Curcumin-Induced Heme Oxygenase-1 Expression Prevents H2O2-Induced Cell Death in Wild Type and Heme Oxygenase-2 Knockout Adipose-Derived Mesenchymal Stem Cells

 and
and
Abstract
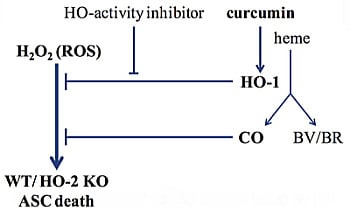
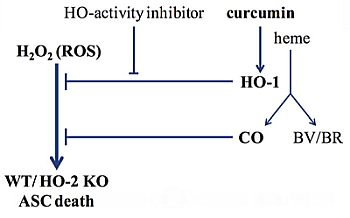
:
1. Introduction
2. Results and Discussion
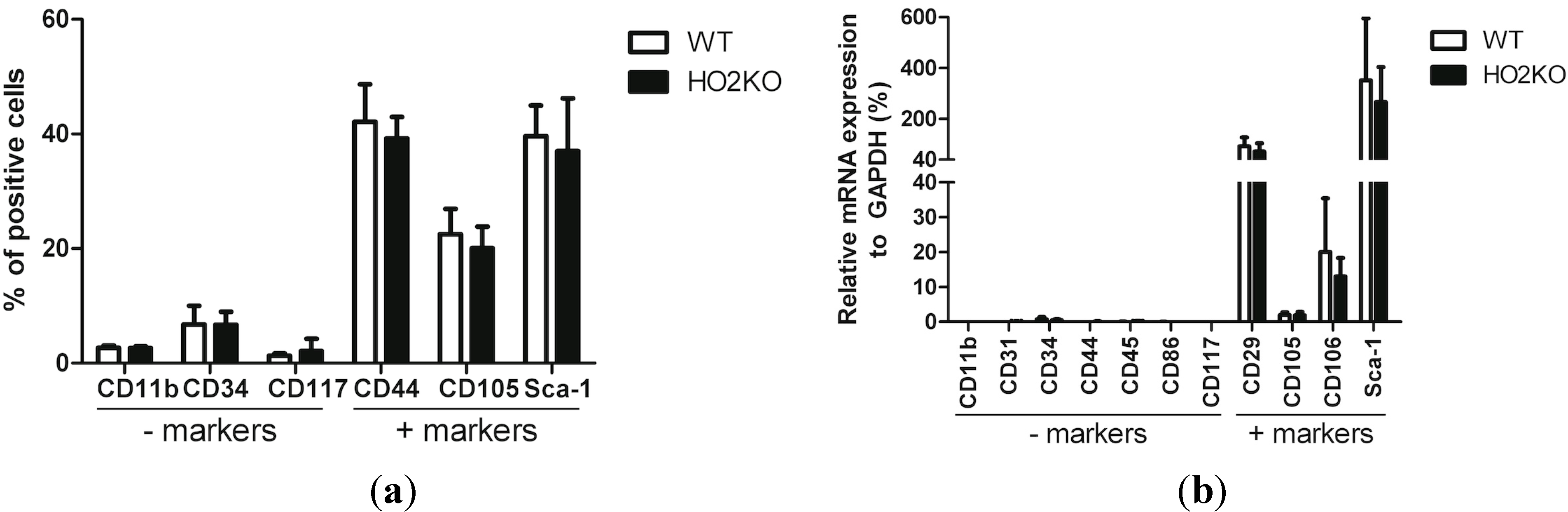
2.1. Isolation and Characterization of Adipose-Derived MSCs (Mesenchymal Stem Cells) (ASCs)

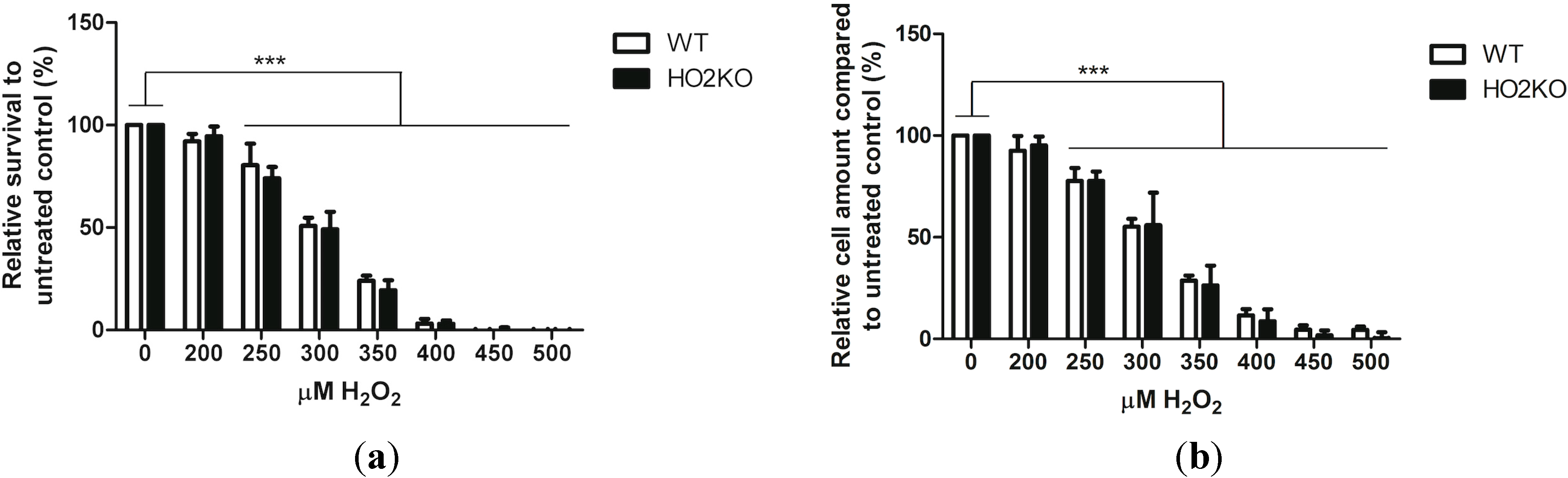
2.2. H2O2 Induces Cell Death in ASCs in a Dose-Dependent Manner Independently from HO-2 Expression

