Loss of Vps54 Function Leads to Vesicle Traffic Impairment, Protein Mis-Sorting and Embryonic Lethality


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
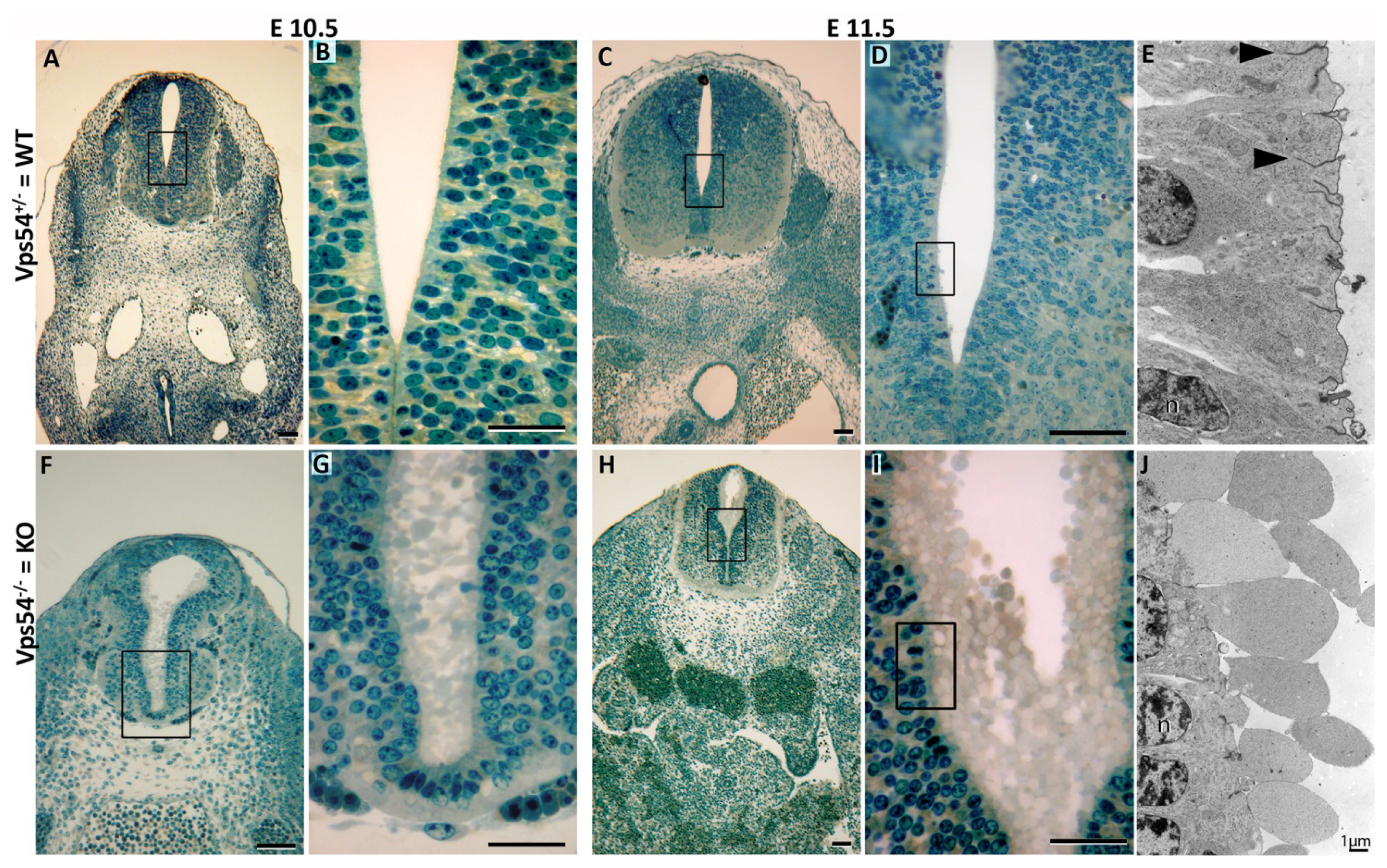
2.1. GARP Mutant Embryos
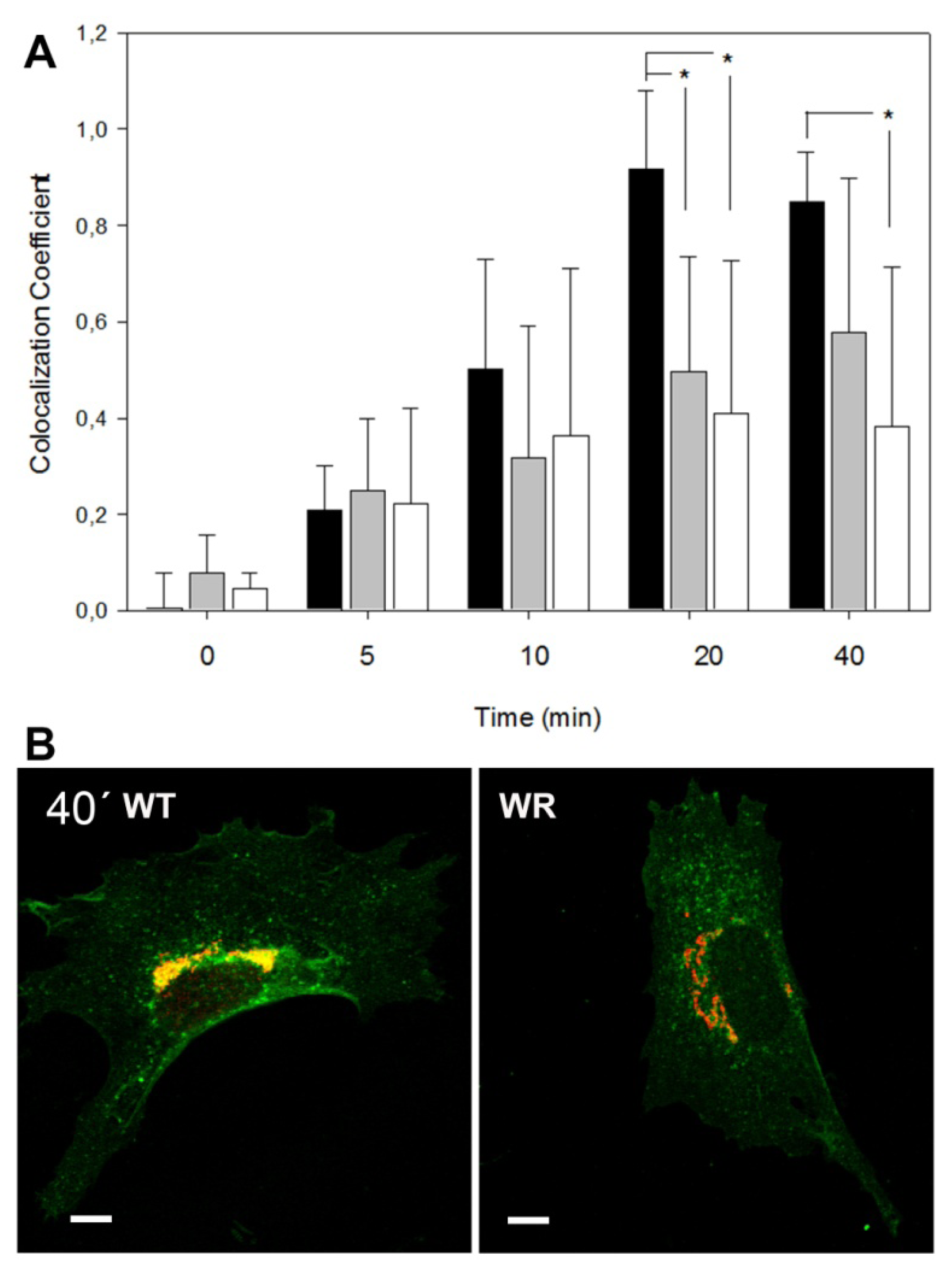
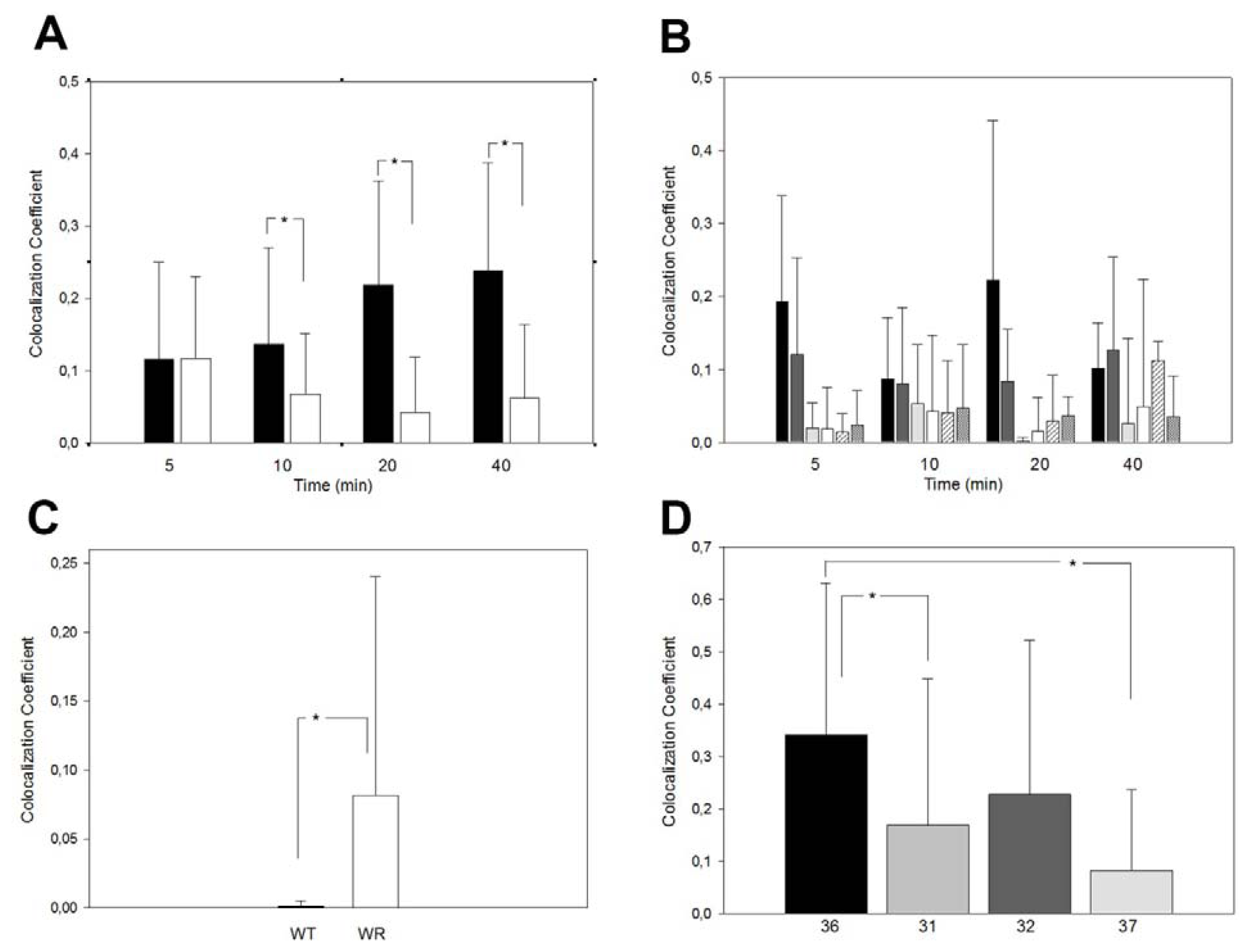
2.2. Retrograde Vesicle Traffic Is Affected in Vps54 Mutant Cells
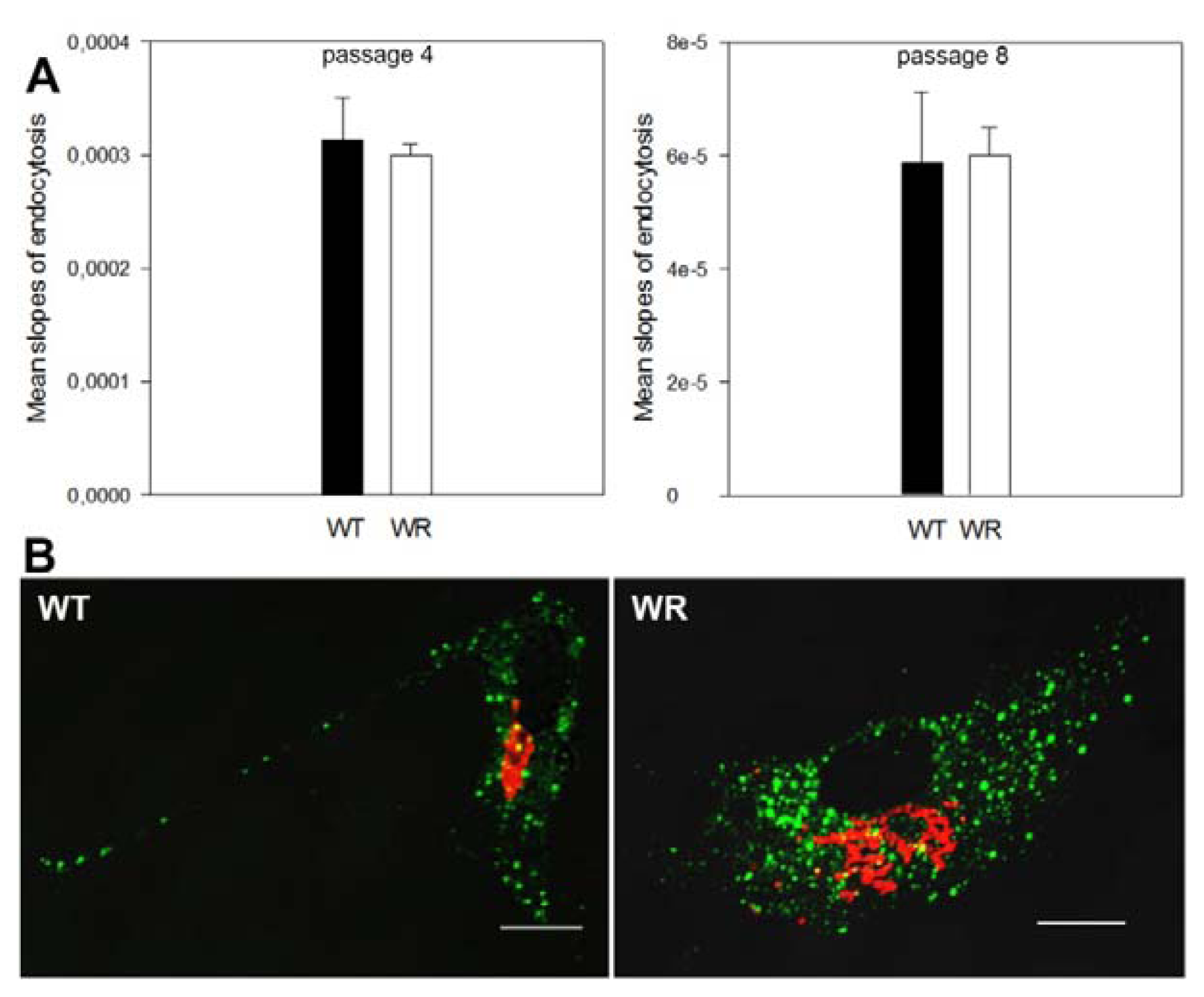
2.3. Endocytosis Is Not Affected in Vps54 Mutant Cells
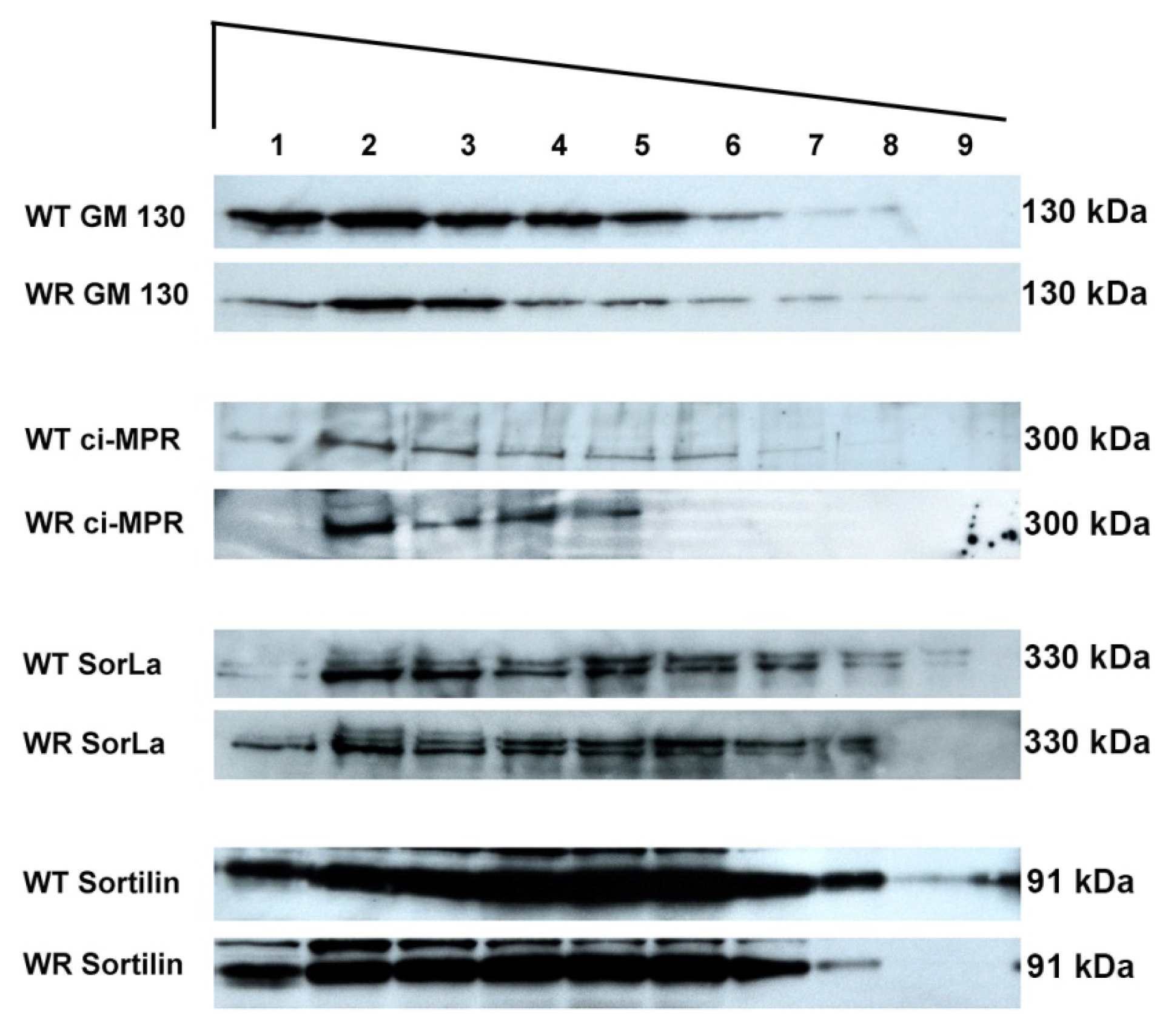
2.4. Protein Mis-Sorting in Vps54 Mutant Cells
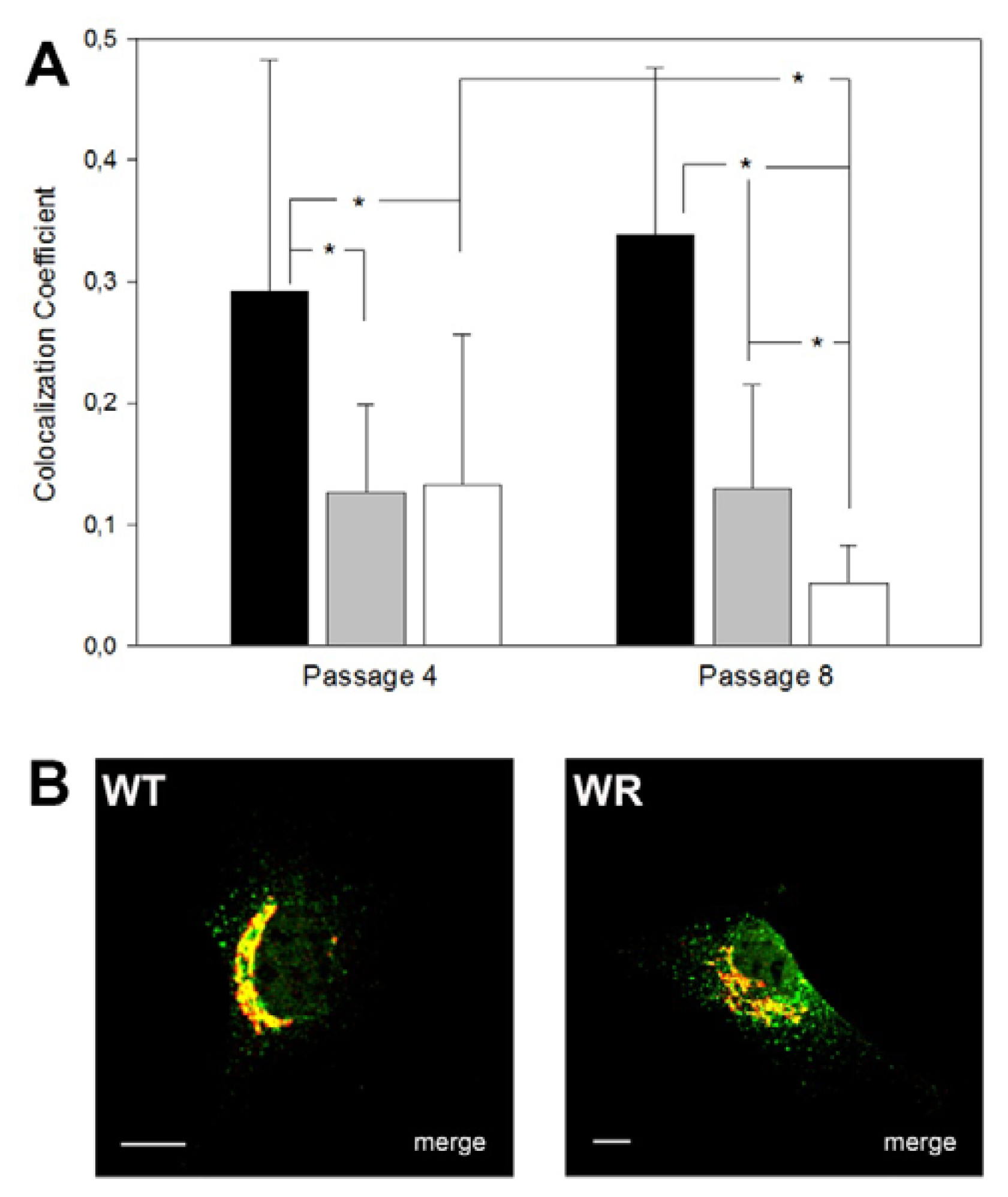
2.5. Use of Skin Fibroblasts for the Analysis of Vesicle Traffic Defects
3. Experimental Section
3.1. Mice and Diagnostics
3.2. EM and Light Microscopy
3.3. Culture of MEFs and SKFs
3.4. Human SKF Lines
3.5. Choleratoxin Transport Assay
3.6. Mannose-6-Phosphate Receptor Assay
3.7. HRP Fluid Phase Endocytosis Assay
3.8. Antibody Co-Internalization
3.9. Cell Fractionation and Western Blotting
3.10. APP Immunostaining
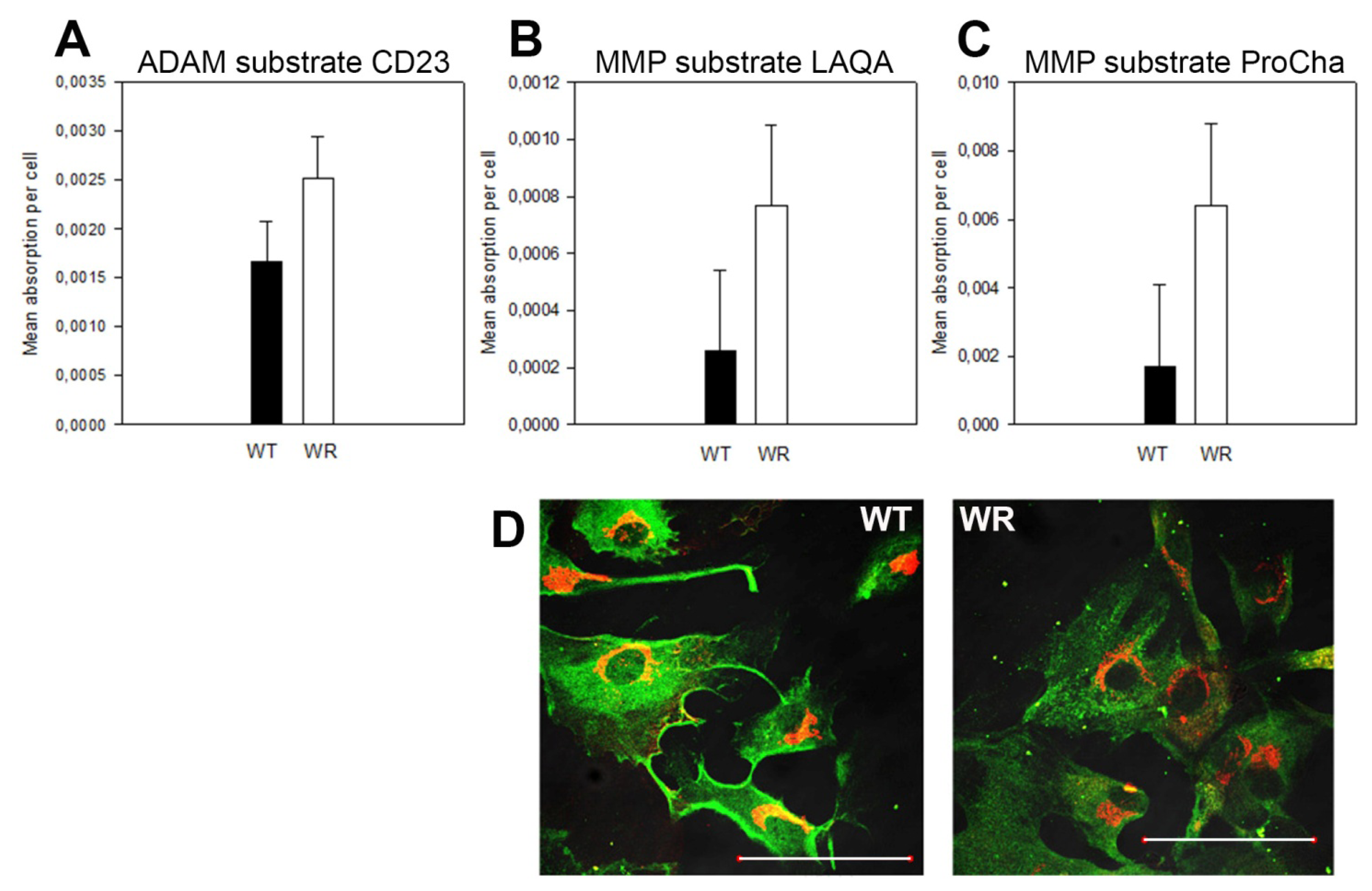
3.11. Quantification of Active Membrane-Bound and Secreted Metalloproteases
4. Conclusions
Supplementary Information
