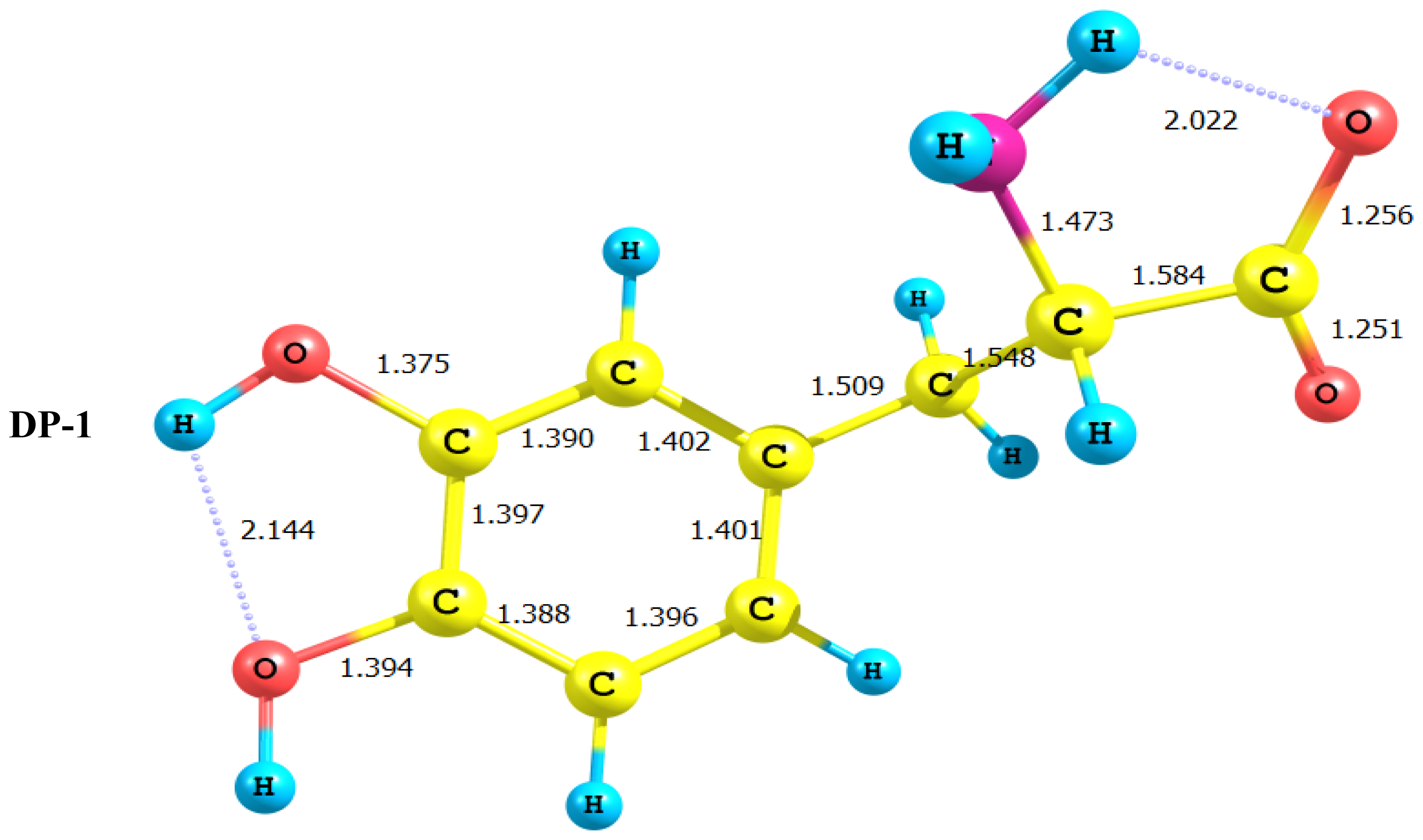
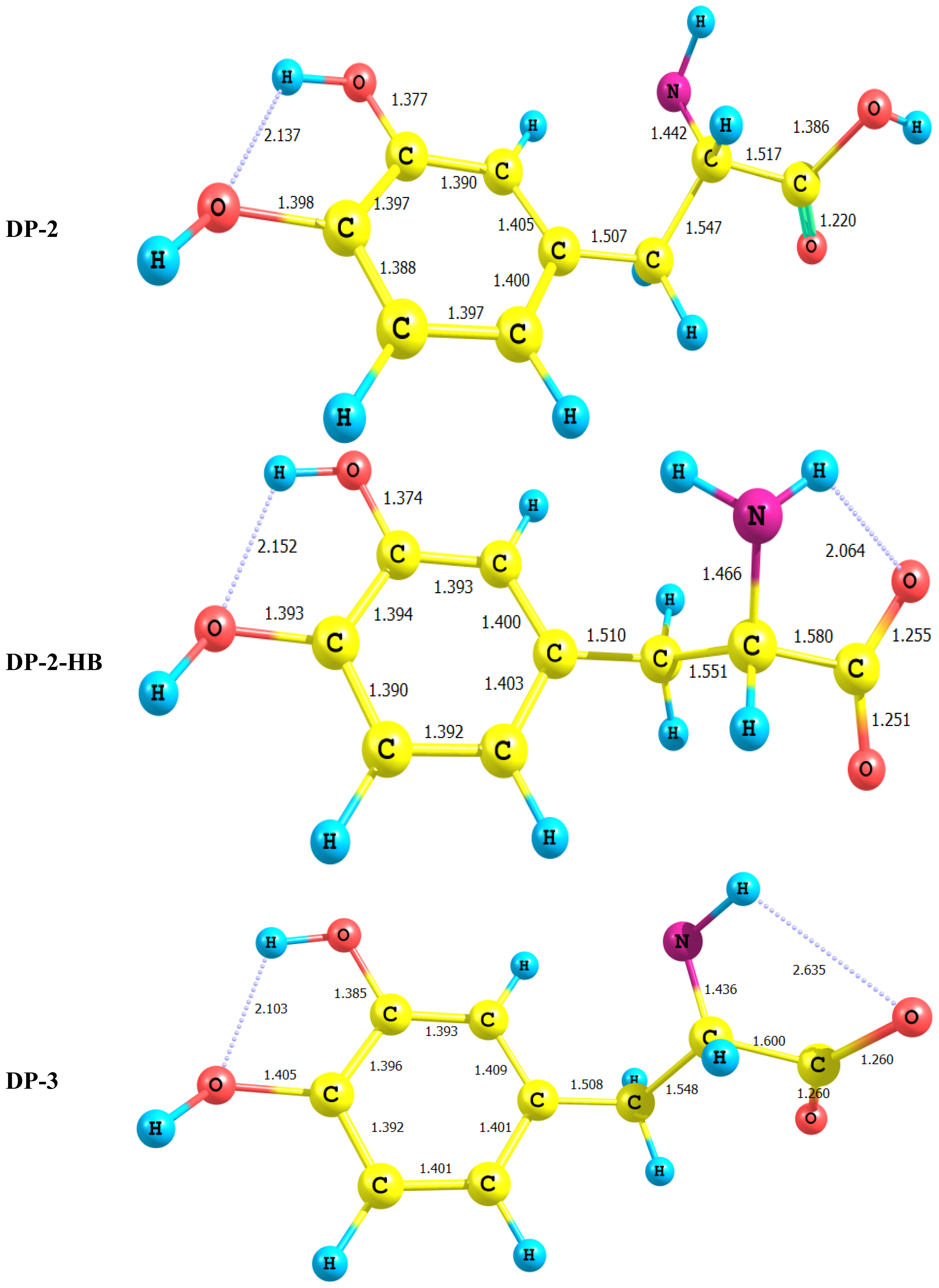
2.1. Equilibrium Geometry of LD and Its Deprotonated Forms
- In all cases studied, deprotonation is local in nature, i.e., its effect is localized in the region where deprotonation took place and is not transmitted through the length of the molecule.
- Deprotonation of the carboxyl group (hydrogen atom H23) causes a delocalization of charge in the O–C=O region. The two RC–O bonds are considerably affected. Thus, the C–O bond length is reduced from a value of 1.359 Ǻ to 1.256 Ǻ which is very close to the other RC–O of 1.251 Ǻ. The C10–C11 bond length is slightly stretched by only 3% of its original value. This is accompanied by widening of the O12–C11–O13 angle to a value of 129.5° to minimize the repulsion due to the accumulation of the charge density in this region. The hybridization scheme of the oxygen atom remains, however, almost unaffected as: core 2s1.7 2p5.02 3p0.01
- It is of special interest to note that the effect of deprotonation of the carboxyl group proton is transmitted to the NH2 group. The charge density on N is markedly increased. This point will be discussed further since it might very well underlay the formation of zwitterions.
- In LD, the aliphatic side chain is tilted out of plane by an angle of 79°. Upon deprotonation of the carboxyl proton, the dihedral angle is reduced to 54° and the side chain is forced back to approach the plane of the rest of the molecule.
- Deprotonation of the amino group has an even more localized effect. Thus, while the C–N bond length is slightly reduced, all other bond lengths are hardly affected. The charge is now accumulated on the N atom. This is not even transmitted to the C–N carbon. This is accompanied by a change in the N atom hybridization scheme from 2s1.402p4.413p0.01 to 2s1.552p4.393p0.01. The major effect is the accumulation of extra 11% e into the 2 s space.
- Deprotonation of the catechol hydroxyl group causes the expected decrease in the phenyl–O bond length and increase of the negative charge on the oxygen atom. However, it seems that a subsequent hydrogen bond is formed with O-catechol hydroxyl H atom. This is stabilized by the increased negative charge on the O atom and the formation of a five-membered ring. The length of this H-bond is typical (1.881 Ǻ).
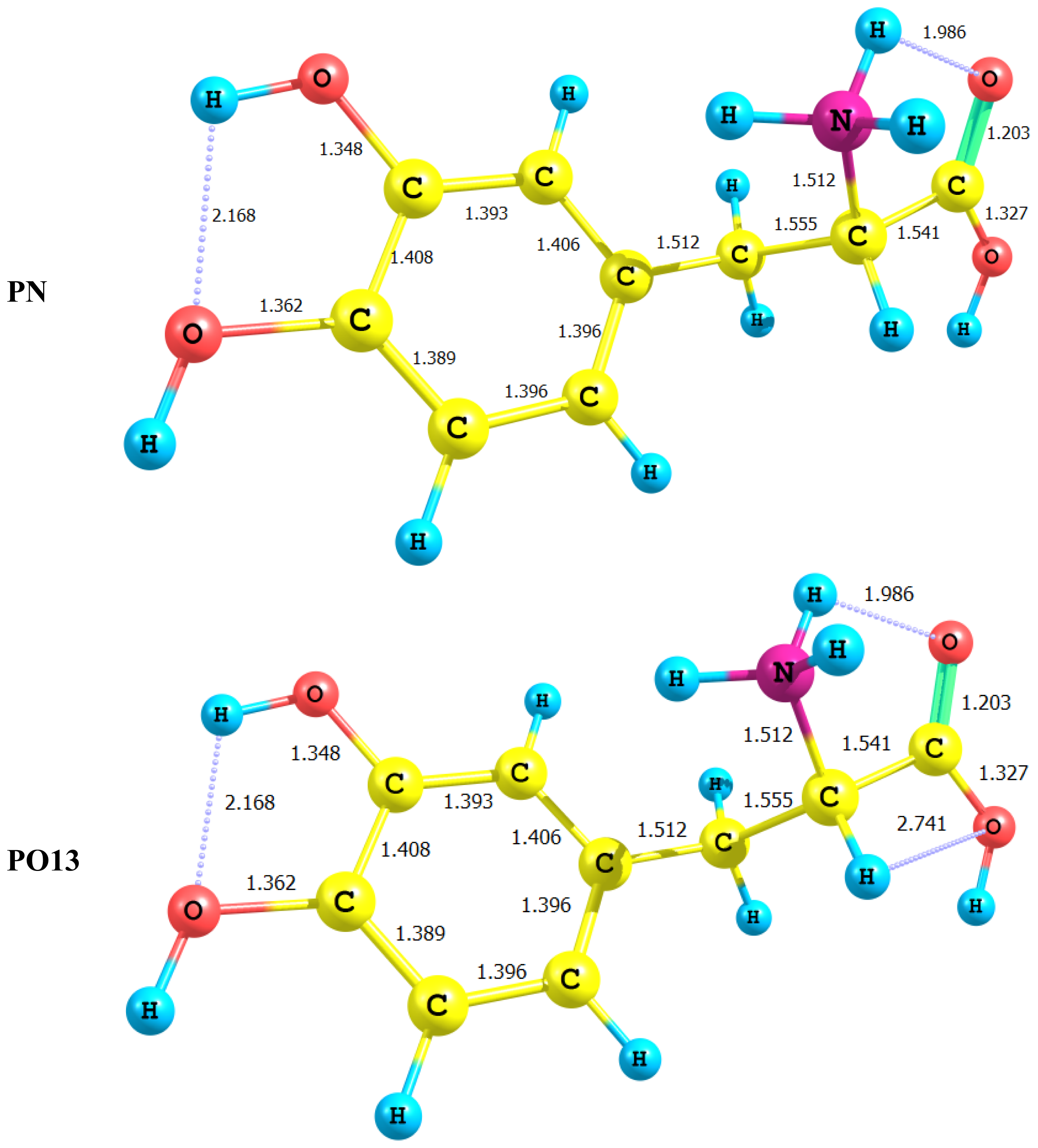
2.2. Energetics of the Protonation/Deprotonation of LD
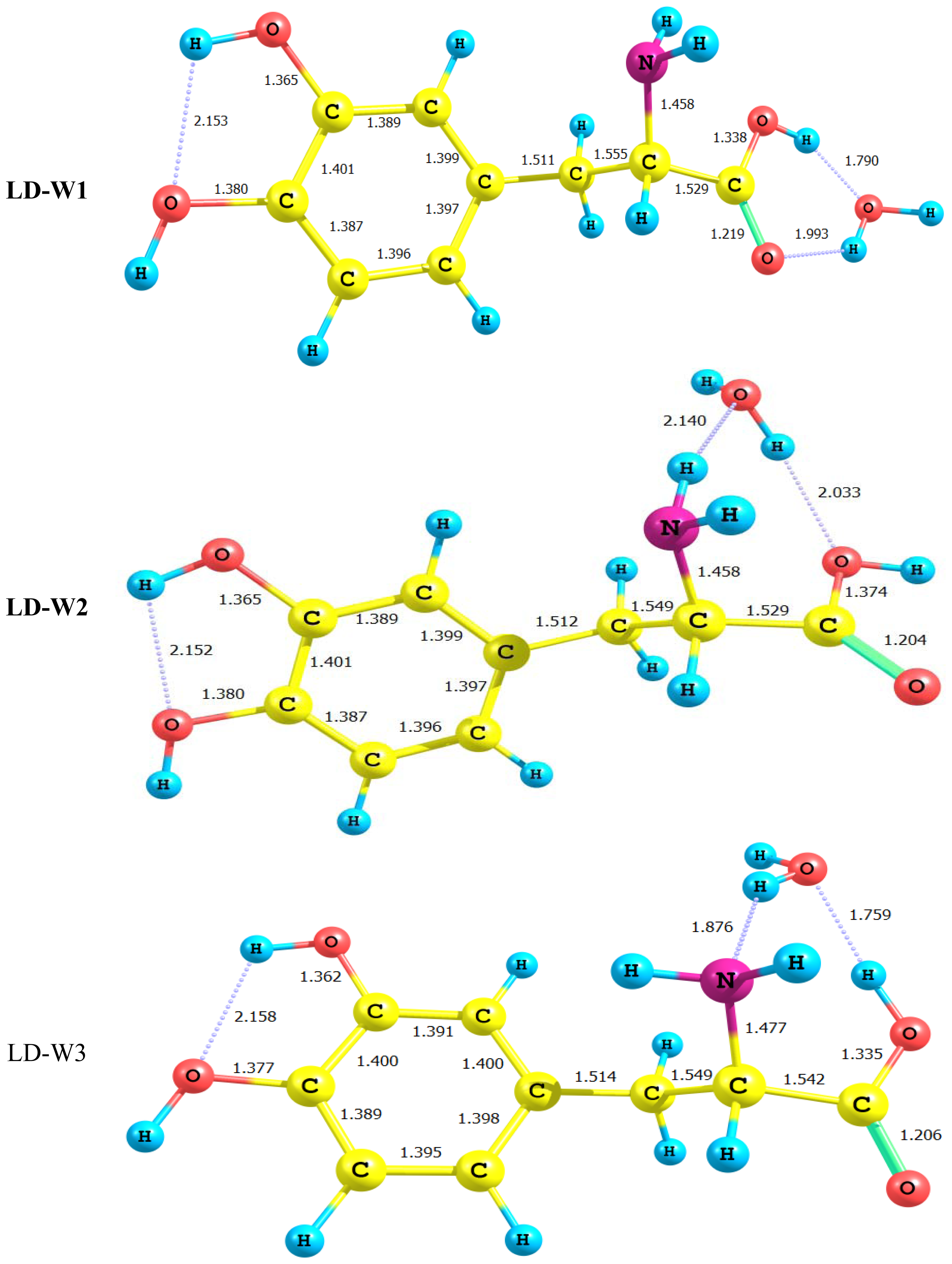
2.3. Water-LD Complexes
2.4. NBO Analysis
- The major D-A interaction in LD is localized on the phenyl ring and involves πC–C−π*C–C.
- Strong interaction between the p lone-pair electrons of the hydroxyl group oxygen atoms and the ring π*C–C. This CT interaction spans a narrow range of 23–26 kcal/mol.
- A very strong interaction involving the p lone pair of the carboxyl group O12 and the π*C11–O13. This interaction amounts to 41.99 kcal/mol and reflects the strong delocalization of the π-electron density in the O–C–O region.
- The p lone pair of O13 is, surprisingly interacting with the Rydberg orbitals of the carbonyl carbon. This lone pair also interacts via CT to π*C–O12 by a considerable amount of energy of 34.54 kcal/mol.
- In conclusion, LD behaves as if it is composed of two non- or at least weekly interacting subsystems, namely the aryl moiety and the amino acid side chain. One can trace, however, week interaction through the σ-framework of the aryl moiety transmitted to σC9–C10 which interacts with σ*C10–N14 and to a lesser extent with σ*C11–O13. Furthermore, σC9–C10 also interacts with the Rydberg type orbitals of C11 and N14. Thus, although, there is no π conjugation extending over the LD framework, yet it seems that the σ framework is able to transmit the interaction between the catechol moiety and the amino acid side chain.
- DP-1, where the carboxyl group proton has been detached, shows a dimensioned interaction across the molecular framework. Thus, the interaction of σC9–C10 with the amino group side chain is reduced to almost half its value in LD. However, it maintains its week interaction with the Rydberge type orbitals on C11 and N14. Furthermore, a new very strong interaction appears between each of the lone pair orbitals on O12 and O13 with the empty antibonding p orbital on C11. This interaction amounts to 191 and 205 kcal/mol for O12 and O13 respectively. This indicates clearly that deprotonation of the carboxyl group has a pronounced effect on the charge density distribution that is localized to a great extent in the O–C=O region.
- For DP-2 where the amino group proton is detached the process has very little effect on the charge density distribution. Thus, the across- subsystem interactions through σC9–C10 is enhanced slightly. A new week interaction emerges between the N14 lone pair and the antibonding σ*C10–C11 orbital. Deprotonation in this case has the subsequent effect of localizing the charge density onto the nitrogen atom which acts as a very poor electron donor.
- For DP-4, where the catechol moeity hydroxyl proton is removed the deprotonation process has strong perturbation effect on the π charge density of the aryl moiety. Thus, a strong interaction appears between the deprotonated O13 lone pair and the π*C3–C6 This interaction (E = 71 kcal/mol) is delocalized over the entire catechol π-framework, and has no marked effect on the interaction between the aryl moiety and the amino acid side chain.
3. Computational Methods
4. Conclusion
- LD is not planar with its side chain acting as a free rotor across several single bonds. However, deprotonation of the carboxyl group or the amino group forces the side chain into the plane of the catechol moiety.
- Deprotonation is local in nature, for all sites considered the changes in geometry and charge density are localized in the region where the deprotonation took place. However, the effect of carboxyl group deprotonation is transmitted to the C–NH2 region and leads to the accumulation of excess charge density on N14. This point is of special importance when considering the formation of zwitterions.
- Deprotonation of the amino group proton lead to the change of the hybridization scheme of N14.
- NBO analysis of LD and its deprotonated forms reveals detailed information of bonding characteristics and interactions across the molecular frame work. Thus, although there is a pronounced cross conjugation in the π-system, the interaction between the catechol moiety and the amino acid side chain is marked through the inductive and CT effects across the σ and nonbonding frameworks.
- Deprotonation of the carboxyl group is more favorable than deprotonation at other sites. However, it should be noted that stabilization of the deprotonated structures by intramolecular H-bonding is visible in the case of the deprotonation of the amino group or the catechol hydroxyl group.
Acknowledgements
References
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanism and models. Neuron 2003, 39, 889–909. [Google Scholar]
- Nutt, J.G.; Fellman, J.H. Pharmacokinetics of levodopa. Clin. Neuropharmacol 1984, 7, 35–49. [Google Scholar]
- Goodal, M.C.; Alton, H. Metabolism of 3,4-dihydroxyphenylalanine (l-dopa) in human subjects. Biochem. Pharmacol 1972, 21, 2401–2408. [Google Scholar]
- Montgomery, E.B. Pharmacokinetics and pharmacodynamics of levodopa. Neurology 1992, 42, 17–22. [Google Scholar]
- Tyce, G.M. Metabolism of 3,4-dihydroxyphenylalanine by isolated perfused rat liver. Biochem. Pharmacol 1971, 20, 3447–3462. [Google Scholar]
- Sasahara, K.; Nitanai, T.; Habara, T.; Morioka, T.; Nakajima, E. Dosage form design for improvement of bioavailability of levodopa V: Absorption andmetabolism of levodopa in intestinal segments of dogs. J. Pharm. Sci 1981, 70, 1157–1160. [Google Scholar]
- Nutt, J.G.; Woodward, W.R.; Hammerstad, J.P.; Carter, J.H.; Anderson, J.L. The “oneoff” phenomenon in Parkinson’s disease. Relation to levodopa absorption and transport. N. Engl. J. Med 1984, 10, 483–488. [Google Scholar]
- Reches, A.; Fahn, S. 3-O-Methyldopa blocks dopa metabolism in rat corpus striatum. Ann. Neurol 1982, 12, 267–2671. [Google Scholar]
- Fujino, K.; Yoshitake, T.; Kehr, J.; Notha, H.; Yamaguchi, M. Simultaneous determination of 5-hydroxyindoles and catechols by high-performanceliquid chromatography with fluorescence detection following derivatization with benzylamine and 1,2-diphenylethylenediamine. J. Chromatogr. A 2003, 1012, 169–177. [Google Scholar]
- Brown, M.J.; Dollery, C.T. A specific radioenzymatic assay for dihydroxyphenylalanine (DOPA). Plasma dopa may be the precursor of urine free dopamine. Br. J. Clin. Pharmacol 1981, 11, 79–83. [Google Scholar]
- Zurcher, G.; da Prada, M. Radioenzymatic assay of femtomole concentrations of DOPA in tissues and body fluids. J. Neurochem 1979, 33, 631–639. [Google Scholar]
- Kim, B.K.; Koda, R.T. Fluorimetric determination of methyldopa in biological fluids. J. Pharm. Sci 1977, 66, 1632–1634. [Google Scholar]
- Mell, L.D.; Gustafson, A.B. Urinary free methyldopa determined by reversed-phase high-performance liquid chromatography. Clin. Chem 1978, 24, 23–26. [Google Scholar]
- Tornkvist, A.; Sjoberg, P.J.; Markides, K.E.; Bergquist, J. Analysis of catecholamines and related substances using porous graphitic carbon as separation media in liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2004, 801, 323–329. [Google Scholar]
- Oliveira, C.H.; Barrientos-Astigarraga, R.E.; Sucupira, M.; Graudenz, G.S.; Muscara, M.N.; De, N.G. Quantification of methyldopa in human plasma by high-performance liquid chromatography-electrospray tandem mass spectrometry: Application to a bioequivalence study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2002, 768, 341–348. [Google Scholar]
- Rondelli, I.; Acerbi, D.; Mariotti, F.; Ventura, P. Simultaneous determination of levodopa methyl ester, levodopa, 3-O-methyldopa and dopamine in plasma by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Appl 1994, 653, 17–23. [Google Scholar]
- Saxer, C.; Niina, M.; Nakashima, A.; Nagae, Y.; Masuda, N. Simultaneous determination of levodopa and 3-O-methyldopa in human plasma by liquid chromatography with electrochemical detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2004, 802, 299–305. [Google Scholar]
- Karimi, M.; Carl, J.L.; Loftin, S.; Perlmutter, J.S. Modified high-performance liquid chromatography with electrochemical detection method for plasma measurement of levodopa, 3-O-methyldopa, dopamine, carbidopa and 3,4-dihydroxyphenyl acetic acid. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2006, 836, 120–123. [Google Scholar]
- Blandini, F.; Martignoni, E.; Pacchetti, C.; Desideri, S.; Pivellini, D.; Nappi, G. Simultaneous determination of l-dopa and 3-O-methyldopa in human platelets and plasma using high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Sci. Appl 1997, 700, 278–282. [Google Scholar]
- Mitsui, A.; Nohta, H.; Ohkura, Y. High-performance liquid chromatography of plasma catecholamines using 1,2-diphenylethylenediamine as precolumn fluorescence derivatization reagent. J. Chromatogr 1985, 344, 61–70. [Google Scholar]
- Kamahori, M.; Taki, M.; Watanabe, Y.; Miura, J. Analysis of plasma catecholamines by high-performance liquid chromatography with fluorescence detection, simple preparation for pre-column fluorescence derivatization. J. Chromatogr 1991, 567, 351–358. [Google Scholar]
- Jeon, H.K.; Nohta, H.; Ohkura, Y. High-performance liquid chromatography determination of catecholamines and their precursors and metabolites in human urine and plasma by postcolumn derivatization involving chemical oxidation followed by fluorescence reaction. Anal. Biochem 1992, 200, 332–338. [Google Scholar]
- Yoshitake, T.; Kehr, J.; Yoshitake, S.; Fujino, K.; Nohta, H.; Yamaguchi, M. Determination of serotonin, noradrenaline, dopamine and their metabolites in rat brain extracts and microdialysis samples by column liquid chromatography with fluorescence detection following derivatization with benzylamine and 1,2-diphenylethylenediamine. J. Chromatogr. B 2004, 807, 177–183. [Google Scholar]
- Nohta, H.; Lee, M.K.; Okhura, Y. Fluorescent products of the reaction for the determination of catecholamines with 1,2-diphenylethylenediamine. Anal. Chim. Acta 1992, 267, 137–139. [Google Scholar]
- Nohta, H.; Yamaguchi, E.; Ohkura, Y.; Watanabe, H. Measurement of catecholamines, their precursors and metabolites in human urine and plasma by by high-performance liquid chromatography with fluorescence derivatization. J. Chromatogr 1989, 493, 15–26. [Google Scholar]
- Rona, K.; Ary, K.; Gaghalyi, B.; Klebovich, I. Determination of a-methyldopa in human plasma by validated high-performance liquid chromatography with fluorescence detection. J. Chromatogr. A 1996, 730, 125–131. [Google Scholar]
- Bahrami, G.; Kiani, A.; Mirzaeei, S. A rapid high performance liquid chromatographic determination of methyldopa in human serum with fluorescence detection and alumina extraction, application to a bioequivalence study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2006, 832, 197–201. [Google Scholar]
- Wood, A.T.; Hall, M.R. Reversed-phase high-performance liquid chromatography of catecholamines and indoleamines using a simple gradient solvent system and native fluorescence detection. J. Chromatogr. B Biomed. Sci. Appl 2000, 744, 221–225. [Google Scholar]
- Frisch, M.J; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C. Gaussian 03; Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys 1970, 19, 553–566. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO, version 3.1; TCI, University of Madison: Madison, WI, USA, 1998. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Ee/au | Deprotonation and protonation energies (DE)/kcal/mol =EDP − Ee | Relative energy kcal/mol |
|---|---|---|---|
| LD | −705.45367 | −2.984 | |
| LD-HB * | −705.45843 | 0 | |
| DP-1 | −704.90918 | −344.653 | |
| DP-2 | −704.83826 | −389.156 | −43.496 |
| DP-2-HB * | −704.90757 | −345.660 | |
| DP-3 | −704.14807 | −822.248 | |
| DP-4 | −704.88462 | −360.067 | −13.719 |
| DP-4-HB * | −704.90648 | −346.348 | |
| PO7 | −705.74793 | 181.6252 | |
| PN | −705.82031 | 227.0671 | |
| PO13 | −705.81224 | 221.9833 |
| Compounds | Ee/au | (ΔEb) kcal/mol | EBSSE | (ΔEb-bsse) kcal/mol | Ec | (ΔEcb) kcal/mol | H\au | ΔH kcal/mol |
|---|---|---|---|---|---|---|---|---|
| L-Dopa | −705.45367 | −705.25677 | −705.24186 | |||||
| LD-W1 | −781.92896 | −10.559 | −781.91194 | −0.11922 | −781.70706 | −8.206 | −781.68921 | −8.788 |
| LD-W2 | −781.91946 | −4.598 | −781.91112 | −0.63377 | −781.69836 | −2.783 | −781.68007 | −3.026 |
| LD-W3 | −781.92633 | −8.905 | −781.90779 | −2.72335 | −781.70373 | −5.939 | −781.68652 | −7.073 |
| Water | −76.45846 | −76.43716 | −76.43339 |
| Compounds | Donor | Acceptor | E(2) |
|---|---|---|---|
| l-Dopa | LP O7 | π*C1–C2 | 26.47 |
| LP O8 | π*C3–C6 | 23.54 | |
| LP O12 | π*C11–O13 | 41.99 | |
| LP O13 | π*C11–O12 | 34.54 | |
| LP N14 | σ*C10–C11 | 8.80 | |
| DP-1 | LP O7 | π*C1–C2 | 23.95 |
| LP O8 | π*C3–C6 | 20.03 | |
| LP O12 | π*C11–O13 | 18.55 | |
| LP O13 | π*C11–O12 | 19.45 | |
| σ*C10–C11 | 18.32 | ||
| LP N14 | π*C9–C10 | 3.63 | |
| DP-2 | LP O7 | π*C1–C2 | 23.07 |
| LP O8 | π*C3–C6 | 19.27 | |
| LP O12 | π*C11–O13 | 28.99 | |
| LP O13 | π*C11–O12 | 33.59 | |
| σ*C10–C11 | 15.74 | ||
| LP N14 | π*C11–O13 | 8.31 | |
| σ*C10–C11 | 10.78 | ||
| DP-3 | LP O7 | π*C1–C2 | 21.90 |
| LP O8 | π*C3–C6 | 12.90 | |
| LP O12 | π*C11–O13 | 17.88 | |
| LP O13 | π*C11–O12 | 17.42 | |
| π*C10–C11 | 18.50 | ||
| LP N14 | σ*C10–H22 | 10.04 | |
| σ*C10–C11 | 8.63 | ||
| DP-4 | LP O7 | π*C1–C2 | 27.89 |
| LP O8 | π*C3–C6 | 71.04 | |
| LP O12 | π*C11–O13 | 38.99 | |
| LP O13 | π*C11–O12 | 34.28 | |
| σ*C10–C11 | 16.05 | ||
| LP N14 | σ*C10–C11 | 8.96 | |
| Occupancy | Type | Atom | Hybridization coefficients | Energy, au |
|---|---|---|---|---|
| (1.97866) | LP (1) | O7 | s(45.05%) p 1.22(54.91%) | −0.60588 |
| (1.87642) | LP (2) | O7 | s(0.00%) p 1.00(99.94%) | −0.32010 |
| (1.97737) | LP (1) | O12 | s(44.86%) p 1.23(55.11%) | −0.64468 |
| (1.82991) | LP (2) | O12 | s(0.00%) p 1.00(99.94%) | −0.35517 |
| (1.97844) | LP (1) | O13 | s(58.78%) p 0.70(41.20%) | −0.71385 |
| (1.94184) | LP (1) | N14 | s(18.42%) p 4.43(81.53%) | −0.31638 |
| (1.98704) | BD (1) | O7-H18 | O(74.69%) H(25.31%) | −0.73784 |
| (1.98734) | BD (1) | O12-H23 | O(74.86%) H(25.14%) | −0.77110 |
| (1.98866) | BD (1) | N14-H24 | N(68.12%) H(31.88%) | −0.62658 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Elroby, S.A.K.; Makki, M.S.I.; Sobahi, T.R.; Hilal, R.H. Toward the Understanding of the Metabolism of Levodopa I. DFT Investigation of the Equilibrium Geometries, Acid-Base Properties and Levodopa-Water Complexes. Int. J. Mol. Sci. 2012, 13, 4321-4339. https://doi.org/10.3390/ijms13044321
Elroby SAK, Makki MSI, Sobahi TR, Hilal RH. Toward the Understanding of the Metabolism of Levodopa I. DFT Investigation of the Equilibrium Geometries, Acid-Base Properties and Levodopa-Water Complexes. International Journal of Molecular Sciences. 2012; 13(4):4321-4339. https://doi.org/10.3390/ijms13044321
Chicago/Turabian StyleElroby, Shabaan A. K., Mohamed S. I. Makki, Tariq R. Sobahi, and Rifaat H. Hilal. 2012. "Toward the Understanding of the Metabolism of Levodopa I. DFT Investigation of the Equilibrium Geometries, Acid-Base Properties and Levodopa-Water Complexes" International Journal of Molecular Sciences 13, no. 4: 4321-4339. https://doi.org/10.3390/ijms13044321
APA StyleElroby, S. A. K., Makki, M. S. I., Sobahi, T. R., & Hilal, R. H. (2012). Toward the Understanding of the Metabolism of Levodopa I. DFT Investigation of the Equilibrium Geometries, Acid-Base Properties and Levodopa-Water Complexes. International Journal of Molecular Sciences, 13(4), 4321-4339. https://doi.org/10.3390/ijms13044321