Urinary Bladder Cancer Susceptibility Markers. What Do We Know about Functional Mechanisms?

Abstract
:1. Introduction
2. Bladder Cancer
3. Genome-Wide Association Studies
3.1. Principle of GWAS
3.2. GWAS for Bladder Cancer Risk
4. Functional Studies of Risk SNPs in Bladder Cancer
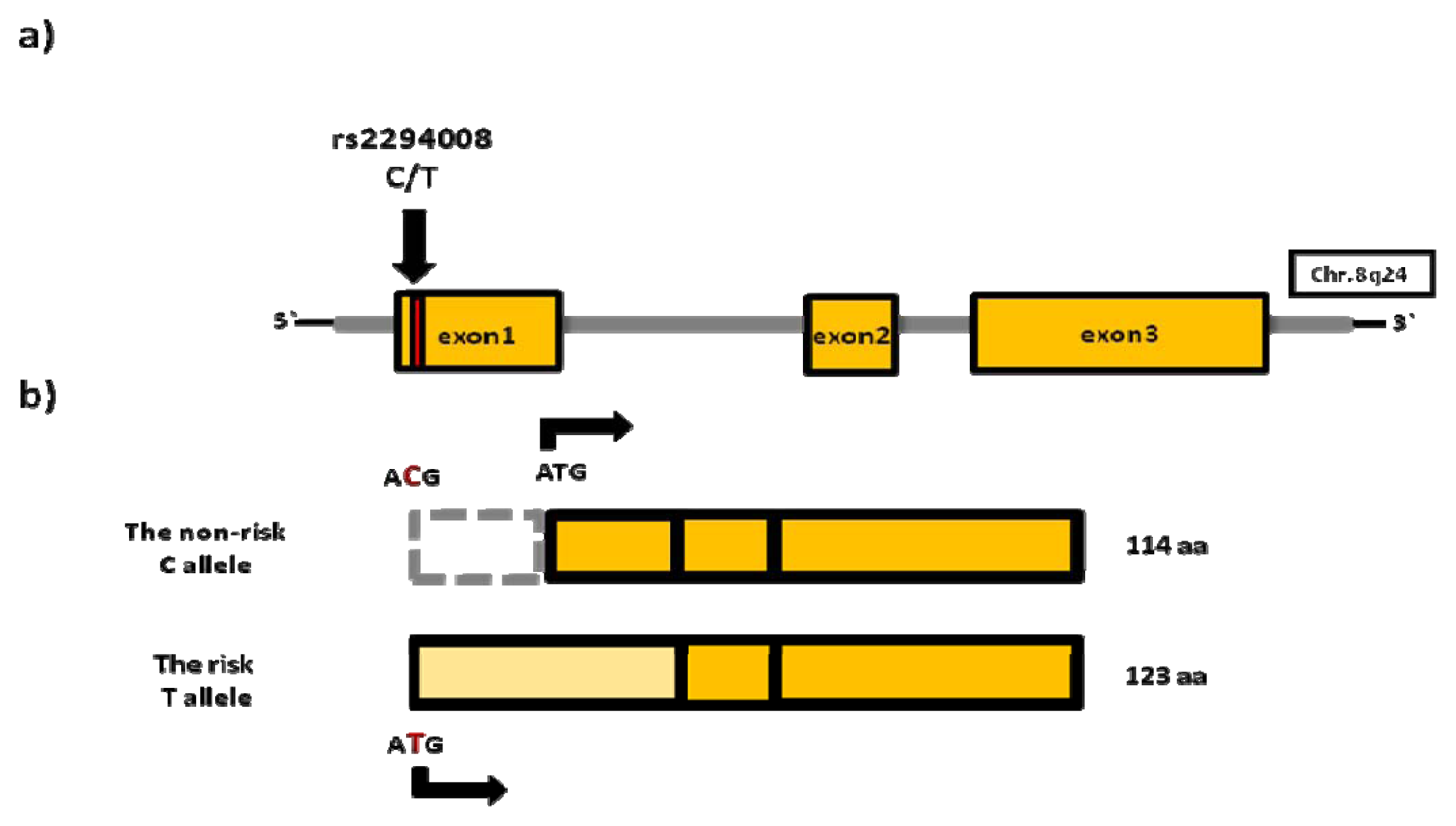
4.1. rs2294008 (PSCA)
4.2. rs17863783 (UGT1A)
4.3. Functional Studies of Risk SNPs in UBC Susceptibility Regions
4.3.1. MYC Locus on 8q24
4.3.2. TERT/CLPTM1L Locus on 5p15
5. Future of Functional Studies
5.1. Possible Role of Variants in Non-Coding Regions
5.2. Rapid Development of New Technologies
5.3. The Impact of Context
6. Conclusions
Conflict of Interest
References
- Chung, C.C.; Magalhaes, W.C.; Gonzalez-Bosquet, J.; Chanock, S.J. Genome-wide association studies in cancer—Current and future directions. Carcinogenesis 2010, 31, 111–120. [Google Scholar]
- Wu, X.; Hildebrandt, M.A.; Chang, D.W. Genome-wide association studies of bladder cancer risk: A field synopsis of progress and potential applications. Cancer Metastasis Rev 2009, 28, 269–280. [Google Scholar]
- Freedman, M.L.; Monteiro, A.N.; Gayther, S.A.; Coetzee, G.A.; Risch, A.; Plass, C.; Casey, G.; de Biasi, M.; Carlson, C.; Duggan, D.; et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet 2011, 43, 513–518. [Google Scholar]
- Burger, M.; Catto, J.W.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; la Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol 2013, 63, 234–241. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Kiemeney, L.A.; Grotenhuis, A.J.; Vermeulen, S.H.; Wu, X. Genome-wide association studies in bladder cancer: First results and potential relevance. Curr. Opin. Urol 2009, 19, 540–546. [Google Scholar]
- Kiltie, A.E. Common predisposition alleles for moderately common cancers: Bladder cancer. Curr. Opin. Genet. Dev 2010, 20, 218–224. [Google Scholar]
- Ploeg, M.; Aben, K.K.; Kiemeney, L.A. The present and future burden of urinary bladder cancer in the world. World J. Urol 2009, 27, 289–293. [Google Scholar]