ijms-14-10908-s001.pdfAcknowledgments
Conflict of Interest
References
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in als. Annu. Rev. Neurosci 2004, 27, 723–749. [Google Scholar]
- Rosen, D.R. Mutations in cu/zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet 2001, 29, 160–165. [Google Scholar]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/rna helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (als4). Am. J. Hum. Genet 2004, 74, 1128–1135. [Google Scholar]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein vapb causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet 2004, 75, 822–831. [Google Scholar]
- Moser, J.M.; Bigini, P.; Schmitt-John, T. The wobbler mouse, an als animal model. Mol. Genet. Genomics 2013. [Google Scholar] [CrossRef]
- Schmitt-John, T.; Drepper, C.; Mussmann, A.; Hahn, P.; Kuhlmann, M.; Thiel, C.; Hafner, M.; Lengeling, A.; Heimann, P.; Jones, J.M.; et al. Mutation of vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet 2005, 37, 1213–1215. [Google Scholar]
- Conibear, E.; Stevens, T.H. Vps52p, vps53p, and vps54p form a novel multisubunit complex required for protein sorting at the yeast late golgi. Mol. Biol. Cell 2000, 11, 305–323. [Google Scholar]
- Liewen, H.; Meinhold-Heerlein, I.; Oliveira, V.; Schwarzenbacher, R.; Luo, G.; Wadle, A.; Jung, M.; Pfreundschuh, M.; Stenner-Liewen, F. Characterization of the human garp (golgi associated retrograde protein) complex. Exp. Cell Res 2005, 306, 24–34. [Google Scholar]
- Perez-Victoria, F.J.; Mardones, G.A.; Bonifacino, J.S. Requirement of the human garp complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin d to lysosomes. Mol. Biol. Cell 2008, 19, 2350–2362. [Google Scholar]
- Perez-Victoria, F.J.; Bonifacino, J.S. Dual roles of the mammalian garp complex in tethering and snare complex assembly at the trans-golgi network. Mol. Cell Biol 2009, 29, 5251–5263. [Google Scholar]
- Sugimoto, M.; Kondo, M.; Hirose, M.; Suzuki, M.; Mekada, K.; Abe, T.; Kiyonari, H.; Ogura, A.; Takagi, N.; Artzt, K.; et al. Molecular identification of t(w5): Vps52 promotes pluripotential cell differentiation through cell-cell interactions. Cell Rep 2012, 2, 1363–1374. [Google Scholar]