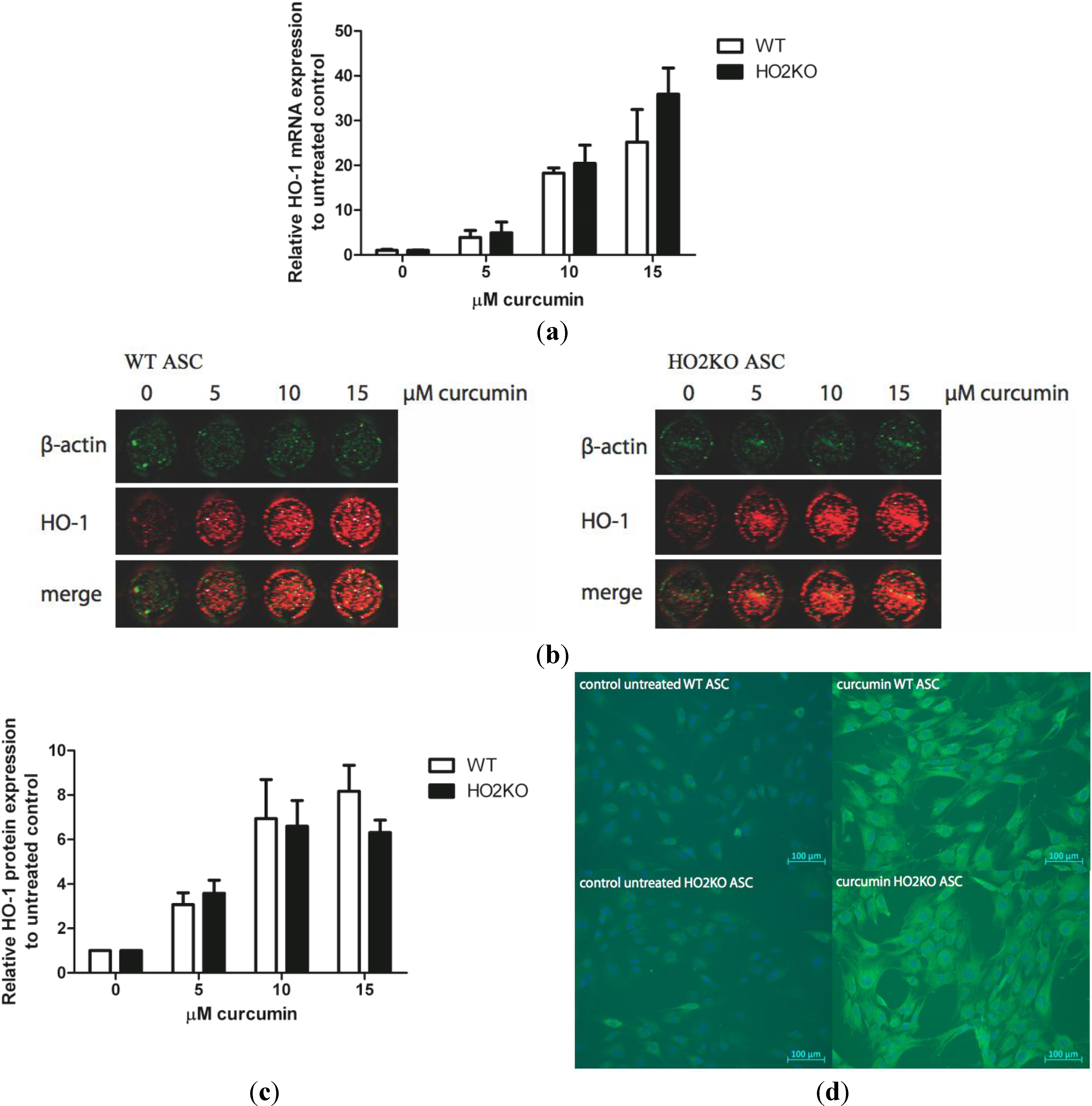
2.3. Can Curcumin Induce HO-1 Expression in ASCs?

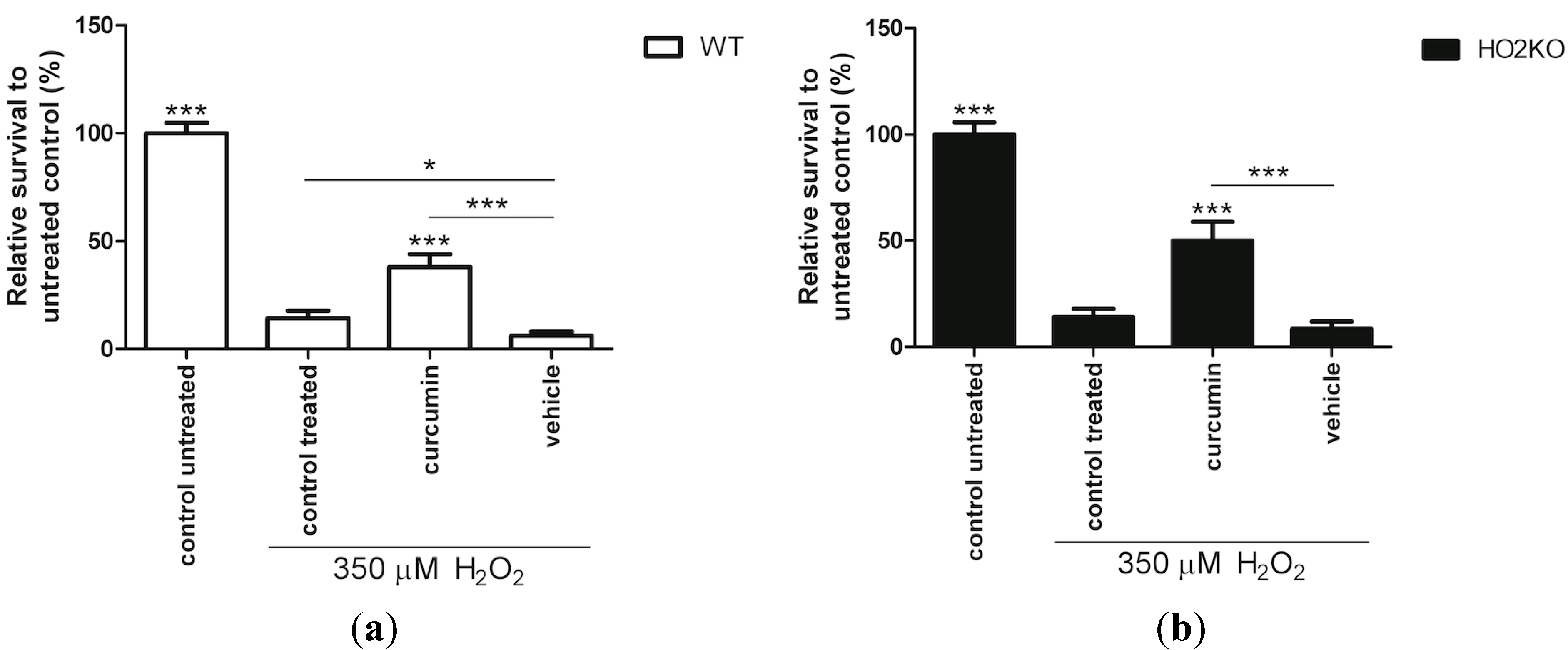
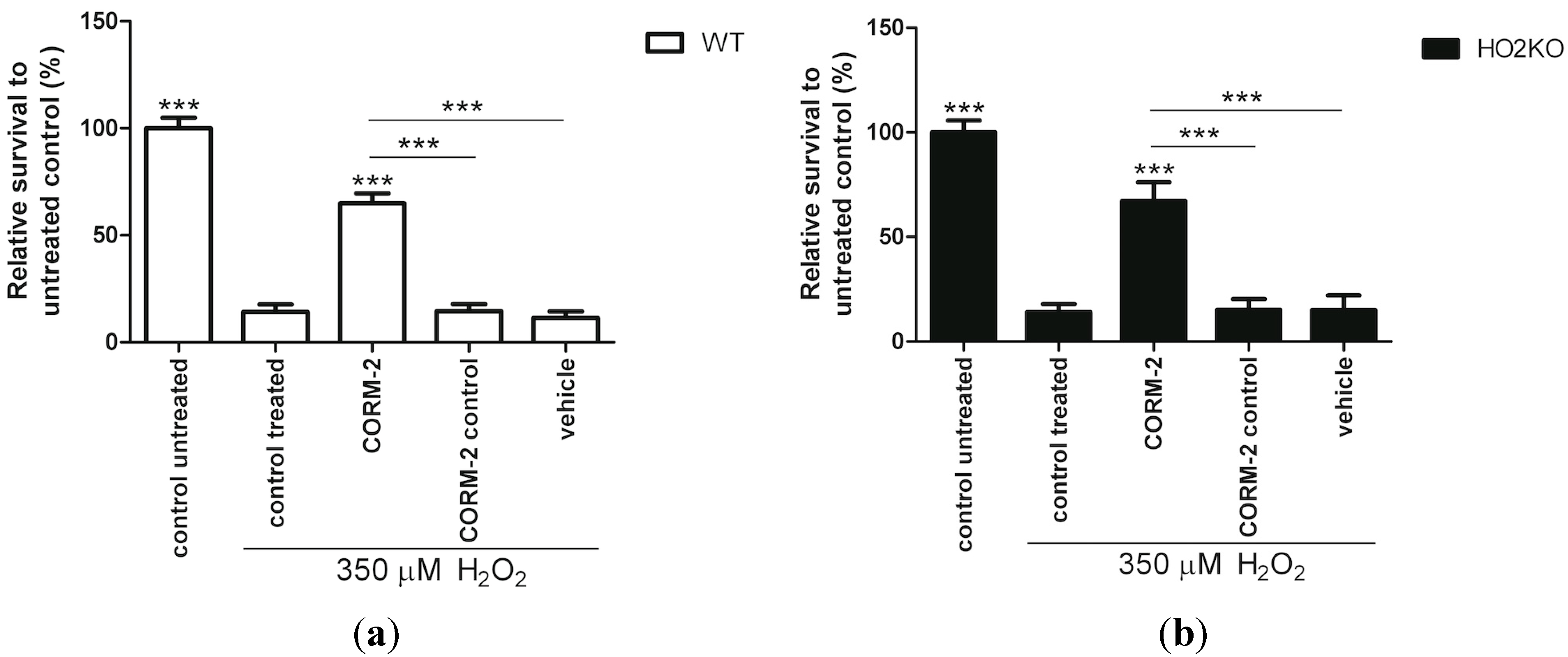
2.4. HO-1 Inducer Curcumin Protects against H2O2-Mediated ASC Death in a HO-2 Independent Manner

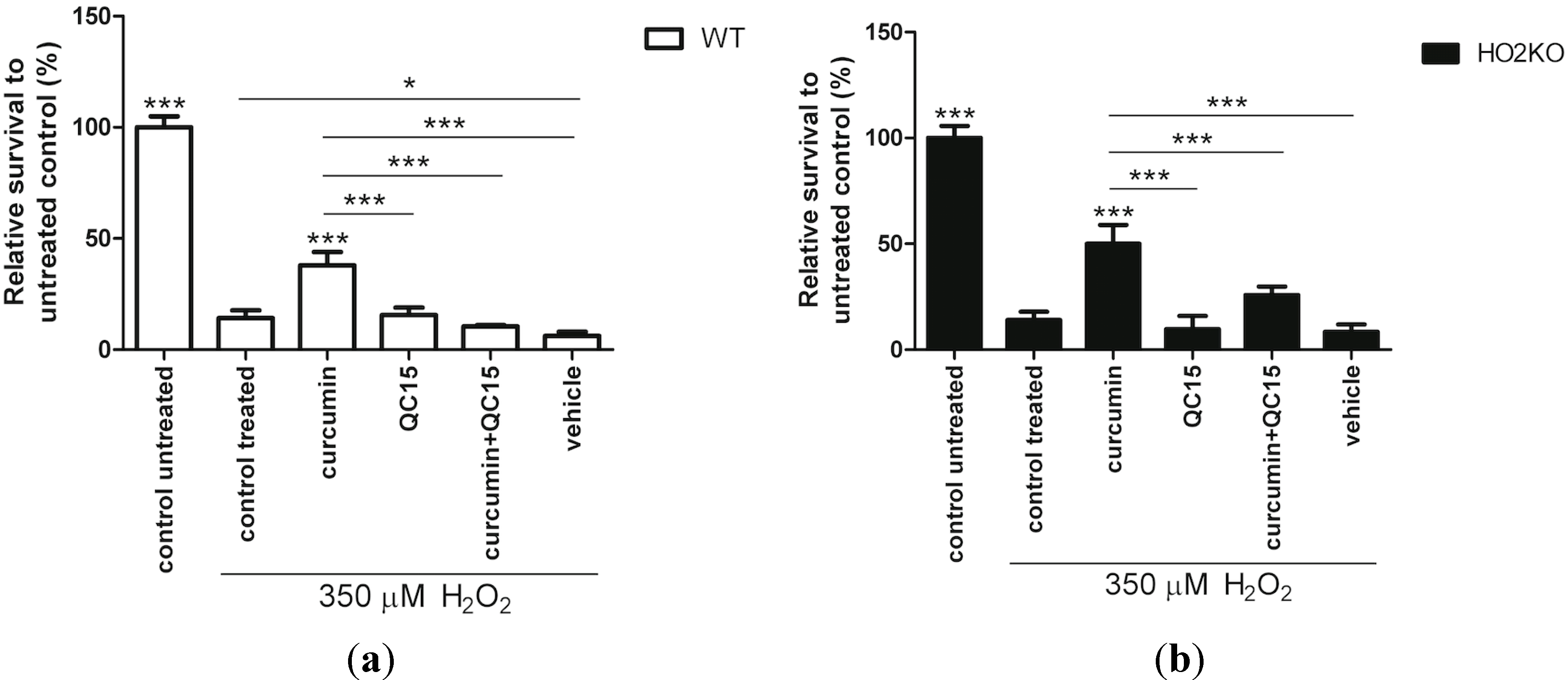
2.5. Is Rescue from H2O2-Induced ASC Death by Curcumin Mediated by HO-Activity?

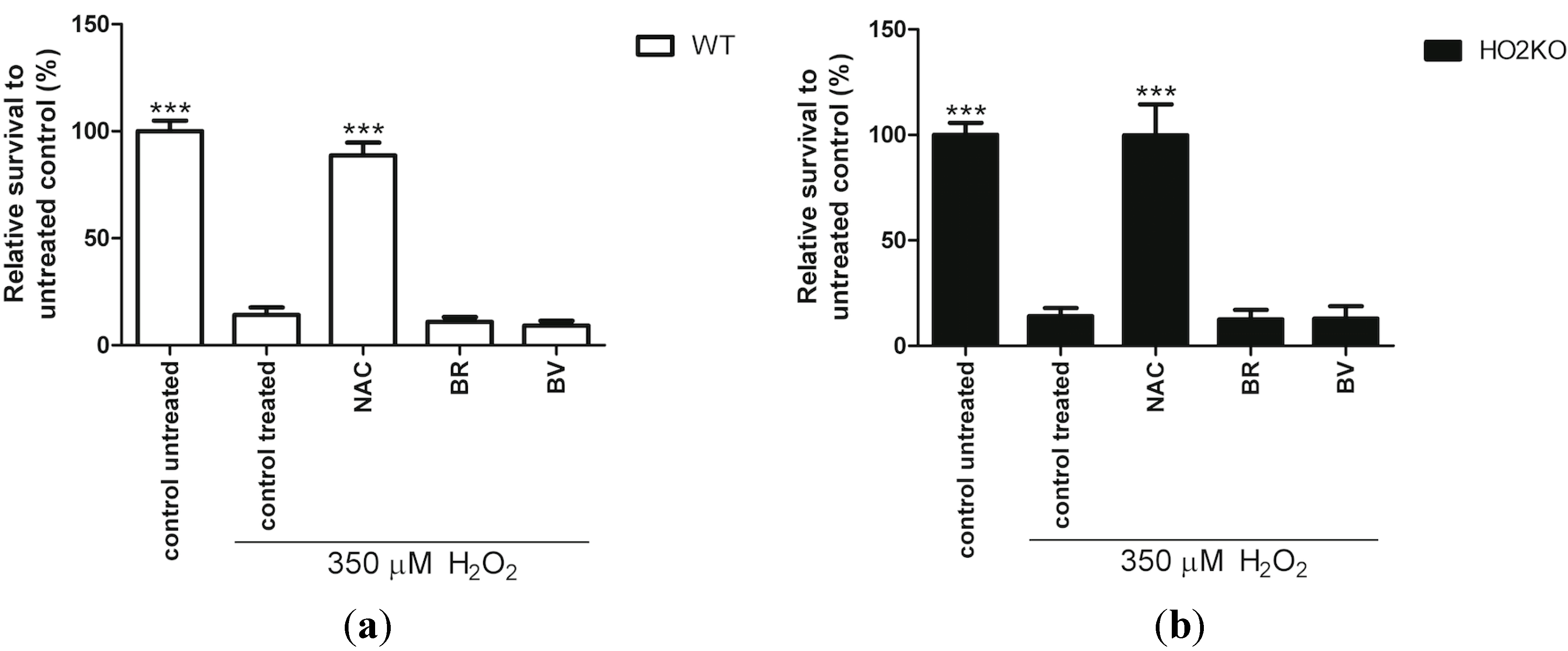
2.6. Do Anti-Oxidants Bilirubin, Biliverdin, and NAC Rescue from H2O2-Induced ASC Death?

2.7. Does HO-Effector Molecule Carbon Monoxide Influence H2O2-Induced ASC Death?

2.8. In Summary
2.9. Clinical Relevance
3. Experimental Section
3.1. Reagents
3.2. Mice
3.3. Isolation and Culture of ASCs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sense | Antisense |
|---|---|---|
| Cell Surface Marker | ||
| CD45 | 5'-GACAGAGTGCAAAGGAGACC-3' | 5'-ATCACTGGGTGTAGGTGTTTG-3' |
| Sca1 | 5'-AGCAGTTATTGTGGATTCTC-3' | 5'-TAGTACCCAGGATCTCCATAC-3' |
| CD105 | 5'-TTGTACCCACAACAGGTCTC-3' | 5'-GGTGGTAAACGTCACCTCAC-3' |
| CD29 | 5'-AAATTGAGATCAGGAGAACCAC-3' | 5'-GGTAATCTTCAGCCCTCTTG-3' |
| CD11b | 5'-CTGGTCACAGCCCTAGCC-3' | 5'-TTTGCATTCTCTTGGAAGGTC-3' |
| CD31 | 5'-CTGGTGCTCTATGCAAGC-3' | 5'-GCTGTTGATGGTGAAGGAG-3' |
| CD34 | 5'-TGAGTCTGCTGCATCTAAATAAC-3' | 5'-CTCATTGGTAGGAACTGATGG-3' |
| CD117 | 5'-GCCAGACAGCCACGTCTC-3' | 5'-CTGATTGTGCTGGATGGATG-3' |
| CD106 | 5'-CGTGGACATCTACTCTTTCC-3' | 5'-TGTAAACTGGGTAAATGTCTGG-3' |
| CD86 | 5'-GTCAGTGATCGCCAACTTC-3' | 5'-TCTTCTTAGGTTTCGGGTGAC-3' |
| Housekeeping Gene | ||
| GAPDH | 5'-GGCAAATTCAACGGCACA-3' | 5'-GTTAGTGGGGTCTCGCTCCTG-3' |
| Cytoprotective Enzymes | ||
| HO-1 | 5'-CAACATTGAGCTGTTTGAGG-3' | 5'-TGGTCTTTGTGTTCCTCTGTC-3' |
| HO-2 | 5'-AAGGAAGGGACCAAGGAAG-3' | 5'-AGTGGTGGCCAGCTTAAATAG-3' |
3.4. Heme Oxygenase Protein Expression by in-Cell Western
3.5. Heme Oxygenase Protein Expression by Immuno-Fluorescent Staining
3.6. mRNA Isolation and Quantitative-Reverse-Transcriptase-PCR
3.7. Determination of Cell Viability
3.8. Determination of Cell Amount
3.9. Statistical Analysis
4. Conclusions
Supplementary Information
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leung, A.; Crombleholme, T.M.; Keswani, S.G. Fetal wound healing: Implications for minimal scar formation. Curr. Opin. Pediatr. 2012, 24, 371–378. [Google Scholar] [CrossRef]
- Meyer, M.; Muller, A.K.; Yang, J.; Sulcova, J.; Werner, S. The role of chronic inflammation in cutaneous fibrosis: Fibroblast growth factor receptor deficiency in keratinocytes as an example. J. Investig. Dermatol. Symp. Proc. 2011, 15, 48–52. [Google Scholar] [CrossRef]
- Butler, K.L.; Goverman, J.; Ma, H.; Fischman, A.; Yu, Y.M.; Bilodeau, M.; Rad, A.M.; Bonab, A.A.; Tompkins, R.G.; Fagan, S.P. Stem cells and burns: Review and therapeutic implications. J. Burn Care Res. 2010, 31, 874–881. [Google Scholar] [CrossRef]
- Chen, F.M.; Wu, L.A.; Zhang, M.; Zhang, R.; Sun, H.H. Homing of endogenous stem/progenitor cells for in situ tissue regeneration: Promises, strategies, and translational perspectives. Biomaterials 2011, 32, 3189–3209. [Google Scholar] [CrossRef]
- Chen, J.S.; Wong, V.W.; Gurtner, G.C. Therapeutic potential of bone marrow-derived mesenchymal stem cells for cutaneous wound healing. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef]
- Maxson, S.; Lopez, E.A.; Yoo, D.; Danilkovitch-Miagkova, A.; Leroux, M.A. Concise review: Role of mesenchymal stem cells in wound repair. Stem Cells Transl. Med. 2012, 1, 142–149. [Google Scholar] [CrossRef]
- Aggarwal, S.; Moggio, A.; Bussolati, B. Concise review: Stem/progenitor cells for renal tissue repair: Current knowledge and perspectives. Stem Cells Transl. Med. 2013, 2, 1011–1019. [Google Scholar]
- Bernstein, H.S.; Srivastava, D. Stem cell therapy for cardiac disease. Pediatr. Res. 2012, 71, 491–499. [Google Scholar] [CrossRef]
- Sinclair, K.; Yerkovich, S.T.; Chambers, D.C. Mesenchymal stem cells and the lung. Respirology 2013, 18, 397–411. [Google Scholar]
- Sasaki, M.; Abe, R.; Fujita, Y.; Ando, S.; Inokuma, D.; Shimizu, H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J. Immunol. 2008, 180, 2581–2587. [Google Scholar] [CrossRef]
- Fu, X.; Fang, L.; Li, X.; Cheng, B.; Sheng, Z. Enhanced wound-healing quality with bone marrow mesenchymal stem cells autografting after skin injury. Wound Repair Regen. 2006, 14, 325–335. [Google Scholar] [CrossRef]
- Liu, Z.J.; Zhuge, Y.; Velazquez, O.C. Trafficking and differentiation of mesenchymal stem cells. J. Cell. Biochem. 2009, 106, 984–991. [Google Scholar] [CrossRef]
- Marconi, S.; Bonaconsa, M.; Scambi, I.; Squintani, G.M.; Rui, W.; Turano, E.; Ungaro, D.; D’Agostino, S.; Barbieri, F.; Angiari, S.; et al. Systemic treatment with adipose-derived mesenchymal stem cells ameliorates clinical and pathological features in the amyotrophic lateral sclerosis murine model. Neuroscience 2013, 248, 333–343. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Lu, D.B.; Jiang, Y.Z.; Zhou, L.N.; Cheng, L.Q.; Chen, B. PGC-1α prevents apoptosis in adipose-derived stem cells by reducing reactive oxygen species production in a diabetic microenvironment. Diabetes Res. Clin. Pract. 2013, 100, 368–375. [Google Scholar] [CrossRef]
- Zuk, P.A. The adipose-derived stem cell: Looking back and looking ahead. Mol. Biol. Cell 2010, 21, 1783–1787. [Google Scholar] [CrossRef]
- Mizuno, H.; Tobita, M.; Uysal, A.C. Concise review: Adipose-derived stem cells as a novel tool for future regenerative medicine. Stem Cells 2012, 30, 804–810. [Google Scholar] [CrossRef]
- Hamedi-Asl, P.; Halabian, R.; Bahmani, P.; Mohammadipour, M.; Mohammadzadeh, M.; Roushandeh, A.M.; Jahanian-Najafabadi, A.; Kuwahara, Y.; Roudkenar, M.H. Adenovirus-mediated expression of the HO-1 protein within MSCs decreased cytotoxicity and inhibited apoptosis induced by oxidative stresses. Cell Stress Chaperones 2012, 17, 181–190. [Google Scholar] [CrossRef]
- Mohammadzadeh, M.; Halabian, R.; Gharehbaghian, A.; Amirizadeh, N.; Jahanian-Najafabadi, A.; Roushandeh, A.M.; Roudkenar, M.H. Nrf-2 overexpression in mesenchymal stem cells reduces oxidative stress-induced apoptosis and cytotoxicity. Cell Stress Chaperones 2012, 17, 553–565. [Google Scholar] [CrossRef]
- Halabian, R.; Tehrani, H.A.; Jahanian-Najafabadi, A.; Habibi Roudkenar, M. Lipocalin-2-mediated upregulation of various antioxidants and growth factors protects bone marrow-derived mesenchymal stem cells against unfavorable microenvironments. Cell Stress Chaperones 2013, 18, 785–800. [Google Scholar] [CrossRef]
- Zhu, W.; Chen, J.; Cong, X.; Hu, S.; Chen, X. Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem cells. Stem Cells 2006, 24, 416–425. [Google Scholar] [CrossRef]
- Yagi, H.; Tan, J.; Tuan, R.S. Polyphenols suppress hydrogen peroxide-induced oxidative stress in human bone-marrow derived mesenchymal stem cells. J. Cell. Biochem. 2013, 114, 1163–1173. [Google Scholar] [CrossRef]
- McGinley, L.; McMahon, J.; Strappe, P.; Barry, F.; Murphy, M.; O’Toole, D.; O’Brien, T. Lentiviral vector mediated modification of mesenchymal stem cells & enhanced survival in an in vitro model of ischaemia. Stem Cell Res. Ther. 2011, 2. [Google Scholar] [CrossRef]
- Xie, X.; Sun, A.; Zhu, W.; Huang, Z.; Hu, X.; Jia, J.; Zou, Y.; Ge, J. Transplantation of mesenchymal stem cells preconditioned with hydrogen sulfide enhances repair of myocardial infarction in rats. Tohoku J. Exp. Med. 2012, 226, 29–36. [Google Scholar] [CrossRef]
- Kongpetch, S.; Kukongviriyapan, V.; Prawan, A.; Senggunprai, L.; Kukongviriyapan, U.; Buranrat, B. Crucial role of heme oxygenase-1 on the sensitivity of cholangiocarcinoma cells to chemotherapeutic agents. PLoS One 2012, 7, e34994. [Google Scholar] [CrossRef]
- Wagener, F.A.; Dankers, A.C.; van Summeren, F.; Scharstuhl, A.; van den Heuvel, J.J.; Koenderink, J.B.; Pennings, S.W.; Russel, F.G.; Masereeuw, R. Heme Oxygenase-1 and breast cancer resistance protein protect against heme-induced toxicity. Curr. Pharm. Des. 2013, 19, 2698–2707. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Ann. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Morse, D.; Choi, A.M. Heme oxygenase-1: From bench to bedside. Am. J. Respir. Crit. Care Med. 2005, 172, 660–670. [Google Scholar] [CrossRef]
- Gozzelino, R.; Soares, M.P. Coupling Heme and Iron Metabolism via Ferritin H Chain. Antioxid. Redox Signal. 2013, 20, 1754–1769. [Google Scholar] [CrossRef]
- Wagener, F.A.; Scharstuhl, A.; Tyrrell, R.M.; von den Hoff, J.W.; Jozkowicz, A.; Dulak, J.; Russel, F.G.; Kuijpers-Jagtman, A.M. The heme–heme oxygenase system in wound healing; implications for scar formation. Curr. Drug Targets 2010, 11, 1571–1585. [Google Scholar] [CrossRef]
- Burgess, A.P.; Vanella, L.; Bellner, L.; Gotlinger, K.; Falck, J.R.; Abraham, N.G.; Schwartzman, M.L.; Kappas, A. Heme oxygenase (HO-1) rescue of adipocyte dysfunction in HO-2 deficient mice via recruitment of epoxyeicosatrienoic acids (EETs) and adiponectin. Cell. Physiol. Biochem. 2012, 29, 99–110. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef]
- Sodhi, K.; Inoue, K.; Gotlinger, K.H.; Canestraro, M.; Vanella, L.; Kim, D.H.; Manthati, V.L.; Koduru, S.R.; Falck, J.R.; Schwartzman, M.L.; et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J. Pharmacol. Exp. Ther. 2009, 331, 906–916. [Google Scholar] [CrossRef]
- Hou, C.; Shen, L.; Huang, Q.; Mi, J.; Wu, Y.; Yang, M.; Zeng, W.; Li, L.; Chen, W.; Zhu, C. The effect of heme oxygenase-1 complexed with collagen on MSC performance in the treatment of diabetic ischemic ulcer. Biomaterials 2013, 34, 112–120. [Google Scholar] [CrossRef]
- Tsubokawa, T.; Yagi, K.; Nakanishi, C.; Zuka, M.; Nohara, A.; Ino, H.; Fujino, N.; Konno, T.; Kawashiri, M.A.; Ishibashi-Ueda, H.; et al. Impact of anti-apoptotic and anti-oxidative effects of bone marrow mesenchymal stem cells with transient overexpression of heme oxygenase-1 on myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1320–H1329. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, L.; Tang, Y.; Ma, G.; Shen, C.; Qi, C.; Zhu, Q.; Yao, Y.; Liu, N. HO-1 gene overexpression enhances the beneficial effects of superparamagnetic iron oxide labeled bone marrow stromal cells transplantation in swine hearts underwent ischemia/reperfusion: An MRI study. Basic Res. Cardiol. 2010, 105, 431–442. [Google Scholar] [CrossRef]
- Yang, J.J.; Yang, X.; Liu, Z.Q.; Hu, S.Y.; Du, Z.Y.; Feng, L.L.; Liu, J.F.; Chen, Y.D. Transplantation of adipose tissue-derived stem cells overexpressing heme oxygenase-1 improves functions and remodeling of infarcted myocardium in rabbits. Tohoku J. Exp. Med. 2012, 226, 231–241. [Google Scholar] [CrossRef]
- Wojakowski, W.; Tendera, M.; Cybulski, W.; Zuba-Surma, E.K.; Szade, K.; Florczyk, U.; Kozakowska, M.; Szymula, A.; Krzych, L.; Paslawska, U.; et al. Effects of intracoronary delivery of allogenic bone marrow-derived stem cells expressing heme oxygenase-1 on myocardial reperfusion injury. Thromb. Haemost. 2012, 108, 464–475. [Google Scholar] [CrossRef]
- Liang, O.D.; Mitsialis, S.A.; Chang, M.S.; Vergadi, E.; Lee, C.; Aslam, M.; Fernandez-Gonzalez, A.; Liu, X.; Baveja, R.; Kourembanas, S. Mesenchymal stromal cells expressing heme oxygenase-1 reverse pulmonary hypertension. Stem Cells 2011, 29, 99–107. [Google Scholar] [CrossRef]
- Zeng, B.; Chen, H.; Zhu, C.; Ren, X.; Lin, G.; Cao, F. Effects of combined mesenchymal stem cells and heme oxygenase-1 therapy on cardiac performance. Eur. J. Cardio-Thorac. Surg. 2008, 34, 850–856. [Google Scholar] [CrossRef]
- Zeng, B.; Lin, G.; Ren, X.; Zhang, Y.; Chen, H. Over-expression of HO-1 on mesenchymal stem cells promotes angiogenesis and improves myocardial function in infarcted myocardium. J. Biomed. Sci. 2010, 17, 80. [Google Scholar]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef]
- Lin, C.Y.; Peng, C.Y.; Huang, T.T.; Wu, M.L.; Lai, Y.L.; Peng, D.H.; Chen, P.F.; Chen, H.F.; Yen, B.L.; Wu, K.K.; et al. Exacerbation of oxidative stress-induced cell death and differentiation in induced pluripotent stem cells lacking heme oxygenase-1. Stem Cells Dev. 2012, 21, 1675–1687. [Google Scholar] [CrossRef]
- Basuroy, S.; Bhattacharya, S.; Tcheranova, D.; Qu, Y.; Regan, R.F.; Leffler, C.W.; Parfenova, H. HO-2 provides endogenous protection against oxidative stress and apoptosis caused by TNF-α in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2006, 291, C897–C908. [Google Scholar] [CrossRef]
- Chen, J.; Regan, R.F. Heme oxygenase-2 gene deletion increases astrocyte vulnerability to hemin. Biochem. Biophys. Res. Commun. 2004, 318, 88–94. [Google Scholar] [CrossRef]
- Rogers, B.; Yakopson, V.; Teng, Z.P.; Guo, Y.; Regan, R.F. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Radic. Biol. Med. 2003, 35, 872–881. [Google Scholar] [CrossRef]
- Chen, J.; Tu, Y.; Connolly, E.C.; Ronnett, G.V. Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr. Neurovasc. Res. 2005, 2, 121–131. [Google Scholar] [CrossRef]
- Chen, J.; Tu, Y.; Moon, C.; Nagata, E.; Ronnett, G.V. Heme oxygenase-1 and heme oxygenase-2 have distinct roles in the proliferation and survival of olfactory receptor neurons mediated by cGMP and bilirubin, respectively. J. Neurochem. 2003, 85, 1247–1261. [Google Scholar] [CrossRef]
- Kim, Y.M.; Choi, B.M.; Kim, Y.S.; Kwon, Y.G.; Kibbe, M.R.; Billiar, T.R.; Tzeng, E. Protective effect of p53 in vascular smooth muscle cells against nitric oxide-induced apoptosis is mediated by up-regulation of heme oxygenase-2. BMB Rep. 2008, 41, 164–169. [Google Scholar] [CrossRef]
- Parfenova, H.; Basuroy, S.; Bhattacharya, S.; Tcheranova, D.; Qu, Y.; Regan, R.F.; Leffler, C.W. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: Contributions of HO-1 and HO-2 to cytoprotection. Am. J. Physiol. Cell Physiol. 2006, 290, C1399–C1410. [Google Scholar] [CrossRef]
- Dore, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef]
- Dore, S.; Goto, S.; Sampei, K.; Blackshaw, S.; Hester, L.D.; Ingi, T.; Sawa, A.; Traystman, R.J.; Koehler, R.C.; Snyder, S.H. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 2000, 99, 587–592. [Google Scholar] [CrossRef]
- Ter Huurne, M.; Schelbergen, R.; Blattes, R.; Blom, A.; de Munter, W.; Grevers, L.C.; Jeanson, J.; Noel, D.; Casteilla, L.; Jorgensen, C.; et al. Antiinflammatory and chondroprotective effects of intraarticular injection of adipose-derived stem cells in experimental osteoarthritis. Arthritis Rheumatol. 2012, 64, 3604–3613. [Google Scholar] [CrossRef]
- Zheng, B.; Cao, B.; Li, G.; Huard, J. Mouse adipose-derived stem cells undergo multilineage differentiation in vitro but primarily osteogenic and chondrogenic differentiation in vivo. Tissue Eng. 2006, 12, 1891–1901. [Google Scholar] [CrossRef]
- Kolf, C.M.; Cho, E.; Tuan, R.S. Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: Regulation of niche, self-renewal and differentiation. Arthritis Res. Ther. 2007, 9. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; MacDougald, O.A. Adipose tissue stem cells meet preadipocyte commitment: Going back to the future. J. Lipid Res. 2012, 53, 227–246. [Google Scholar] [CrossRef]
- Yamamoto, N.; Akamatsu, H.; Hasegawa, S.; Yamada, T.; Nakata, S.; Ohkuma, M.; Miyachi, E.; Marunouchi, T.; Matsunaga, K. Isolation of multipotent stem cells from mouse adipose tissue. J. Dermatol. Sci. 2007, 48, 43–52. [Google Scholar] [CrossRef]
- Nakagami, H.; Maeda, K.; Morishita, R.; Iguchi, S.; Nishikawa, T.; Takami, Y.; Kikuchi, Y.; Saito, Y.; Tamai, K.; Ogihara, T.; et al. Novel autologous cell therapy in ischemic limb disease through growth factor secretion by cultured adipose tissue-derived stromal cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2542–2547. [Google Scholar] [CrossRef]
- Sung, J.H.; Yang, H.M.; Park, J.B.; Choi, G.S.; Joh, J.W.; Kwon, C.H.; Chun, J.M.; Lee, S.K.; Kim, S.J. Isolation and characterization of mouse mesenchymal stem cells. Transplant. Proc. 2008, 40, 2649–2654. [Google Scholar] [CrossRef]
- Wang, L.; Huang, H.; Fan, Y.; Kong, B.; Hu, H.; Hu, K.; Guo, J.; Mei, Y.; Liu, W.L. Effects of Downregulation of MicroRNA-181a on H2O2-Induced H9c2 Cell Apoptosis via the Mitochondrial Apoptotic Pathway. Oxid. Med. Cell. Longev. 2014, 2014, 960362. [Google Scholar] [CrossRef]
- Kou, X.; Shen, K.; An, Y.; Qi, S.; Dai, W.X.; Yin, Z. Ampelopsin inhibits H2O2-induced apoptosis by ERK and Akt signaling pathways and up-regulation of heme oxygenase-1. Phytother. Res. PTR 2012, 26, 988–994. [Google Scholar] [CrossRef]
- Wang, F.W.; Wang, Z.; Zhang, Y.M.; Du, Z.X.; Zhang, X.L.; Liu, Q.; Guo, Y.J.; Li, X.G.; Hao, A.J. Protective effect of melatonin on bone marrow mesenchymal stem cells against hydrogen peroxide-induced apoptosis in vitro. J. Cell. Biochem. 2013, 114, 2346–2355. [Google Scholar] [CrossRef]
- Scharstuhl, A.; Mutsaers, H.A.; Pennings, S.W.; Szarek, W.A.; Russel, F.G.; Wagener, F.A. Curcumin-induced fibroblast apoptosis and in vitro wound contraction are regulated by antioxidants and heme oxygenase: Implications for scar formation. J. Cell. Mol. Med. 2009, 13, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Ren, X.; Lin, G.; Zhu, C.; Chen, H.; Yin, J.; Jiang, H.; Yang, B.; Ding, D. Paracrine action of HO-1-modified mesenchymal stem cells mediates cardiac protection and functional improvement. Cell Biol. Int. 2008, 32, 1256–1264. [Google Scholar] [CrossRef]
- Masoud, M.S.; Anwar, S.S.; Afzal, M.Z.; Mehmood, A.; Khan, S.N.; Riazuddin, S. Pre-conditioned mesenchymal stem cells ameliorate renal ischemic injury in rats by augmented survival and engraftment. J. Transl. Med. 2012, 10. [Google Scholar] [CrossRef]
- Loboda, A.; Was, H.; Jozkowicz, A.; Dulak, J. Janus face of Nrf2-HO-1 axis in cancer—friend in chemoprevention, foe in anticancer therapy. Lung Cancer 2008, 60, 1–3. [Google Scholar] [CrossRef]
- Alcaraz, M.J.; Habib, A.; Lebret, M.; Creminon, C.; Levy-Toledano, S.; Maclouf, J. Enhanced expression of haem oxygenase-1 by nitric oxide and antiinflammatory drugs in NIH 3T3 fibroblasts. Br. J. Pharmacol. 2000, 130, 57–64. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Zhou, Z.S.; Lu, G.S.; Song, B.; Guo, J.X. Melatonin improves bladder symptoms and may ameliorate bladder damage via increasing HO-1 in rats. Inflammation 2013, 36, 651–657. [Google Scholar] [CrossRef]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. CMLS 2008, 65, 1631–1652. [Google Scholar] [CrossRef]
- Epstein, J.; Sanderson, I.R.; Macdonald, T.T. Curcumin as a therapeutic agent: The evidence from in vitro, animal and human studies. Br. J. Nutr. 2010, 103, 1545–1557. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R.; Bassi, R.; Green, C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med. 2000, 28, 1303–1312. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The Indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K.; Aggarwal, B.B. Curcumin, a component of golden spice: From bedside to bench and back. Biotechnol. Adv. 2014, 32, 1053–1064. [Google Scholar] [CrossRef]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of curcumin. Curr. Drug Targets 2011, 12, 332–347. [Google Scholar] [CrossRef]
- Sugishima, M.; Higashimoto, Y.; Oishi, T.; Takahashi, H.; Sakamoto, H.; Noguchi, M.; Fukuyama, K. X-ray crystallographic and biochemical characterization of the inhibitory action of an imidazole-dioxolane compound on heme oxygenase. Biochemistry 2007, 46, 1860–1867. [Google Scholar] [CrossRef]
- Kinobe, R.T.; Vlahakis, J.Z.; Vreman, H.J.; Stevenson, D.K.; