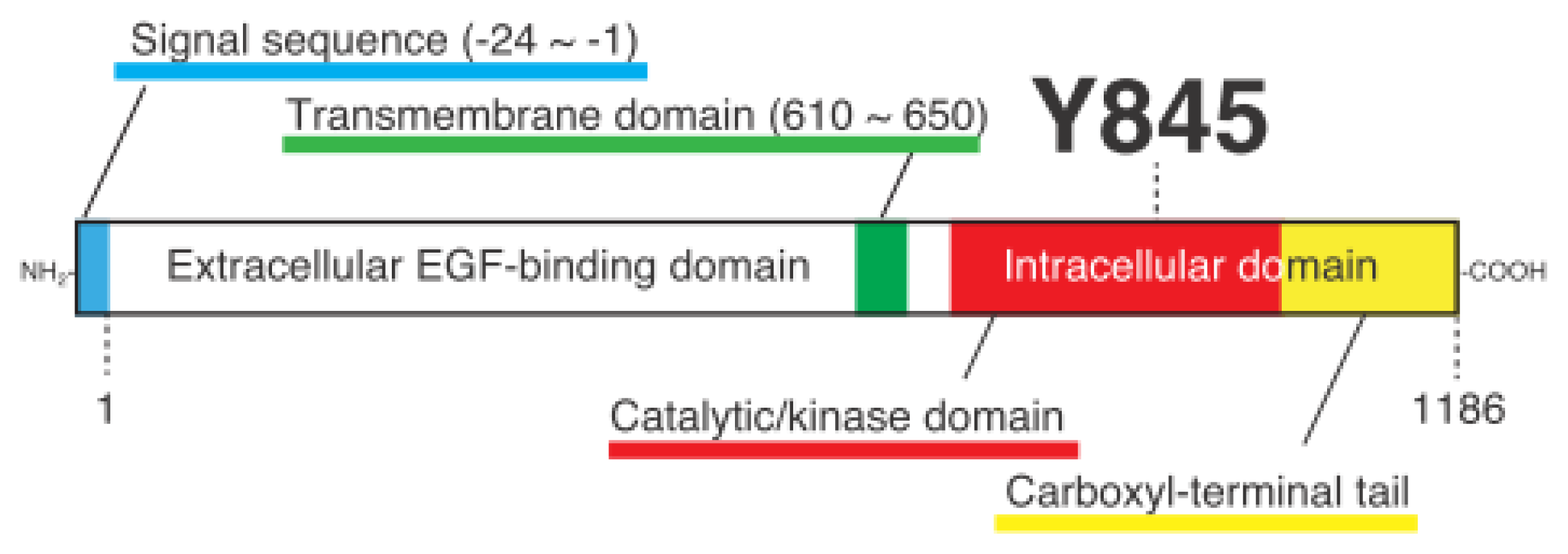
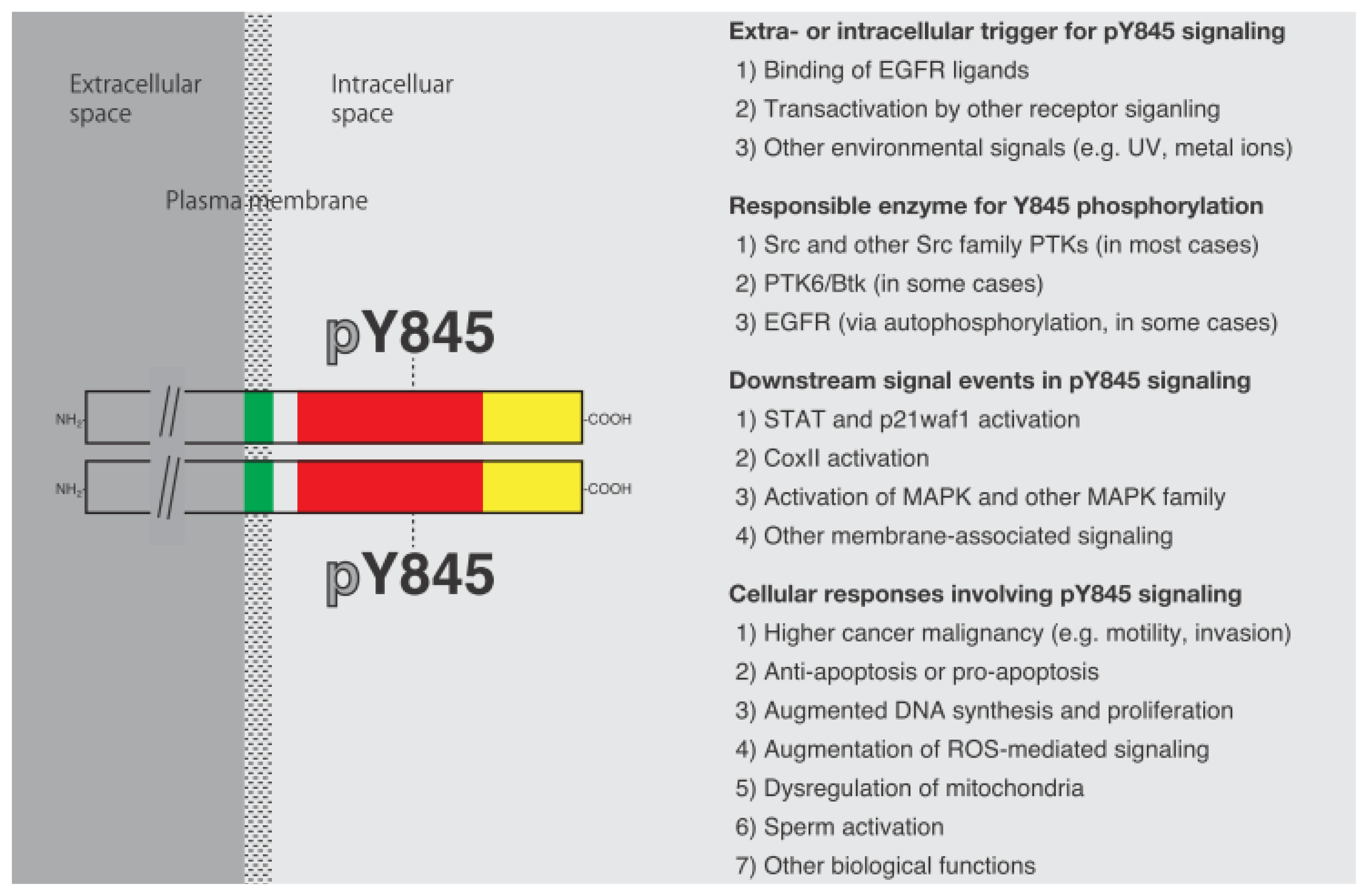
Cellular Functions Regulated by Phosphorylation of EGFR on Tyr845

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Discovery of Src and EGFR, and Their Crosstalk
1.2. Auto- and Trans-Phosphorylation of EGFR
1.3. Discovery of Y845 Phosphorylation of EGFR by Src
2. Y845 Phosphorylation and Cancer Cell Functions
2.1. Cooperation of EGFR and Src in Cancer Cells Involving Y845 Phosphorylation
2.2. Y845 Phosphorylation in Several Types of Cancer Cells
2.3. Y845 Phosphorylation as a Diagnostic Marker for Cancer Treatment
3. Y845 Phosphorylation in a Variety of Cellular Functions
3.1. Y845 Phosphorylation in Transactivated EGFR
3.2. Reactive Oxygen Species and Y845 Phosphorylation
3.3. Involvement of Y845 Phosphorylation in Cell Cycle Control and Cell Viability
3.4. Cell Adhesion Signaling and Y845 Phosphorylation
3.5. Sperm Functions and Y845 Phosphorylation
3.6. Other Cellular Functions and Y845 Phosphorylation
4. Molecular Insights into Y845 Phosphorylation and Its Applications
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol 2009, 21, 140–146. [Google Scholar]
- Jove, R.; Hanafusa, H. Cell transformation by the viral src oncogene. Annu. Rev. Cell Biol 1987, 3, 31–56. [Google Scholar]
- Abram, C.L.; Courtneidge, S.A. Src family tyrosine kinases and growth factor signaling. Exp. Cell Res 2000, 254, 1–13. [Google Scholar]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol 1997, 13, 513–609. [Google Scholar]
- Brown, M.T.; Cooper, J.A. Regulation, substrates and functions of Src. Biochim. Biophys. Acta 1996, 1287, 121–149. [Google Scholar]
- Downward, J.; Yarden, Y.; Mayes, E.; Scrace, G.; Totty, N.; Stockwell, P.; Ullrich, A.; Schlessinger, J.; Waterfield, M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 1984, 307, 521–527. [Google Scholar]
- Carpenter, G.; Cohen, S. Epidermal growth factor. J. Biol. Chem 1990, 265, 7709–7712. [Google Scholar]
- Yamamoto, T.; Hihara, H.; Nishida, T.; Kawai, S.; Toyoshima, K. A new avian erythroblastosis virus, AEV-H, carries erbB gene responsible for the induction of both erythroblastosis and sarcomas. Cell 1983, 34, 225–232. [Google Scholar]
- Hackel, P.O.; Zwick, E.; Prenzel, N.; Ullrich, A. Epidermal growth factor receptors: Critical mediators of multiple receptor pathways. Curr. Opin. Cell Biol 1999, 11, 184–189. [Google Scholar]
- Kypta, R.M.; Goldberg, Y.; Ulug, E.T.; Courtneidge, S.A. Association between the PDGF receptor and members of the src family of tyrosine kinases. Cell 1990, 62, 481–492. [Google Scholar]
- Kremer, N.E.; D’Arcangelo, G.; Thomas, S.M.; DeMarco, M.; Brugge, J.S.; Halegoua, S. Signal transduction by nerve growth factor and fibroblast growth factor in PC12 cells requires a sequence of src and ras actions. J. Cell Biol 1991, 115, 809–819. [Google Scholar]
- Courtneidge, S.A.; Dhand, R.; Pilat, D.; Twamley, G.M.; Waterfield, M.D.; Roussel, M.F. Activation of Src family kinases by colony stimulating factor-1, and their association with its receptor. EMBO J 1993, 12, 943–950. [Google Scholar]
- Zhan, X.; Plourde, C.; Hu, X.; Friesel, R.; Maciag, T. Association of fibroblast growth factor receptor-1 with c-Src correlates with association between c-Src and cortactin. J. Biol. Chem 1994, 269, 20221–20224. [Google Scholar]
- Muthuswamy, S.K.; Siegel, P.M.; Dankort, D.L.; Webster, M.A.; Muller, W.J. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol. Cell. Biol 1994, 14, 735–743. [Google Scholar]
- Luttrell, D.K.; Luttrell, L.M.; Parsons, S.J. Augmented mitogenic responsiveness to epidermal growth factor in murine fibroblasts that overexpress pp60c-src. Mol. Cell. Biol 1988, 8, 497–501. [Google Scholar]
- Carpenter, G. The biochemistry and physiology of the receptor-kinase for epidermal growth factor. Mol. Cell. Endocrinol 1983, 31, 1–19. [Google Scholar]
- Rozakis-Adcock, M.; Fernley, R.; Wade, J.; Pawson, T.; Bowtell, D. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 1993, 363, 83–85. [Google Scholar]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun 1995, 215, 1078–1087. [Google Scholar]
- Biscardi, J.S.; Maa, M.C.; Tice, D.A.; Cox, M.E.; Leu, T.H.; Parsons, S.J. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem 1999, 274, 8335–8343. [Google Scholar]
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420. [Google Scholar]
- Downward, J.; Parker, P.; Waterfield, M.D. Autophosphorylation sites on the epidermal growth factor receptor. Nature 1984, 311, 483–485. [Google Scholar]
- Gill, G.N.; Bertics, P.J.; Santon, J.B. Epidermal growth factor and its receptor. Mol. Cell. Endocrinol 1987, 51, 169–186. [Google Scholar]
- Gill, G.N.; Santon, J.B.; Bertics, P.J. Regulatory features of the epidermal growth factor receptor. J. Cell Physiol. Suppl 1987, 5, 35–41. [Google Scholar]
- Margolis, B.L.; Lax, I.; Kris, R.; Dombalagian, M.; Honegger, A.M.; Howk, R.; Givol, D.; Ullrich, A.; Schlessinger, J. All autophosphorylation sites of epidermal growth factor (EGF) receptor and HER2/neu are located in their carboxyl-terminal tails. Identification of a novel site in EGF receptor. J. Biol. Chem 1989, 264, 10667–10671. [Google Scholar]
- Walton, G.M.; Chen, W.S.; Rosenfeld, M.G.; Gill, G.N. Analysis of deletions of the carboxyl terminus of the epidermal growth factor receptor reveals self-phosphorylation at tyrosine 992 and enhanced in vivo tyrosine phosphorylation of cell substrates. J. Biol. Chem 1990, 265, 1750–1754. [Google Scholar]
- Stover, D.R.; Becker, M.; Liebetanz, J.; Lydon, N.B. Src phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85 α. J. Biol. Chem 1995, 270, 15591–15597. [Google Scholar]
- Ravid, T.; Sweeney, C.; Gee, P.; Carraway, K.L., III; Goldkorn, T. Epidermal growth factor receptor activation under oxidative stress fails to promote c-Cbl mediated down-regulation. J. Biol. Chem. 2002, 277, 31214–31219. [Google Scholar]
- Wright, J.D.; Reuter, C.W.; Weber, M.J. Identification of sites on epidermal growth factor receptors which are phosphorylated by pp60src in vitro. Biochim. Biophys. Acta 1996, 1312, 85–93. [Google Scholar]
- Hunter, T.; Ling, N.; Cooper, J.A. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature 1984, 311, 480–483. [Google Scholar]
- Heisermann, G.J.; Gill, G.N. Epidermal growth factor receptor threonine and serine residues phosphorylated in vivo. J. Biol. Chem 1988, 263, 13152–13158. [Google Scholar]
- Kuppuswamy, D.; Dalton, M.; Pike, L.J. Serine 1002 is a site of in vivo and in vitro phosphorylation of the epidermal growth factor receptor. J. Biol. Chem 1993, 268, 19134–19142. [Google Scholar]
- Fukami, Y.; Tokmakov, A.A.; Konaka, K.; Sato, K. Peptide inhibitors of the mitogen-activated protein kinase pathway: A structure-mimetic peptide corresponding to the conserved inter-DFG-APE region in the kinase domain. Pharmacol. Ther 1999, 82, 399–407. [Google Scholar]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar]
- Cola, C.; Brunati, A.M.; Borin, G.; Ruzza, P.; Calderan, A.; de Castiglione, R.; Pinna, L.A. Synthetic peptides reproducing the EGF-receptor segment homologous to the pp60v-src phosphoacceptor site. Phosphorylation by tyrosine protein kinases. Biochim. Biophys. Acta 1989, 1012, 191–195. [Google Scholar]
- Gotoh, N.; Tojo, A.; Hino, M.; Yazaki, Y.; Shibuya, M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun 1992, 186, 768–774. [Google Scholar]
- Wilson, L.K.; Luttrell, D.K.; Parsons, J.T.; Parsons, S.J. pp60c-src tyrosine kinase, myristylation, and modulatory domains are required for enhanced mitogenic responsiveness to epidermal growth factor seen in cells overexpressing c-. src. Mol. Cell. Biol 1989, 9, 1536–1544. [Google Scholar]
- Wilson, L.K.; Parsons, S.J. Enhanced EGF mitogenic response is associated with enhanced tyrosine phosphorylation of specific cellular proteins in fibroblasts overexpressing c-src. Oncogene 1990, 5, 1471–1480. [Google Scholar]
- Wasilenko, W.J.; Payne, D.M.; Fitzgerald, D.L.; Weber, M.J. Phosphorylation and activation of epidermal growth factor receptors in cells transformed by the src oncogene. Mol. Cell. Biol 1991, 11, 309–321. [Google Scholar]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. Site-specific association of c-Src with epidermal growth factor receptor in A431 cells. Biochem. Biophys. Res. Commun 1995, 210, 844–851. [Google Scholar]
- Fukami, Y.; Sato, K.; Ikeda, K.; Kamisango, K.; Koizumi, K.; Matsuno, T. Evidence for autoinhibitory regulation of the c-src gene product. A possible interaction between the src homology 2 domain and autophosphorylation site. J. Biol. Chem 1993, 268, 1132–1140. [Google Scholar]
- Maa, M.C.; Leu, T.H.; McCarley, D.J.; Schatzman, R.C.; Parsons, S.J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc. Natl. Acad. Sci. USA 1995, 92, 6981–6985. [Google Scholar]
- Belsches, A.P.; Haskell, M.D.; Parsons, S.J. Role of c-Src tyrosine kinase in EGF-induced mitogenesis. Front. Biosci 1997, 2, d501–d518. [Google Scholar]
- Biscardi, J.S.; Belsches, A.P.; Parsons, S.J. Characterization of human epidermal growth factor receptor and c-Src interactions in human breast tumor cells. Mol. Carcinog 1998, 21, 261–272. [Google Scholar]
- Biscardi, J.S.; Ishizawar, R.C.; Silva, C.M.; Parsons, S.J. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res 2000, 2, 203–210. [Google Scholar]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar]
- Kloth, M.T.; Laughlin, K.K.; Biscardi, J.S.; Boerner, J.L.; Parsons, S.J.; Silva, C.M. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J. Biol. Chem 2003, 278, 1671–1679. [Google Scholar]
- Sato, K.; Nagao, T.; Kakumoto, M.; Kimoto, M.; Otsuki, T.; Iwasaki, T.; Tokmakov, A.A.; Owada, K.; Fukami, Y. Adaptor protein Shc is an isoform-specific direct activator of the tyrosine kinase c-Src. J. Biol. Chem 2002, 277, 29568–29576. [Google Scholar]
- Sato, K.; Nagao, T.; Iwasaki, T.; Nishihira, Y.; Fukami, Y. Src-dependent phosphorylation of the EGF receptor Tyr-845 mediates Stat-p21waf1 pathway in A431 cells. Genes Cells 2003, 8, 995–1003. [Google Scholar]
- Boerner, J.L.; Biscardi, J.S.; Silva, C.M.; Parsons, S.J. Transactivating agonists of the EGF receptor require Tyr 845 phosphorylation for induction of DNA synthesis. Mol. Carcinog 2005, 44, 262–273. [Google Scholar]
- Haskell, M.D.; Slack, J.K.; Parsons, J.T.; Parsons, S.J. c-Src tyrosine phosphorylation of epidermal growth factor receptor, P190 RhoGAP, and focal adhesion kinase regulates diverse cellular processes. Chem. Rev 2001, 101, 2425–2440. [Google Scholar]
- Sato, K.; Kimoto, M.; Kakumoto, M.; Horiuchi, D.; Iwasaki, T.; Tokmakov, A.A.; Fukami, Y. Adaptor protein Shc undergoes translocation and mediates up-regulation of the tyrosine kinase c-Src in EGF-stimulated A431 cells. Genes Cells 2000, 5, 749–764. [Google Scholar]
- Xi, G.; Shen, X.; Clemmons, D.R. p66shc inhibits insulin-like growth factor-I signaling via direct binding to Src through its polyproline and Src homology 2 domains, resulting in impairment of Src kinase activation. J. Biol. Chem 2010, 285, 6937–6951. [Google Scholar]
- MacLeod, C.L.; Luk, A.; Castagnola, J.; Cronin, M.; Mendelsohn, J. EGF induces cell cycle arrest of A431 human epidermoid carcinoma cells. J. Cell. Physiol 1986, 127, 175–182. [Google Scholar]
- Gulli, L.F.; Palmer, K.C.; Chen, Y.Q.; Reddy, K.B. Epidermal growth factor-induced apoptosis in A431 cells can be reversed by reducing the tyrosine kinase activity. Cell Growth Differ 1996, 7, 173–178. [Google Scholar]
- Boerner, J.L.; Demory, M.L.; Silva, C.; Parsons, S.J. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol. Cell. Biol 2004, 24, 7059–7071. [Google Scholar]
- Demory, M.L.; Boerner, J.L.; Davidson, R.; Faust, W.; Miyake, T.; Lee, I.; Hüttemann, M.; Douglas, R.; Haddad, G.; Parsons, S.J. Epidermal growth factor receptor translocation to the mitochondria: Regulation and effect. J. Biol. Chem 2009, 284, 36592–36604. [Google Scholar]
- Arachiche, A.; Augereau, O.; Decossas, M.; Pertuiset, C.; Gontier, E.; Letellier, T.; Dachary-Prigent, J. Localization of PTP-1B, SHP-2, and Src exclusively in rat brain mitochondria and functional consequences. J. Biol. Chem 2008, 283, 24406–24411. [Google Scholar]
- Hebert-Chatelain, E.; Jose, C.; Gutierrez Cortes, N.; Dupuy, J.W.; Rocher, C.; Dachary-Prigent, J.; Letellier, T. Preservation of NADH ubiquinone-oxidoreductase activity by Src kinase-mediated phosphorylation of NDUFB10. Biochim. Biophys. Acta 2012, 1817, 718–725. [Google Scholar]
- Miyazaki, T.; Neff, L.; Tanaka, S.; Horne, W.C.; Baron, R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J. Cell Biol 2003, 160, 709–718. [Google Scholar]
- Itoh, S.; Lemay, S.; Osawa, M.; Che, W.; Duan, Y.; Tompkins, A.; Brookes, P.S.; Sheu, S.S.; Abe, J. Mitochondrial Dok-4 recruits Src kinase and regulates NF-kappaB activation in endothelial cells. J. Biol. Chem 2005, 280, 26383–26396. [Google Scholar]
- Salvi, M.; Brunati, A.M.; Bordin, L.; la Rocca, N.; Clari, G.; Toninello, A. Characterization and location of Src-dependent tyrosine phosphorylation in rat brain mitochondria. Biochim. Biophys. Acta 2002, 1589, 181–195. [Google Scholar]
- Tibaldi, E.; Brunati, A.M.; Massimino, M.L.; Stringaro, A.; Colone, M.; Agostinelli, E.; Arancia, G.; Toninello, A. Src-Tyrosine kinases are major agents in mitochondrial tyrosine phosphorylation. J. Cell. Biochem 2008, 104, 840–849. [Google Scholar]
- Tibaldi, E.; Venerando, A.; Zonta, F.; Bidoia, C.; Magrin, E.; Marin, O.; Toninello, A.; Bordin, L.; Martini, V.; Pagano, M.A.; et al. Interaction between the SH3 domain of Src family kinases and the proline-rich motif of HTLV-1 p13: A novel mechanism underlying delivery of Src family kinases to mitochondria. Biochem. J 2011, 439, 505–516. [Google Scholar]
- Hebert-Chatelain, E. Src kinases are important regulators of mitochondrial functions. Int. J. Biochem. Cell Biol. 2013, 45, 90–98. [Google Scholar]
- Ogura, M.; Yamaki, J.; Homma, M.K.; Homma, Y. Mitochondrial c-Src regulates cell survival through phosphorylation of respiratory chain components. Biochem. J 2012, 447, 281–289. [Google Scholar]
- Cvrljevic, A.N.; Akhavan, D.; Wu, M.; Martinello, P.; Furnari, F.B.; Johnston, A.J.; Guo, D.; Pike, L.; Cavenee, W.K.; Scott, A.M.; et al. Activation of Src induces mitochondrial localisation of de2–7EGFR (EGFRvIII) in glioma cells: Implications for glucose metabolism. J. Cell Sci 2011, 124, 2938–2950. [Google Scholar]
- Miyake, T.; Parsons, S.J. Functional interactions between choline kinase α, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene 2012, 31, 1431–1441. [Google Scholar]
- Kannangai, R.; Sahin, F.; Torbenson, M.S. EGFR is phosphorylated at Ty845 in hepatocellular carcinoma. Mod. Pathol 2006, 19, 1456–1461. [Google Scholar]
- Jung, O.; Choi, Y.J.; Kwak, T.K.; Kang, M.; Lee, M.S.; Ryu, J.; Kim, H.J.; Lee, J.W. The COOH-terminus of TM4SF5 in hepatoma cell lines regulates c-Src to form invasive protrusions via EGFR Tyr845 phosphorylation. Biochim. Biophys. Acta 2013, 1833, 629–642. [Google Scholar]
- Kim, M.; Jung, J.; Lee, K. Roles of ERK, PI3 kinase, and PLCγ pathways induced by overexpression of translationally controlled tumor protein in HeLa cells. Arch. Biochem. Biophys 2009, 485, 82–87. [Google Scholar]
- Jung, J.; Kim, H.Y.; Kim, M.; Sohn, K.; Kim, M.; Lee, K. Translationally controlled tumor protein induces human breast epithelial cell transformation through the activation of Src. Oncogene 2011, 30, 2264–2274. [Google Scholar]
- Vieira, A.V.; Lamaze, C.; Schmid, S.L. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 1996, 274, 2086–2089. [Google Scholar]
- Ware, M.F.; Tice, D.A.; Parsons, S.J.; Lauffenburger, D.A. Overexpression of cellular Src in fibroblasts enhances endocytic internalization of epidermal growth factor receptor. J. Biol. Chem 1997, 272, 30185–30190. [Google Scholar]
- Medts, T.; de Diesbach, P.; Cominelli, A.; N’Kuli, F.; Tyteca, D.; Courtoy, P.J. Acute ligand-independent Src activation mimics low EGF-induced EGFR surface signalling and redistribution into recycling endosomes. Exp. Cell Res 2010, 316, 3239–3253. [Google Scholar]
- Hu, J.; Jo, M.; Cavenee, W.K.; Furnari, F.; VandenBerg, S.R.; Gonias, S.L. Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc. Natl. Acad. Sci. USA 2011, 108, 15984–15989. [Google Scholar]
- Huang, P.H.; Miraldi, E.R.; Xu, A.M.; Kundukulam, V.A.; Del Rosario, A.M.; Flynn, R.A.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Phosphotyrosine signaling analysis of site-specific mutations on EGFRvIII identifies determinants governing glioblastoma cell growth. Mol. Biosyst 2010, 6, 1227–1237. [Google Scholar]
- Ishizawar, R.C.; Miyake, T.; Parsons, S.J. c-Src modulates ErbB2 and ErbB3 heterocomplex formation and function. Oncogene 2007, 26, 3503–3510. [Google Scholar]
- Gschwantler-Kaulich, D.; Hudelist, G.; Koestler, W.J.; Czerwenka, K.; Mueller, R.; Helmy, S.; Ruecklinger, E.; Kubista, E.; Singer, C.F. EGFR activity in HER-2 over-expressing metastatic breast cancer: Evidence for simultaneous phosphorylation of Her-2/neu and EGFR. Oncol. Rep 2005, 14, 305–311. [Google Scholar]
- Chen, Y.R.; Fu, Y.N.; Lin, C.H.; Yang, S.T.; Hu, S.F.; Chen, Y.T.; Tsai, S.F.; Huang, S.F. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene 2006, 25, 1205–1215. [Google Scholar]
- Koizumi, F.; Shimoyama, T.; Taguchi, F.; Saijo, N.; Nishio, K. Establishment of a human non-small cell lung cancer cell line resistant to gefitinib. Int. J. Cancer 2005, 116, 36–44. [Google Scholar]
- Chung, B.M.; Dimri, M.; George, M.; Reddi, A.L.; Chen, G.; Band, V.; Band, H. The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene 2009, 28, 1821–1832. [Google Scholar]
- Chun, P.Y.; Feng, F.Y.; Scheurer, A.M.; Davis, M.A.; Lawrence, T.S.; Nyati, M.K. Synergistic effects of gemcitabine and gefitinib in the treatment of head and neck carcinoma. Cancer Res 2006, 66, 981–988. [Google Scholar]
- Endoh, H.; Ishibashi, Y.; Yamaki, E.; Yoshida, T.; Yajima, T.; Kimura, H.; Kosaka, T.; Onozato, R.; Tanaka, S.; Mitsudomi, T.; et al. Immunohistochemical analysis of phosphorylated epidermal growth factor receptor might provide a surrogate marker of EGFR mutation. Lung Cancer 2009, 63, 241–246. [Google Scholar]
- Fink-Retter, A.; Gschwantler-Kaulich, D.; Hudelist, G.; Mueller, R.; Kubista, E.; Czerwenka, K.; Singer, C.F. Differential spatial expression and activation pattern of EGFR and HER2 in human breast cancer. Oncol. Rep 2007, 18, 299–304. [Google Scholar]
- Sonnweber, B.; Dlaska, M.; Skvortsov, S.; Dirnhofer, S.; Schmid, T.; Hilbe, W. High predictive value of epidermal growth factor receptor phosphorylation but not of EGFRvIII mutation in resected stage I non-small cell lung cancer (NSCLC). J. Clin. Pathol 2006, 59, 255–259. [Google Scholar]
- Fu, Y.N.; Yeh, C.L.; Cheng, H.H.; Yang, C.H.; Tsai, S.F.; Huang, S.F.; Chen, Y.R. EGFR mutants found in non-small cell lung cancer show different levels of sensitivity to suppression of Src: Implications in targeting therapy. Oncogene 2008, 27, 957–965. [Google Scholar]
- Mueller, K.L.; Hunter, L.A.; Ethier, S.P.; Boerner, J.L. Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res 2008, 68, 3314–3322. [Google Scholar]
- Yamamoto, N.; Mammadova, G.; Song, R.X.; Fukami, Y.; Sato, K. Tyrosine phosphorylation of p145met mediated by EGFR and Src is required for serum-independent survival of human bladder carcinoma cells. J. Cell Sci 2006, 119, 4623–4633. [Google Scholar]
- Kihira, S.; Yoshida, J.; Kawada, Y.; Hitomi, Y.; Asada, T.; Hisatomi, R.; Ohta, A.; Iwasaki, T.; Mahbub Hasan, A.K.; Fukami, Y.; et al. Membrane microdomain-associated uroplakin IIIa contributes to Src-dependent mechanisms of anti-apoptotic proliferation in human bladder carcinoma cells. Biol. Open 2012, 1, 1024–1034. [Google Scholar]
- Reznik, T.E.; Sang, Y.; Ma, Y.; Abounader, R.; Rosen, E.M.; Xia, S.; Laterra, J. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol. Cancer Res 2008, 6, 139–150. [Google Scholar]
- Knowlden, J.M.; Hutcheson, I.R.; Barrow, D.; Gee, J.M.; Nicholson, R.I. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: A supporting role to the epidermal growth factor receptor. Endocrinology 2005, 146, 4609–4618. [Google Scholar]
- Hall, E.H.; Gurel, V.; Dahlberg, A.E.; McMichael, J.; Brautigan, D.L. Inhibition of human breast cancer matrigel invasion by streptolysin O activation of the EGF receptor ErbB1. Cell Signal 2011, 23, 1972–1977. [Google Scholar]
- Iwasaki, T.; Mukai, M.; Tsujimura, T.; Tatsuta, M.; Nakamura, H.; Terada, N.; Akedo, H. Ipriflavone inhibits osteolytic bone metastasis of human breast cancer cells in a nude mouse model. Int. J. Cancer 2002, 100, 381–387. [Google Scholar]
- Aquino, G.; Pannone, G.; Santoro, A.; Liguori, G.; Franco, R.; Serpico, R.; Florio, G.; de Rosa, A.; Mattoni, M.; Cozza, V.; et al. pEGFR-Tyr 845 expression as prognostic factors in oral squamous cell carcinoma: A tissue-microarray study with clinic-pathological correlations. Cancer Biol. Ther 2012, 13, 967–977. [Google Scholar]
- Dhupkar, P.; Dowling, M.; Cengel, K.; Chen, B. Effects of anti-EGFR antibody cetuximab on androgen-independent prostate cancer cells. Anticancer Res 2010, 30, 1905–1910. [Google Scholar]
- Jones, H.E.; Gee, J.M.; Barrow, D.; Tonge, D.; Holloway, B.; Nicholson, R.I. Inhibition of insulin receptor isoform-A signalling restores sensitivity to gefitinib in previously de novo resistant colon cancer cells. Br. J. Cancer 2006, 95, 172–180. [Google Scholar]
- Polychronis, A.; Sinnett, H.D.; Hadjiminas, D.; Singhal, H.; Mansi, J.L.; Shivapatham, D.; Shousha, S.; Jiang, J.; Peston, D.; Barrett, N.; et al. Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: A double-blind placebo-controlled phase II randomised trial. Lancet Oncol 2005, 6, 383–391. [Google Scholar]
- Kandala, P.K.; Wright, S.E.; Srivastava, S.K. Blocking epidermal growth factor receptor activation by 3,3′-diindolylmethane suppresses ovarian tumor growth in vitro and in vivo. J. Pharmacol. Exp. Ther 2012, 341, 24–32. [Google Scholar]
- Kong, L.; Deng, Z.; Shen, H.; Zhang, Y. Src family kinase inhibitor PP2 efficiently inhibits cervical cancer cell proliferation through down-regulating phospho-Src-Y416 and phospho-EGFR-Y1173. Mol. Cell Biochem 2011, 348, 11–19. [Google Scholar]
- Li, Z.; Hosoi, Y.; Cai, K.; Tanno, Y.; Matsumoto, Y.; Enomoto, A.; Morita, A.; Nakagawa, K.; Miyagawa, K. Src tyrosine kinase inhibitor PP2 suppresses ERK1/2 activation and epidermal growth factor receptor transactivation by X-irradiation. Biochem. Biophys. Res. Commun 2006, 341, 363–368. [Google Scholar]
- Maruko, A.; Ohtake, Y.; Kawaguchi, M.; Kobayashi, T.; Baba, T.; Kuwahara, Y.; Nakagawa, H.; Shimura, T.; Fukumoto, M.; Ohkubo, Y. X-radiation-induced down-regulation of the EGF receptor in primary cultured rat hepatocytes. Radiat. Res 2010, 173, 620–628. [Google Scholar]
- Lu, Y.; Li, X.; Liang, K.; Luwor, R.; Siddik, Z.H.; Mills, G.B.; Mendelsohn, J.; Fan, Z. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res 2007, 67, 8240–8247. [Google Scholar]
- Li, X.; Lu, Y.; Liang, K.; Hsu, J.M.; Albarracin, C.; Mills, G.B.; Hung, M.C.; Fan, Z. Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene 2012, 31, 4372–4383. [Google Scholar]
- Morgan, M.A.; Parsels, L.A.; Kollar, L.E.; Normolle, D.P.; Maybaum, J.; Lawrence, T.S. The combination of epidermal growth factor receptor inhibitors with gemcitabine and radiation in pancreatic cancer. Clin. Cancer Res 2008, 14, 5142–5149. [Google Scholar]
- Selvendiran, K.; Bratasz, A.; Tong, L.; Ignarro, L.J.; Kuppusamy, P. NCX-4016, a nitro-derivative of aspirin, inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in cisplatin-resistant human ovarian cancer cells and xenografts. Cell Cycle 2008, 7, 81–88. [Google Scholar]
- Narayanan, B.A.; Reddy, B.S.; Bosland, M.C.; Nargi, D.; Horton, L.; Randolph, C.; Narayanan, N.K. Exisulind in combination with celecoxib modulates epidermal growth factor receptor, cyclooxygenase-2, and cyclin D1 against prostate carcinogenesis: In vivo evidence. Clin. Cancer Res 2007, 13, 5965–5973. [Google Scholar]
- Hudelist, G.; Köstler, W.J.; Czerwenka, K.; Kubista, E.; Attems, J.; Müller, R.; Gschwantler-Kaulich, D.; Manavi, M.; Huber, I.; Hoschützky, H.; et al. Her-2/neu and EGFR tyrosine kinase activation predict the efficacy of trastuzumab-based therapy in patients with metastatic breast cancer. Int. J. Cancer 2006, 118, 1126–1134. [Google Scholar]
- Thelemann, A.; Petti, F.; Griffin, G.; Iwata, K.; Hunt, T.; Settinari, T.; Fenyo, D.; Gibson, N.; Haley, J.D. Phosphotyrosine signaling networks in epidermal growth factor receptor overexpressing squamous carcinoma cells. Mol. Cell Proteomics 2005, 4, 356–376. [Google Scholar]
- Fischgräbe, J.; Götte, M.; Michels, K.; Kiesel, L.; Wülfing, P. Targeting endothelin A receptor enhances anti-proliferative and anti-invasive effects of the HER2 antibody trastuzumab in HER2-overexpressing breast cancer cells. Int. J. Cancer 2010, 127, 696–706. [Google Scholar]
- Park, Y.J.; Lee, H.; Lee, J.H. Macrophage inhibitory cytokine-1 transactivates ErbB family receptors via the activation of Src in SK-BR-3 human breast cancer cells. BMB Rep 2010, 43, 91–96. [Google Scholar]
- Drube, S.; Stirnweiss, J.; Valkova, C.; Liebmann, C. Ligand-independent and EGF receptor-supported transactivation: Lessons from β2-adrenergic receptor signalling. Cell Signal 2006, 18, 1633–1646. [Google Scholar]
- Du, T.; Li, B.; Li, H.; Li, M.; Hertz, L.; Peng, L. Signaling pathways of isoproterenol-induced ERK1/2 phosphorylation in primary cultures of astrocytes are concentration-dependent. J. Neurochem 2010, 115, 1007–1023. [Google Scholar]
- Du, T.; Li, B.; Liu, S.; Zang, P.; Prevot, V.; Hertz, L.; Peng, L. ERK phosphorylation in intact, adult brain by α2-adrenergic transactivation of EGF receptors. Neurochem. Int 2009, 55, 593–600. [Google Scholar]
- Amos, S.; Martin, P.M.; Polar, G.A.; Parsons, S.J.; Hussaini, I.M. Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase Cδ/c-Src pathways in glioblastoma cells. J. Biol. Chem 2005, 280, 7729–7738. [Google Scholar]
- Lin, C.R.; Chen, W.S.; Lazar, C.S.; Carpenter, C.D.; Gill, G.N.; Evans, R.M.; Rosenfeld, M.G. Protein kinase C phosphorylation at Thr 654 of the unoccupied EGF receptor and EGF binding regulate functional receptor loss by independent mechanisms. Cell 1986, 44, 839–848. [Google Scholar]
- Van Baal, J.; de Widt, J.; Divecha, N.; van Blitterswijk, W.J. Diacylglycerol kinase θ counteracts protein kinase C-mediated inactivation of the EGF receptor. Int. J. Biochem. Cell Biol 2012, 44, 1791–1799. [Google Scholar]
- Reinehr, R.; Sommerfeld, A.; Häussinger, D. Insulin induces swelling-dependent activation of the epidermal growth factor receptor in rat liver. J. Biol. Chem 2010, 285, 25904–25912. [Google Scholar]
- Chen, J.; Chen, J.K.; Harris, R.C. Angiotensin II induces epithelial-to-mesenchymal transition in renal epithelial cells through reactive oxygen species/Src/caveolin-mediated activation of an epidermal growth factor receptor-extracellular signal-regulated kinase signaling pathway. Mol. Cell. Biol 2012, 32, 981–991. [Google Scholar]
- Kim, J.; Ahn, S.; Rajagopal, K.; Lefkowitz, R.J. Independent β-arrestin2 and Gq/protein kinase Cζ pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J. Biol. Chem 2009, 284, 11953–11962. [Google Scholar]
- Nair, V.D.; Sealfon, S.C. Agonist-specific transactivation of phosphoinositide 3-kinase signaling pathway mediated by the dopamine D2 receptor. J. Biol. Chem 2003, 278, 47053–47061. [Google Scholar]
- Santiskulvong, C.; Rozengurt, E. Galardin (GM 6001), a broad-spectrum matrix metalloproteinase inhibitor, blocks bombesin- and LPA-induced EGF receptor transactivation and DNA synthesis in rat-1 cells. Exp. Cell Res 2003, 290, 437–446. [Google Scholar]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol. Cancer 2008, 7, 69. [Google Scholar]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. The radioprotector Bowman-Birk proteinase inhibitor stimulates DNA repair via epidermal growth factor receptor phosphorylation and nuclear transport. Radiother. Oncol 2008, 86, 375–382. [Google Scholar]
- Iordanov, M.S.; Choi, R.J.; Ryabinina, O.P.; Dinh, T.H.; Bright, R.K.; Magun, B.E. The UV (Ribotoxic) stress response of human keratinocytes involves the unexpected uncoupling of the Ras-extracellular signal-regulated kinase signaling cascade from the activated epidermal growth factor receptor. Mol. Cell. Biol 2002, 22, 5380–5394. [Google Scholar]
- Gardner, O.S.; Dewar, B.J.; Earp, H.S.; Samet, J.M.; Graves, L.M. Dependence of peroxisome proliferator-activated receptor ligand-induced mitogen-activated protein kinase signaling on epidermal growth factor receptor transactivation. J. Biol. Chem 2003, 278, 46261–46269. [Google Scholar]
- Mueller, K.L.; Powell, K.; Madden, J.M.; Eblen, S.T.; Boerner, J.L. EGFR tyrosine 845 phosphorylation-dependent proliferation and transformation of breast cancer cells require activation of p38 MAPK. Transl. Oncol 2012, 5, 327–334. [Google Scholar]
- Samarakoon, R.; Higgins, C.E.; Higgins, S.P.; Higgins, P.J. Differential requirement for MEK/ERK and SMAD signaling in PAI-1 and CTGF expression in response to microtubule disruption. Cell. Signal 2009, 21, 986–995. [Google Scholar]
- Jo, M.; Thomas, K.S.; Marozkina, N.; Amin, T.J.; Silva, C.M.; Parsons, S.J.; Gonias, S.L. Dynamic assembly of the urokinase-type plasminogen activator signaling receptor complex determines the mitogenic activity of urokinase-type plasminogen activator. J. Biol. Chem 2005, 280, 17449–17457. [Google Scholar]
- Jo, M.; Thomas, K.S.; Takimoto, S.; Gaultier, A.; Hsieh, E.H.; Lester, R.D.; Gonias, S.L. Urokinase receptor primes cells to proliferate in response to epidermal growth factor. Oncogene 2007, 26, 2585–2594. [Google Scholar]
- Monaghan-Benson, E.; McKeown-Longo, P.J. Urokinase-type plasminogen activator receptor regulates a novel pathway of fibronectin matrix assembly requiring Src-dependent transactivation of epidermal growth factor receptor. J. Biol. Chem 2006, 281, 9450–9459. [Google Scholar]
- Goldshmit, Y.; Walters, C.E.; Scott, H.J.; Greenhalgh, C.J.; Turnley, A.M. SOCS2 induces neurite outgrowth by regulation of epidermal growth factor receptor activation. J. Biol. Chem 2004, 279, 16349–16355. [Google Scholar]
- Tseng, H.Y.; Liu, Z.M.; Huang, H.S. NADPH oxidase-produced superoxide mediates EGFR transactivation by c-Src in arsenic trioxide-stimulated human keratinocytes. Arch. Toxicol 2012, 86, 935–945. [Google Scholar]
- Rodríguez-Fragoso, L.; Melendez, K.; Hudson, L.G.; Lauer, F.T.; Burchiel, S.W. EGF-receptor phosphorylation and downstream signaling are activated by benzo[a]pyrene 3,6-quinone and benzo[a]pyrene 1,6-quinone in human mammary epithelial cells. Toxicol. Appl. Pharmacol 2009, 235, 321–328. [Google Scholar]
- Martínez Flores, K.; Uribe Marín, B.; Souza Arroyo, V.; Bucio Ortiz, L.; López Reyes, A.; Gómez-Quiroz, L.E.; Rojas del Castillo, E.; Gutiérrezruiz, M. Hepatocytes display a compensatory survival response against cadmium toxicity by a mechanism mediated by EGFR and Src. Toxicol. In Vitro 2013, in press. [Google Scholar]
- Godek, J.; Sargiannidou, I.; Patel, S.; Hurd, L.; Rothman, V.L.; Tuszynski, G.P. Angiocidin inhibits breast cancer proliferation through activation of epidermal growth factor receptor and nuclear factor kappa (NF-κB). Exp. Mol. Pathol 2011, 90, 244–251. [Google Scholar]
- Liu, A.; Garg, P.; Yang, S.; Gong, P.; Pallero, M.A.; Annis, D.S.; Liu, Y.; Passaniti, A.; Mann, D.; Mosher, D.F.; et al. Epidermal growth factor-like repeats of thrombospondins activate phospholipase Cγ and increase epithelial cell migration through indirect epidermal growth factor receptor activation. J. Biol. Chem 2009, 284, 6389–6402. [Google Scholar]
- Westover, E.J.; Covey, D.F.; Brockman, H.L.; Brown, R.E.; Pike, L.J. Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. Studies with cholesterol enantiomers. J. Biol. Chem 2003, 278, 51125–51133. [Google Scholar]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol 1998, 14, 111–136. [Google Scholar]
- Anderson, R.G. The caveolae membrane system. Annu. Rev. Biochem 1998, 67, 199–225. [Google Scholar]
- Mahbub Hasan, A.K.; Fukami, Y.; Sato, K. Gamete membrane microdomains and their associated molecules in fertilization signaling. Mol. Reprod. Dev 2011, 78, 814–830. [Google Scholar]
- Mahbub Hasan, A.K.; Ijiri, T.; Sato, K. Involvement of Src in the adaptation of cancer cells under microenvironmental stresses. J. Signal Transduct 2012, 2012. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar]
- Liu, Y.T.; Song, L.; Templeton, D.M. Heparin suppresses lipid raft-mediated signaling and ligand-independent EGF receptor activation. J. Cell. Physiol 2007, 211, 205–212. [Google Scholar]
- Higgins, S.P.; Samarakoon, R.; Higgins, C.E.; Freytag, J.; Wilkins-Port, C.E.; Higgins, P.J. TGF-β1-induced expression of the anti-apoptotic PAI-1 protein requires EGFR signaling. Cell Commun. Insights 2009, 2, 1–11. [Google Scholar]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. TGF-β1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60c-src/EGFR(Y845) and Rho/ROCK signaling. J. Mol. Cell Cardiol 2008, 44, 527–538. [Google Scholar]
- Kim, J.H.; Xu, C.; Keum, Y.S.; Reddy, B.; Conney, A.; Kong, A.N. Inhibition of EGFR signaling in human prostate cancer PC-3 cells by combination treatment with β-phenylethyl isothiocyanate and curcumin. Carcinogenesis 2006, 27, 475–482. [Google Scholar]
- Amorino, G.P.; Deeble, P.D.; Parsons, S.J. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene 2007, 26, 745–756. [Google Scholar]
- Ray, R.M.; Bhattacharya, S.; Johnson, L.R. EGFR plays a pivotal role in the regulation of polyamine-dependent apoptosis in intestinal epithelial cells. Cell. Signal 2007, 19, 2519–2527. [Google Scholar]
- Reinehr, R.; Becker, S.; Wettstein, M.; Häussinger, D. Involvement of the Src family kinase yes in bile salt-induced apoptosis. Gastroenterology 2004, 127, 1540–1557. [Google Scholar]
- Reinehr, R.; Becker, S.; Eberle, A.; Grether-Beck, S.; Häussinger, D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J. Biol. Chem 2005, 280, 27179–27194. [Google Scholar]
- Wang, Z.; Wang, M.; Carr, B.I. Integrin α5-induced EGFR activation by prothrombin triggers hepatocyte apoptosis via the JNK signaling pathway. J. Cell. Physiol 2008, 216, 551–557. [Google Scholar]
- Moro, L.; Dolce, L.; Cabodi, S.; Bergatto, E.; Boeri Erba, E.; Smeriglio, M.; Turco, E.; Retta, S.F.; Giuffrida, M.G.; Venturino, M.; et al. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem 2002, 277, 9405–9414. [Google Scholar]
- Wang, X.Q.; Sun, P.; Paller, A.S. Ganglioside GM3 blocks the activation of epidermal growth factor receptor induced by integrin at specific tyrosine sites. J. Biol. Chem 2003, 278, 48770–48778. [Google Scholar]
- Balanis, N.; Yoshigi, M.; Wendt, M.K.; Schiemann, W.P.; Carlin, C.R. β3 integrin-EGF receptor cross-talk activates p190RhoGAP in mouse mammary gland epithelial cells. Mol. Biol. Cell 2011, 22, 4288–4301. [Google Scholar]
- Balanis, N.; Carlin, C.R. Mutual cross-talk between fibronectin integrins and the EGF receptor: Molecular basis and biological significance. Cell Logist 2012, 2, 46–51. [Google Scholar]
- Perrais, M.; Chen, X.; Perez-Moreno, M.; Gumbiner, B.M. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell 2007, 18, 2013–2025. [Google Scholar]
- Riggins, R.B.; Thomas, K.S.; Ta, H.Q.; Wen, J.; Davis, R.J.; Schuh, N.R.; Donelan, S.S.; Owen, K.A.; Gibson, M.A.; Shupnik, M.A.; et al. Physical and functional interactions between Cas and c-Src induce tamoxifen resistance of breast cancer cells through pathways involving epidermal growth factor receptor and signal transducer and activator of transcription 5b. Cancer Res 2006, 66, 7007–7015. [Google Scholar]
- Pan, Q.; Qiu, W.Y.; Huo, Y.N.; Yao, Y.F.; Lou, M.F. Low levels of hydrogen peroxide stimulate corneal epithelial cell adhesion, migration, and wound healing. Invest. Ophthalmol. Vis. Sci 2011, 52, 1723–1734. [Google Scholar]
- Daniel, L.; Etkovitz, N.; Weiss, S.R.; Rubinstein, S.; Ickowicz, D.; Breitbart, H. Regulation of the sperm EGF receptor by ouabain leads to initiation of the acrosome reaction. Dev. Biol 2010, 344, 650–657. [Google Scholar]
- Luna, C.; Colás, C.; Pérez-Pé, R.; Cebrián-Pérez, J.A.; Muiño-Blanco, T. A novel epidermal growth factor-dependent extracellular signal-regulated MAP kinase cascade involved in sperm functionality in sheep. Biol. Reprod 2012, 87, 93. [Google Scholar]
- Fassett, J.; Tobolt, D.; Hansen, L.K. Type I collagen structure regulates cell morphology and EGF signaling in primary rat hepatocytes through cAMP-dependent protein kinase A. Mol. Biol. Cell 2006, 17, 345–356. [Google Scholar]
- Luo, L.; Yano, N.; Luo, J.Z. The molecular mechanism of EGF receptor activation in pancreatic β-cells by thyrotropin-releasing hormone. Am. J Physiol. Endocrinol. MeTable 2006, 290, E889–E899. [Google Scholar]
- McEneaney, V.; Harvey, B.J.; Thomas, W. Aldosterone rapidly activates protein kinase D via a mineralocorticoid receptor/EGFR trans-activation pathway in the M1 kidney CCD cell line. J. Steroid Biochem. Mol. Biol 2007, 107, 180–190. [Google Scholar]
- Wu, W.; Graves, L.M.; Gill, G.N.; Parsons, S.J.; Samet, J.M. Src-dependent phosphorylation of the epidermal growth factor receptor on tyrosine 845 is required for zinc-induced Ras activation. J. Biol. Chem 2002, 277, 24252–24257. [Google Scholar]
- Samet, J.M.; Dewar, B.J.; Wu, W.; Graves, L.M. Mechanisms of Zn2+-induced signal initiation through the epidermal growth factor receptor. Toxicol. Appl. Pharmacol 2003, 191, 86–93. [Google Scholar]
- Pontillo, C.A.; García, M.A.; Peña, D.; Cocca, C.; Chiappini, F.; Alvarez, L.; Kleiman de Pisarev, D.; Randi, A.S. Activation of c-Src/HER1/STAT5b and HER1/ERK1/2 signaling pathways and cell migration by hexachlorobenzene in MDA-MB-231 human breast cancer cell line. Toxicol. Sci 2011, 120, 284–296. [Google Scholar]
- Randi, A.S.; Sanchez, M.S.; Alvarez, L.; Cardozo, J.; Pontillo, C.; Kleiman de Pisarev, D.L. Hexachlorobenzene triggers AhR translocation to the nucleus, c-Src activation and EGFR transactivation in rat liver. Toxicol. Lett 2008, 177, 116–122. [Google Scholar]
- Churg, A.; Xie, C.; Wang, X.; Vincent, R.; Wang, R.D. Air pollution particles activate NF-κB on contact with airway epithelial cell surfaces. Toxicol. Appl. Pharmacol 2005, 208, 37–45. [Google Scholar]
- Xu, K.P.; Yin, J.; Yu, F.S. SRC-family tyrosine kinases in wound- and ligand-induced epidermal growth factor receptor activation in human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci 2006, 47, 2832–2839. [Google Scholar]
- Peng, Z.; Raufman, J.P.; Xie, G. Src-mediated cross-talk between farnesoid X and epidermal growth factor receptors inhibits human intestinal cell proliferation and tumorigenesis. PLoS One 2012, 7, e48461. [Google Scholar]
- Xie, G.; Peng, Z.; Raufman, J.P. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol 2012, 302, G1006–G1015. [Google Scholar]
- Allahverdian, S.; Wang, A.; Singhera, G.K.; Wong, B.W.; Dorscheid, D.R. Sialyl Lewis X modification of the epidermal growth factor receptor regulates receptor function during airway epithelial wound repair. Clin. Exp. Allergy 2010, 40, 607–618. [Google Scholar]
- Hyun, S.W.; Anglin, I.E.; Liu, A.; Yang, S.; Sorkin, J.D.; Lillehoj, E.; Tonks, N.K.; Passaniti, A.; Goldblum, S.E. Diverse injurious stimuli reduce protein tyrosine phosphatase-μ expression and enhance epidermal growth factor receptor signaling in human airway epithelia. Exp. Lung Res 2011, 37, 327–343. [Google Scholar]
- Matus, C.E.; Ehrenfeld, P.; Pavicic, F.; Sarmiento, J.M.; Astroza, A.; Sanchez, T.; Salem, C.; Concha, M.; Vidal, M.A.; Gonzalez, C.B.; et al. Activation of kinin B receptor triggers differentiation of cultured human keratinocytes. Br. J. Dermatol 2008, 159, 792–803. [Google Scholar]
- Kansra, S.; Stoll, S.W.; Johnson, J.L.; Elder, J.T. Src family kinase inhibitors block amphiregulin-mediated autocrine ErbB signaling in normal human keratinocytes. Mol. Pharmacol 2005, 67, 1145–1157. [Google Scholar]
- Hur, E.M.; Park, Y.S.; Lee, B.D.; Jang, I.H.; Kim, H.S.; Kim, T.D.; Suh, P.G.; Ryu, S.H.; Kim, K.T. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. Implications for a role of lipid microdomains. J. Biol. Chem 2004, 279, 5852–5860. [Google Scholar]
- Gao, Z.; Yang, J.; Huang, Y.; Yu, Y. N-methyl-N′-nitro-N-nitrosoguanidine interferes with the epidermal growth factor receptor-mediated signaling pathway. Mutat. Res 2005, 570, 175–184. [Google Scholar]
- Iida, M.; Brand, T.M.; Campbell, D.A.; Li, C.; Wheeler, D.L. Yes and Lyn play a role in nuclear translocation of the epidermal growth factor receptor. Oncogene 2012, 32, 759–767. [Google Scholar]
- De Bondt, H.L.; Rosenblatt, J.; Jancark, J.; Jones, H.D.; Morgan, D.O.; Kim, S.-U. Crystal structure of cyclin-dependent kinase 2. Nature 1993, 363, 595–602. [Google Scholar]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massague, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cylin A-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar]
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar]
- Sicheri, F.; Moarefi, I.; Kuriyan, J. Crystal structure of the Src family kinase Hck. Nature 1997, 385, 602–609. [Google Scholar]
- Taylor, S.S.; Yang, J.; Wu, J.; Haste, N.M.; Radzio-Andzelm, E.; Anand, G. PKA: A portrait of protein kinase dynamics. Biochim. Biophys. Acta 2004, 1697, 259–269. [Google Scholar]
- Xu, W.; Harrison, S.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-Src. Nature 1997, 835, 595–602. [Google Scholar]
- Zhang, F.; Strand, A.; Robbins, D.; Cobb, M.H.; Goldsmith, E.J. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature 1994, 367, 704–711. [Google Scholar]
- Choi, S.H.; Mendrola, J.M.; Lemmon, M.A. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene 2007, 26, 1567–1576. [Google Scholar]
- Yang, S.; Park, K.; Turkson, J.; Arteaga, C.L. Ligand-independent phosphorylation of Y869 (Y845) links mutant EGFR signaling to stat-mediated gene expression. Exp. Cell Res 2008, 314, 413–419. [Google Scholar]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar]
- Timms, J.F.; Noble, M.E.; Gregoriou, M. An investigation of the role of Glu-842, Glu-844 and His-846 in the function of the cytoplasmic domain of the epidermal growth factor receptor. Biochem. J 1995, 308, 219–229. [Google Scholar]
- Poppleton, H.M.; Wiepz, G.J.; Bertics, P.J.; Patel, T.B. Modulation of the protein tyrosine kinase activity and autophosphorylation of the epidermal growth factor receptor by its juxtamembrane region. Arch. Biochem. Biophys 1999, 363, 227–236. [Google Scholar]
- Qiu, C.; Tarrant, M.K.; Boronina, T.; Longo, P.A.; Kavran, J.M.; Cole, R.N.; Cole, P.A.; Leahy, D.J. In vitro enzymatic characterization of near full length EGFR in activated and inhibited states. Biochemistry 2009, 48, 6624–6632. [Google Scholar]
- Buerger, C.; Nagel-Wolfrum, K.; Kunz, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. Sequence-specific peptide aptamers, interacting with the intracellular domain of the epidermal growth factor receptor, interfere with Stat3 activation and inhibit the growth of tumor cells. J. Biol. Chem 2003, 278, 37610–37621. [Google Scholar]
- Kim, S.K.; Huang, L. Nanoparticle delivery of a peptide targeting EGFR signaling. J. Control. Release 2012, 157, 279–286. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sato, K.-i. Cellular Functions Regulated by Phosphorylation of EGFR on Tyr845. Int. J. Mol. Sci. 2013, 14, 10761-10790. https://doi.org/10.3390/ijms140610761
Sato K-i. Cellular Functions Regulated by Phosphorylation of EGFR on Tyr845. International Journal of Molecular Sciences. 2013; 14(6):10761-10790. https://doi.org/10.3390/ijms140610761
Chicago/Turabian StyleSato, Ken-ichi. 2013. "Cellular Functions Regulated by Phosphorylation of EGFR on Tyr845" International Journal of Molecular Sciences 14, no. 6: 10761-10790. https://doi.org/10.3390/ijms140610761