- Perez-Victoria, F.J.; Abascal-Palacios, G.; Tascon, I.; Kajava, A.; Magadan, J.G.; Pioro, E.P.; Bonifacino, J.S.; Hierro, A. Structural basis for the wobbler mouse neurodegenerative disorder caused by mutation in the vps54 subunit of the garp complex. Proc. Natl. Acad. Sci. USA 2010, 107, 12860–12865. [Google Scholar]
- Palmisano, R.; Golfi, P.; Heimann, P.; Shaw, C.E.; Troakes, C.; Schmitt-John, T.; Bartsch, J.-W. Endosomal accumulation of app in wobbler motor neurons reflects impaired vesicle trafficking: Implications for human motor neuron disease pathologies. BMC Neurosci 2011, 12, 24. [Google Scholar]
- Meisler, M.H.; Russ, C.; Montgomery, K.T.; Greenway, M.; Ennis, S.; Hardiman, O.; Figlewicz, D.A.; Quenneville, N.R.; Conibear, E.; Brown, R.H., Jr. Evaluation of the golgi trafficking protein vps54 (wobbler) as a candidate for als. Amyotroph. Lateral Scler. 2008, 9, 141–148. [Google Scholar]
- Nieto-Gonzalez, J.; Moser, J.M.; Laurtizen, M.; Schmitt-John, T.; Jensen, K. Reduced gabaergic inhibition underlies cortical hyperexcitability in the wobbler mouse model of als. Cerebr. Cortex 2011, 21, 625–635. [Google Scholar]
- Bonifacino, J.S.; Hierro, A. Transport according to garp: Receiving retrograde cargo at the trans-golgi network. Trends Cell Biol 2011, 21, 159–167. [Google Scholar]