- Ploeg, M.; Aben, K.K.; Hulsbergen-van de Kaa, C.A.; Schoenberg, M.P.; Witjes, J.A.; Kiemeney, L.A. Clinical epidemiology of nonurothelial bladder cancer: Analysis of the Netherlands cancer registry. J. Urol 2010, 183, 915–920. [Google Scholar]
- Amin, M.B. Histological variants of urothelial carcinoma: Diagnostic, therapeutic and prognostic implications. Mod. Pathol 2009, 22, S96–S118. [Google Scholar]
- Babjuk, M.; Oosterlinck, W.; Sylvester, R.; Kaasinen, E.; Bohle, A.; Palou-Redorta, J.; Roupret, M. European Association of, U. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder, the 2011 update. Eur. Urol 2011, 59, 997–1008. [Google Scholar]
- Van den Bosch, S.; Alfred Witjes, J. Long-term cancer-specific survival in patients with high-risk, non-muscle-invasive bladder cancer and tumour progression: A systematic review. Eur. Urol 2011, 60, 493–500. [Google Scholar]
- Sawhney, R.; Bourgeois, D.; Chaudhary, U.B. Neo-adjuvant chemotherapy for muscle-invasive bladder cancer: A look ahead. Ann. Oncol 2006, 17, 1360–1369. [Google Scholar]
- Merseburger, A.S.; Kuczyk, M.A. The value of bladder-conserving strategies in muscle-invasive bladder carcinoma compared with radical surgery. Curr. Opin. Urol 2007, 17, 358–362. [Google Scholar]
- Murta-Nascimento, C.; Schmitz-Drager, B.J.; Zeegers, M.P.; Steineck, G.; Kogevinas, M.; Real, F.X.; Malats, N. Epidemiology of urinary bladder cancer: From tumor development to patient’s death. World J. Urol 2007, 25, 285–295. [Google Scholar]
- Volanis, D.; Kadiyska, T.; Galanis, A.; Delakas, D.; Logotheti, S.; Zoumpourlis, V. Environmental factors and genetic susceptibility promote urinary bladder cancer. Toxicol. Lett 2010, 193, 131–137. [Google Scholar]
- Kiriluk, K.J.; Prasad, S.M.; Patel, A.R.; Steinberg, G.D.; Smith, N.D. Bladder cancer risk from occupational and environmental exposures. Urol. Oncol 2012, 30, 199–211. [Google Scholar]
- Chang, D.W.; Gu, J.; Wu, X. Germline prognostic markers for urinary bladder cancer: Obstacles and opportunities. Urol. Oncol 2012, 30, 524–532. [Google Scholar]
- Division of Genomic Medicine. A Catalog of Published Genome-Wide Association Studies. Available online: http://www.genome.gov/gwastudies (accessed on 11 April 2013).