- Rathke-Hartlieb, S.; Budde, P.; Ewert, S.; Schlomann, U.; Staege, M.S.; Jockusch, H.; Bartsch, J.W.; Frey, J. Elevated expression of membrane type 1 metalloproteinase (mt1-mmp) in reactive astrocytes following neurodegeneration in mouse central nervous system. FEBS Lett 2000, 481, 227–234. [Google Scholar]
- Bartsch, J.W.; Wildeboer, D.; Koller, G.; Naus, S.; Rittger, A.; Moss, M.L.; Minai, Y.; Jockusch, H. Tumor necrosis factor-alpha (tnf-alpha) regulates shedding of tnf-alpha receptor 1 by the metalloprotease-disintegrin adam8: Evidence for a protease-regulated feedback loop in neuroprotection. J. Neurosci 2010, 30, 12210–12218. [Google Scholar]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked sod1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet 2007, 16, 2720–2728. [Google Scholar]
- Zhang, F.; Strom, A.L.; Fukada, K.; Lee, S.; Hayward, L.J.; Zhu, H. Interaction between familial amyotrophic lateral sclerosis (als)-linked sod1 mutants and the dynein complex. J. Biol. Chem 2007, 282, 16691–16699. [Google Scholar]
- Kirby, J.; Menzies, F.M.; Cookson, M.R.; Bushby, K.; Shaw, P.J. Differential gene expression in a cell culture model of sod1-related familial motor neurone disease. Hum. Mol. Genet 2002, 11, 2061–2075. [Google Scholar]
- Sakurai, T.; Kamiyoshi, A.; Watanabe, S.; Sato, M.; Shindo, T. Rapid zygosity determination in mice by sybr green real-time genomic pcr of a crude DNA solution. Transgenic Res 2008, 17, 149–155. [Google Scholar]
- Schlomann, U.; Wildeboer, D.; Webster, A.; Antropova, O.; Zeuschner, D.; Knight, C.G.; Docherty, A.J.; Lambert, M.; Skelton, L.; Jockusch, H.; et al. The metalloprotease disintegrin adam8. Processing by autocatalysis is required for proteolytic activity and cell adhesion. J. Biol. Chem 2002, 277, 48210–48219. [Google Scholar]
- Rathke-Hartlieb, S.; Schmidt, V.C.; Jockusch, H.; Schmitt-John, T.; Bartsch, J.W. Spatiotemporal progression of neurodegeneration and glia activation in the wobbler neuropathy of the mouse. Neuroreport 1999, 10, 3411–3416. [Google Scholar]
- Nagy, A.; Gertsenstein, M.; Vintersten, K.; Behringer, R. Manipulating the Mouse Embryo: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2003. [Google Scholar]
- Kristensen, B.; Birkelund, S.; Jorgensen, P.L. Trafficking of na,k-atpase fused to enhanced green fluorescent protein is mediated by protein kinase a or c. J. Membr. Biol 2003, 191, 25–36. [Google Scholar]
- Tancini, B.; Magini, A.; Latterini, L.; Urbanelli, L.; Ciccarone, V.; Elisei, F.; Emiliani, C. Occurrence of an anomalous endocytic compartment in fibroblasts from sandhoff disease patients. Mol. Cell Biochem 2010, 335, 273–282. [Google Scholar]
- Andersen, O.M.; Reiche, J.; Schmidt, V.; Gotthardt, M.; Spoelgen, R.; Behlke, J.; von Arnim, C.A.; Breiderhoff, T.; Jansen, P.; Wu, X.; et al. Neuronal sorting protein-related receptor sorla/lr11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13461–13466. [Google Scholar]
- Moss, M.L.; Rasmussen, F.H. Fluorescent substrates for the proteinases adam17, adam10, adam8, and adam12 useful for high-throughput inhibitor screening. Anal. Biochem 2007, 366, 144–148. [Google Scholar]







© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Karlsson, P.; Droce, A.; Moser, J.M.; Cuhlmann, S.; Padilla, C.O.; Heimann, P.; Bartsch, J.W.; Füchtbauer, A.; Füchtbauer, E.-M.; Schmitt-John, T. Loss of Vps54 Function Leads to Vesicle Traffic Impairment, Protein Mis-Sorting and Embryonic Lethality. Int. J. Mol. Sci. 2013, 14, 10908-10925. https://doi.org/10.3390/ijms140610908
Karlsson P, Droce A, Moser JM, Cuhlmann S, Padilla CO, Heimann P, Bartsch JW, Füchtbauer A, Füchtbauer E-M, Schmitt-John T. Loss of Vps54 Function Leads to Vesicle Traffic Impairment, Protein Mis-Sorting and Embryonic Lethality. International Journal of Molecular Sciences. 2013; 14(6):10908-10925. https://doi.org/10.3390/ijms140610908
Chicago/Turabian StyleKarlsson, Páll, Aida Droce, Jakob M. Moser, Simon Cuhlmann, Carolina Ortiz Padilla, Peter Heimann, Jörg W. Bartsch, Annette Füchtbauer, Ernst-Martin Füchtbauer, and Thomas Schmitt-John. 2013. "Loss of Vps54 Function Leads to Vesicle Traffic Impairment, Protein Mis-Sorting and Embryonic Lethality" International Journal of Molecular Sciences 14, no. 6: 10908-10925. https://doi.org/10.3390/ijms140610908
APA StyleKarlsson, P., Droce, A., Moser, J. M., Cuhlmann, S., Padilla, C. O., Heimann, P., Bartsch, J. W., Füchtbauer, A., Füchtbauer, E.-M., & Schmitt-John, T. (2013). Loss of Vps54 Function Leads to Vesicle Traffic Impairment, Protein Mis-Sorting and Embryonic Lethality. International Journal of Molecular Sciences, 14(6), 10908-10925. https://doi.org/10.3390/ijms140610908