- Chung, C. C.; Chanock, S. J. Current status of genome-wide association studies in cancer. Hum. Genet 2011, 130, 59–78. [Google Scholar]
- Kiemeney, L.A.; Thorlacius, S.; Sulem, P.; Geller, F.; Aben, K.K.; Stacey, S.N.; Gudmundsson, J.; Jakobsdottir, M.; Bergthorsson, J.T.; Sigurdsson, A.; et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat. Genet 2008, 40, 1307–1312. [Google Scholar]
- Kiemeney, L.A.; Sulem, P.; Besenbacher, S.; Vermeulen, S.H.; Sigurdsson, A.; Thorleifsson, G.; Gudbjartsson, D.F.; Stacey, S.N.; Gudmundsson, J.; Zanon, C.; et al. A sequence variant at 4p16.3 confers susceptibility to urinary bladder cancer. Nat. Genet 2010, 42, 415–419. [Google Scholar] [Green Version]
- Wu, X.; Ye, Y.; Kiemeney, L.A.; Sulem, P.; Rafnar, T.; Matullo, G.; Seminara, D.; Yoshida, T.; Saeki, N.; Andrew, A.S.; et al. Genetic variation in the prostate stem cell antigen gene PSCA confers susceptibility to urinary bladder cancer. Nat. Genet 2009, 41, 991–995. [Google Scholar]
- Rothman, N.; Garcia-Closas, M.; Chatterjee, N.; Malats, N.; Wu, X.; Figueroa, J.D.; Real, F.X.; van Den Berg, D.; Matullo, G.; et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat. Genet 2010, 42, 978–984. [Google Scholar]
- Rafnar, T.; Vermeulen, S.H.; Sulem, P.; Thorleifsson, G.; Aben, K.K.; Witjes, J.A.; Grotenhuis, A.J.; Verhaegh, G.W.; Hulsbergen-van de Kaa, C.A.; Besenbacher, S.; et al. European genome-wide association study identifies SLC14A1 as a new urinary bladder cancer susceptibility gene. Hum. Mol. Genet 2011, 20, 4268–4281. [Google Scholar]
- Garcia-Closas, M.; Ye, Y.; Rothman, N.; Figueroa, J.D.; Malats, N.; Dinney, C.P.; Chatterjee, N.; Prokunina-Olsson, L.; Wang, Z.; Lin, J.; et al. A genome-wide association study of bladder cancer identifies a new susceptibility locus within SLC14A1, a urea transporter gene on chromosome 18q12.3. Hum. Mol. Genet 2011, 20, 4282–4289. [Google Scholar]
- Rafnar, T.; Sulem, P.; Stacey, S.N.; Geller, F.; Gudmundsson, J.; Sigurdsson, A.; Jakobsdottir, M.; Helgadottir, H.; Thorlacius, S.; Aben, K.K.; et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet 2009, 41, 221–227. [Google Scholar]
- Garcia-Closas, M.; Malats, N.; Silverman, D.; Dosemeci, M.; Kogevinas, M.; Hein, D.W.; Tardon, A.; Serra, C.; Carrato, A.; Garcia-Closas, R.; et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: Results from the Spanish bladder cancer study and meta-analyses. Lancet 2005, 366, 649–659. [Google Scholar]
- Ono, H.; Hiraoka, N.; Lee, Y.S.; Woo, S.M.; Lee, W.J.; Choi, I.J.; Saito, A.; Yanagihara, K.; Kanai, Y.; Ohnami, S.; et al. Prostate stem cell antigen, a presumable organ-dependent tumor suppressor gene, is down-regulated in gallbladder carcinogenesis. Genes Chromos. Cancer 2012, 51, 30–41. [Google Scholar]
- Amara, N.; Palapattu, G.S.; Schrage, M.; Gu, Z.; Thomas, G.V.; Dorey, F.; Said, J.; Reiter, R.E. Prostate stem cell antigen is overexpressed in human transitional cell carcinoma. Cancer Res 2001, 61, 4660–4665. [Google Scholar]
- Zhigang, Z.; Wenlv, S. Prostate stem cell antigen (PSCA) expression in human prostate cancer tissues and its potential role in prostate carcinogenesis and progression of prostate cancer. World J. Surg. Oncol 2004, 2, 13. [Google Scholar]
- Saffran, D.C.; Raitano, A.B.; Hubert, R.S.; Witte, O.N.; Reiter, R.E.; Jakobovits, A. Anti-PSCA mAbs inhibit tumor growth and metastasis formation and prolong the survival of mice bearing human prostate cancer xenografts. Proc. Natl. Acad. Sci. USA 2001, 98, 2658–2663. [Google Scholar]
- Sakamoto, H.; Yoshimura, K.; Saeki, N.; Katai, H.; Shimoda, T.; Matsuno, Y.; Saito, D.; Sugimura, H.; Tanioka, F.; et al. Study Group of Millennium Genome Project for Cancer. Genetic variation in PSCA is associated with susceptibility to diffuse-type gastric cancer. Nat. Genet. 2008, 40, 730–740. [Google Scholar]
- Tanikawa, C.; Urabe, Y.; Matsuo, K.; Kubo, M.; Takahashi, A.; Ito, H.; Tajima, K.; Kamatani, N.; Nakamura, Y.; Matsuda, K. A genome-wide association study identifies two susceptibility loci for duodenal ulcer in the Japanese population. Nat. Genet 2012, 44, 430–434, , S1–S2.. [Google Scholar]
- Fu, Y.P.; Kohaar, I.; Rothman, N.; Earl, J.; Figueroa, J.D.; Ye, Y.; Malats, N.; Tang, W.; Liu, L.; Garcia-Closas, M.; et al. Common genetic variants in the PSCA gene influence gene expression and bladder cancer risk. Proc. Natl. Acad. Sci. USA 2012, 109, 4974–4979. [Google Scholar]
- Kohaar, I.; Porter-Gill, P.; Lenz, P.; Fu, Y.P.; Mumy, A.; Tang, W.; Apolo, A.B.; Rothman, N.; Baris, D.; Schned, A.R.; et al. Genetic variant as a selection marker for anti-prostate stem cell antigen immunotherapy of bladder cancer. J. Natl. Cancer Instit 2013, 105, 69–73. [Google Scholar]
- Shariat, S.F.; Sfakianos, J.P.; Droller, M.J.; Karakiewicz, P.I.; Meryn, S.; Bochner, B.H. The effect of age and gender on bladder cancer: A critical review of the literature. BJU Int 2010, 105, 300–308. [Google Scholar]
- Miyamoto, H.; Yang, Z.; Chen, Y.T.; Ishiguro, H.; Uemura, H.; Kubota, Y.; Nagashima, Y.; Chang, Y.J.; Hu, Y.C.; Tsai, M.Y.; et al. Promotion of bladder cancer development and progression by androgen receptor signals. J. Natl. Cancer Instit 2007, 99, 558–568. [Google Scholar]
- Miyamoto, H.; Yao, J.L.; Chaux, A.; Zheng, Y.; Hsu, I.; Izumi, K.; Chang, C.; Messing, E.M.; Netto, G.J.; Yeh, S. Expression of androgen and oestrogen receptors and its prognostic significance in urothelial neoplasm of the urinary bladder. BJU Int 2012, 109, 1716–1726. [Google Scholar]
- Gakis, G.; Stenzl, A. Gender-specific differences in muscle-invasive bladder cancer: The concept of sex steroid sensitivity. World J. Urol. 2013. [Google Scholar] [CrossRef]
- Miyamoto, H.; Zheng, Y.; Izumi, K. Nuclear hormone receptor signals as new therapeutic targets for urothelial carcinoma. Curr. Cancer Drug Targets 2012, 12, 14–22. [Google Scholar]
- Bellemare, J.; Rouleau, M.; Harvey, M.; Guillemette, C. Modulation of the human glucuronosyltransferase UGT1A pathway by splice isoform polypeptides is mediated through protein-protein interactions. J. Biol. Chem 2010, 285, 3600–3607. [Google Scholar]
- Nakamura, A.; Nakajima, M.; Yamanaka, H.; Fujiwara, R.; Yokoi, T. Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab. Dispos. Biol. Fate Chem 2008, 36, 1461–1464. [Google Scholar]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Ann. Rev. Pharmacol. Toxicol 2000, 40, 581–616. [Google Scholar]
- Ciotti, M.; Lakshmi, V.M.; Basu, N.; Davis, B.B.; Owens, I.S.; Zenser, T.V. Glucuronidation of benzidine and its metabolites by cDNA-expressed human UDP-glucuronosyltransferases and pH stability of glucuronides. Carcinogenesis 1999, 20, 1963–1969. [Google Scholar]
- Zenser, T.V.; Lakshmi, V.M.; Hsu, F.F.; Davis, B.B. Metabolism of N-acetylbenzidine and initiation of bladder cancer. Mutat. Res. 2002, 506–507, 29–40. [Google Scholar]
- Levesque, E.; Belanger, A.S.; Harvey, M.; Couture, F.; Jonker, D.; Innocenti, F.; Cecchin, E.; Toffoli, G.; Guillemette, C. Refining the UGT1A haplotype associated with irinotecan-induced hematological toxicity in metastatic colorectal cancer patients treated with 5-fluorouracil/irinotecan-based regimens. J. Pharmacol. Exp. Ther 2013, 345, 95–101. [Google Scholar]
- Tang, W.; Fu, Y.P.; Figueroa, J.D.; Malats, N.; Garcia-Closas, M.; Chatterjee, N.; Kogevinas, M.; Baris, D.; Thun, M.; Hall, J.L.; et al. Mapping of the UGT1A locus identifies an uncommon coding variant that affects mRNA expression and protects from bladder cancer. Hum. Mol. Genet 2012, 21, 1918–1930. [Google Scholar]
- Wu, X.; Ros, M.M.; Gu, J.; Kiemeney, L. Epidemiology and genetic susceptibility to bladder cancer. BJU Int 2008, 102, 1207–1215. [Google Scholar]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev 2008, 22, 2755–2766. [Google Scholar]
- Felsher, D.W. MYC inactivation elicits oncogene addiction through both tumor cell-intrinsic and host-dependent mechanisms. Genes Cancer 2010, 1, 597–604. [Google Scholar]
- Nilsson, J.A.; Cleveland, J.L. Myc pathways provoking cell suicide and cancer. Oncogene 2003, 22, 9007–9021. [Google Scholar]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar]
- Theodoratou, E.; Montazeri, Z.; Hawken, S.; Allum, G.C.; Gong, J.; Tait, V.; Kirac, I.; Tazari, M.; Farrington, S.M.; Demarsh, A.; et al. Systematic meta-analyses and field synopsis of genetic association studies in colorectal cancer. J. Natl. Cancer Instit 2012, 104, 1433–1457. [Google Scholar]
- Al Olama, A.A.; Kote-Jarai, Z.; Giles, G.G.; Guy, M.; Morrison, J.; Severi, G.; Leongamornlert, D.A.; Tymrakiewicz, M.; Jhavar, S.; Saunders, E.; et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat. Genet 2009, 41, 1058–1060. [Google Scholar]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar]
- Wang, M.; Wang, M.; Zhang, W.; Yuan, L.; Fu, G.; Wei, Q.; Zhang, Z. Common genetic variants on 8q24 contribute to susceptibility to bladder cancer in a Chinese population. Carcinogenesis 2009, 30, 991–996. [Google Scholar]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar]
- Jia, L.; Landan, G.; Pomerantz, M.; Jaschek, R.; Herman, P.; Reich, D.; Yan, C.; Khalid, O.; Kantoff, P.; Oh, W.; et al. Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet 2009, 5, e1000597. [Google Scholar]
- Ting, M.C.; Liao, C.P.; Yan, C.; Jia, L.; Groshen, S.; Frenkel, B.; Roy-Burman, P.; Coetzee, G.A.; Maxson, R. An enhancer from the 8q24 prostate cancer risk region is sufficient to direct reporter gene expression to a subset of prostate stem-like epithelial cells in transgenic mice. Dis. Models Mech 2012, 5, 366–374. [Google Scholar]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymaki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet 2009, 41, 885–890. [Google Scholar]
- Pomerantz, M.M.; Ahmadiyeh, N.; Jia, L.; Herman, P.; Verzi, M.P.; Doddapaneni, H.; Beckwith, C.A.; Chan, J.A.; Hills, A.; Davis, M.; et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet 2009, 41, 882–884. [Google Scholar]
- Wright, J.B.; Brown, S.J.; Cole, M.D. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell. Biol 2010, 30, 1411–1420. [Google Scholar]
- Wasserman, N.F.; Aneas, I.; Nobrega, M.A. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res 2010, 20, 1191–1197. [Google Scholar]
- Pomerantz, M.M.; Beckwith, C.A.; Regan, M.M.; Wyman, S.K.; Petrovics, G.; Chen, Y.; Hawksworth, D.J.; Schumacher, F.R.; Mucci, L.; Penney, K.L.; et al. Evaluation of the 8q24 prostate cancer risk locus and MYC expression. Cancer Res 2009, 69, 5568–5574. [Google Scholar]
- Sur, I.K.; Hallikas, O.; Vaharautio, A.; Yan, J.; Turunen, M.; Enge, M.; Taipale, M.; Karhu, A.; Aaltonen, L.A.; Taipale, J. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 2012, 338, 1360–1363. [Google Scholar]
- Mhawech-Fauceglia, P.; Cheney, R.T.; Schwaller, J. Genetic alterations in urothelial bladder carcinoma: An updated review. Cancer 2006, 106, 1205–1216. [Google Scholar]
- Ponzielli, R.; Katz, S.; Barsyte-Lovejoy, D.; Penn, L.Z. Cancer therapeutics: Targeting the dark side of Myc. Eur. J. Cancer 2005, 41, 2485–2501. [Google Scholar]
- Baird, D.M. Variation at the TERT locus and predisposition for cancer. Expert Rev. Mol. Med 2010, 12, e16. [Google Scholar]
- Daniel, M.; Peek, G.W.; Tollefsbol, T.O. Regulation of the human catalytic subunit of telomerase (hTERT). Gene 2012, 498, 135–146. [Google Scholar]
- Cifuentes-Rojas, C.; Shippen, D.E. Telomerase regulation. Mutat. Res 2012, 730, 20–27. [Google Scholar]
- Hou, L.; Zhang, X.; Gawron, A.J.; Liu, J. Surrogate tissue telomere length and cancer risk: Shorter or longer? Cancer Lett 2012, 319, 130–135. [Google Scholar]
- Cao, Y.; Bryan, T.M.; Reddel, R.R. Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci 2008, 99, 1092–1099. [Google Scholar]
- Yamamoto, K.; Okamoto, A.; Isonishi, S.; Ochiai, K.; Ohtake, Y. A novel gene, CRR9, which was up-regulated in CDDP-resistant ovarian tumor cell line, was associated with apoptosis. Biochem. and Biophys. Res. Commun 2001, 280, 1148–1154. [Google Scholar]
- Folgueira, M.A.A.K.; Carraro, D.M.; Brentani, H.; Patra, D.F.D.; Barbosa, E.M.; Netto, M.M.; Caldeira, J.R.F.; Katayama, M.L.H.; Soares, F.A.; Oliveira, C.T.; et al. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin. Cancer Res 2005, 11, 7434–7443. [Google Scholar]
- Kote-Jarai, Z.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Dadaev, T.; Jugurnauth-Little, S.; Ross-Adams, H.; al Olama, A.A.; Benlloch, S.; Halim, S.; et al. Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum. Mol. Genet 2013, 22, 2520–2528. [Google Scholar]
- McGrath, M.; Wong, J.Y.; Michaud, D.; Hunter, D.J.; de Vivo, I. Telomere length, cigarette smoking, and bladder cancer risk in men and women. Cancer Epidemiol. Biomark. Prev 2007, 16, 815–819. [Google Scholar]
- Gu, J.; Chen, M.; Shete, S.; Amos, C.I.; Kamat, A.; Ye, Y.; Lin, J.; Dinney, C.P.; Wu, X. A genome-wide association study identifies a locus on chromosome 14q21 as a predictor of leukocyte telomere length and as a marker of susceptibility for bladder cancer. Cancer Prev. Res 2011, 4, 514–521. [Google Scholar]
- Lan, Q.; Cawthon, R.; Gao, Y.; Hu, W.; Hosgood, H.D., III; Barone-Adesi, F.; Ji, B.T.; Bassig, B.; Chow, W.H.; Shu, X.; et al. Longer telomere length in peripheral white blood cells is associated with risk of lung cancer and the rs2736100 (CLPTM1L-TERT) polymorphism in a prospective cohort study among women in China. PLoS One 2013, 8, e59230. [Google Scholar]
- Bojesen, S.E.; Pooley, K.A.; Johnatty, S.E.; Beesley, J.; Michailidou, K.; Tyrer, J.P.; Edwards, S.L.; Pickett, H.A.; Shen, H.C.; Smart, C.E.; et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet 2013, 45, 371–384. [Google Scholar]
- Zienolddiny, S.; Skaug, V.; Landvik, N.E.; Ryberg, D.; Phillips, D.H.; Houlston, R.; Haugen, A. The TERT-CLPTM1L lung cancer susceptibility variant associates with higher DNA adduct formation in the lung. Carcinogenesis 2009, 30, 1368–1371. [Google Scholar]
- James, M.A.; Wen, W.D.; Wang, Y.A.; Byers, L.A.; Heymach, J.V.; Coombes, K.R.; Girard, L.; Minna, J.; You, M. Functional characterization of CLPTM1L as a lung cancer risk candidate gene in the 5p15.33 Locus. PLoS One 2012, 7, e36116. [Google Scholar]
- Rahman, R.; Grundy, R. Histone deacetylase inhibition as an anticancer telomerase-targeting strategy. Int. J. Cancer 2011, 129, 2765–2774. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar]
- Kompier, L.C.; Lurkin, I.; van der Aa, M.N.; van Rhijn, B.W.; van der Kwast, T.H.; Zwarthoff, E.C. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS One 2010, 5, e13821. [Google Scholar]
- Consortium, E.P.; Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar]
- Cheung, V.G.; Spielman, R.S. Genetics of human gene expression: Mapping DNA variants that influence gene expression. Nat. Rev. Genet 2009, 10, 595–604. [Google Scholar]
- Pittman, A.M.; Naranjo, S.; Jalava, S.E.; Twiss, P.; Ma, Y.; Olver, B.; Lloyd, A.; Vijayakrishnan, J.; Qureshi, M.; Broderick, P.; et al. Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLoS Genet 2010, 6, e1001126. [Google Scholar]
- Akamatsu, S.; Takata, R.; Ashikawa, K.; Hosono, N.; Kamatani, N.; Fujioka, T.; Ogawa, O.; Kubo, M.; Nakamura, Y.; Nakagawa, H. A functional variant in NKX3.1 associated with prostate cancer susceptibility down-regulates NKX3.1 expression. Hum. Mol. Genet 2010, 19, 4265–4272. [Google Scholar]
- Schodel, J.; Bardella, C.; Sciesielski, L.K.; Brown, J.M.; Pugh, C.W.; Buckle, V.; Tomlinson, I.P.; Ratcliffe, P.J.; Mole, D.R. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat. Genet 2012, 44, 420–425, , S1–S2.. [Google Scholar]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol 2013, 26, 155–165. [Google Scholar]
- Kumar, V.; Westra, H.J.; Karjalainen, J.; Zhernakova, D.V.; Esko, T.; Hrdlickova, B.; Almeida, R.; Zhernakova, A.; Reinmaa, E.; Vosa, U.; et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet 2013, 9, e1003201. [Google Scholar]
- Jendrzejewski, J.; He, H.; Radomska, H.S.; Li, W.; Tomsic, J.; Liyanarachchi, S.; Davuluri, R.V.; Nagy, R.; de la Chapelle, A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. USA 2012, 109, 8646–8651. [Google Scholar]
- Lee, Y.; Gamazon, E.R.; Rebman, E.; Lee, Y.; Lee, S.; Dolan, M.E.; Cox, N.J.; Lussier, Y.A. Variants affecting exon skipping contribute to complex traits. PLoS Genet 2012, 8, e1002998. [Google Scholar]
- Kim, Y.J.; Kim, H.S. Alternative splicing and its impact as a cancer diagnostic marker. Genomics Inf 2012, 10, 74–80. [Google Scholar]
- Narla, G.; Difeo, A.; Reeves, H.L.; Schaid, D.J.; Hirshfeld, J.; Hod, E.; Katz, A.; Isaacs, W.B.; Hebbring, S.; Komiya, A.; et al. A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res 2005, 65, 1213–1222. [Google Scholar]
- Gong, J.; Tong, Y.; Zhang, H.M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.Y. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat 2012, 33, 254–263. [Google Scholar]
- Kim, H.K.; Prokunina-Olsson, L.; Chanock, S.J. Common genetic variants in miR-1206 (8q24.2) and miR-612 (11q13.3) affect biogenesis of mature miRNA forms. PloS One 2012, 7, e47454. [Google Scholar]
- Wang, K.; Li, J.; Guo, H.; Xu, X.; Xiong, G.; Guan, X.; Liu, B.; Li, J.; Chen, X.; Yang, K.; et al. MiR-196a binding-site SNP regulates RAP1A expression contributing to esophageal squamous cell carcinoma risk and metastasis. Carcinogenesis 2012, 33, 2147–2154. [Google Scholar]
- Fitze, G.; Schierz, M.; Kuhlisch, E.; Schreiber, M.; Ziegler, A.; Roesner, D.; Schackert, H.K. Novel intronic polymorphisms in the RET proto-oncogene and their association with Hirschsprung disease. Hum. Mutat 2003, 22, 177. [Google Scholar]
- Hienonen, T.; Sammalkorpi, H.; Enholm, S.; Alhopuro, P.; Barber, T.D.; Lehtonen, R.; Nupponen, N.N.; Lehtonen, H.; Salovaara, R.; Mecklin, J.P.; et al. Mutations in two short noncoding mononucleotide repeats in most microsatellite-unstable colorectal cancers. Cancer Res 2005, 65, 4607–4613. [Google Scholar]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar]
- Epstein, D.J. Cis-regulatory mutations in human disease. Brief. Funct. Genomics Proteomics 2009, 8, 310–316. [Google Scholar]
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol 2012, 30, 1095–1106. [Google Scholar]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar]
- Genomes Project, C.; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [Green Version]
- Cirulli, E.T.; Goldstein, D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet 2010, 11, 415–425. [Google Scholar]
- Park, J.H.; Wacholder, S.; Gail, M.H.; Peters, U.; Jacobs, K.B.; Chanock, S.J.; Chatterjee, N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet 2010, 42, 570–575. [Google Scholar]
- McCarroll, S.A. Extending genome-wide association studies to copy-number variation. Hum. Mol. Genet 2008, 17, R135–R142. [Google Scholar]
- Easton, D.F.; Eeles, R.A. Genome-wide association studies in cancer. Hum. Mol. Genet 2008, 17, R109–R115. [Google Scholar]
- Garcia-Closas, M.; Couch, F.J.; Lindstrom, S.; Michailidou, K.; Schmidt, M.K.; Brook, M.N.; Orr, N.; Rhie, S.K.; Riboli, E.; Feigelson, H.S.; et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat. Genet 2013, 45, 392–398. [Google Scholar]
- Goebell, P.J.; Knowles, M.A. Bladder cancer or bladder cancers? Genetically distinct malignant conditions of the urothelium. Urol. Oncol 2010, 28, 409–428. [Google Scholar]
- Kraft, P.; Haiman, C.A. GWAS identifies a common breast cancer risk allele among BRCA1 carriers. Nat. Genet 2010, 42, 819–820. [Google Scholar]
- Le Marchand, L.; Wilkens, L.R. Design considerations for genomic association studies: Importance of gene-environment interactions. Cancer Epidemiol. Biomark. Prev 2008, 17, 263–267. [Google Scholar]
- Garcia-Closas, M.; Rothman, N.; Figueroa, J.D.; Prokunina-Olsson, L.; Han, S.S.; Baris, D.; Jacobs, E.J.; Malats, N.; de Vivo, I.; Albanes, D.; et al. Common genetic polymorphisms modify the effect of smoking on absolute risk of bladder cancer. Cancer Res 2013, 73, 2211–2220. [Google Scholar]
- Hindorff, L.A.; Gillanders, E.M.; Manolio, T.A. Genetic architecture of cancer and other complex diseases: Lessons learned and future directions. Carcinogenesis 2011, 32, 945–954. [Google Scholar]
- Stenzl, A.; Hennenlotter, J.; Schilling, D. Can we still afford bladder cancer? Curr. Opin. Urol 2008, 18, 488–492. [Google Scholar]
- Larre, S.; Catto, J.W.; Cookson, M.S.; Messing, E.M.; Shariat, S.F.; Soloway, M.S.; Svatek, R.S.; Lotan, Y.; Zlotta, A.R.; Grossman, H.B. Screening for bladder cancer: Rationale, limitations, whom to target, and perspectives. Eur. Urol 2013, 63, 1049–1058. [Google Scholar]
- Gibson, G. Hints of hidden heritability in GWAS. Nat. Genet 2010, 42, 558–560. [Google Scholar]



{kind=link}
{kind=link}
| Chrom. | Gene | SNP (identified allele) | Location | MAF | OR (95% CI) | Study type | Ref. |
|---|---|---|---|---|---|---|---|
| 8q24 | MYC | rs9642880 [T] | Intergenic | 0.45 | 1.22 (1.15–1.29) | GWAS | [21] |
| 3q28 | TP63 | rs710521 [A] | Intergenic | 0.73 | 1.19 (1.12–1.27) | GWAS | [21] |
| 5p15 | CLPTM1L | rs401681[C] | Intronic | 0.53 | 1.12 (1.06–1.18) | Follow-up GWAS | [27] |
| 5p15 | TERT | rs2736098 [A] | Exonic | 0.25 | 1.16 (1.08–1.23) | Follow-up GWAS | [27] |
| 8q24 | PSCA | rs2294008 [T] | Exonic | 0.46 | 1.15 (1.10–1.20) | GWAS | [23] |
| 4p16 | FGFR3/TACC3 | rs798766 [T] | Intronic | 0.19 | 1.24 (1.17–1.32) | GWAS | [22] |
| 22q13 | CBX6/APOBEC3A | rs1014971 [C] | Intergenic | 0.38 | 0.88 (0.85–0.91) | GWAS | [24] |
| 19q12 | CCNE1 | rs8102137 [C] | Intergenic | 0.33 | 1.13 (1.09–1.17) | GWAS | [24] |
| 2q37 | UGT1A | rs11892031 [C] | Intronic | 0.08 | 0.84 (0.79–0.89) | GWAS | [24] |
| 18q12 | SLC14A1 | rs17674580 [T] | Intronic | 0.33 | 1.17 (1.11–1.22) | GWAS | [25] |
| 18q12 | SLC14A1 | rs7238033 [T] | Intronic | 0.43 | 1.20 (1.13–1.28) | Meta-analysis | [26] |
| 1p13 | GSTM1 | Null * | – | 0.51 | 1.5 (1.3–1.6) | Candidate gene, meta-analysis | [28] |
| 8p22 | NAT2 | Slow acetylator * | – | 0.56 | 1.4 (1.2–1.6) | Candidate gene, meta-analysis | [28] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dudek, A.M.; Grotenhuis, A.J.; Vermeulen, S.H.; Kiemeney, L.A.L.M.; Verhaegh, G.W. Urinary Bladder Cancer Susceptibility Markers. What Do We Know about Functional Mechanisms? Int. J. Mol. Sci. 2013, 14, 12346-12366. https://doi.org/10.3390/ijms140612346
Dudek AM, Grotenhuis AJ, Vermeulen SH, Kiemeney LALM, Verhaegh GW. Urinary Bladder Cancer Susceptibility Markers. What Do We Know about Functional Mechanisms? International Journal of Molecular Sciences. 2013; 14(6):12346-12366. https://doi.org/10.3390/ijms140612346
Chicago/Turabian StyleDudek, Aleksandra M., Anne J. Grotenhuis, Sita H. Vermeulen, Lambertus A. L. M. Kiemeney, and Gerald W. Verhaegh. 2013. "Urinary Bladder Cancer Susceptibility Markers. What Do We Know about Functional Mechanisms?" International Journal of Molecular Sciences 14, no. 6: 12346-12366. https://doi.org/10.3390/ijms140612346
APA StyleDudek, A. M., Grotenhuis, A. J., Vermeulen, S. H., Kiemeney, L. A. L. M., & Verhaegh, G. W. (2013). Urinary Bladder Cancer Susceptibility Markers. What Do We Know about Functional Mechanisms? International Journal of Molecular Sciences, 14(6), 12346-12366. https://doi.org/10.3390/ijms140612346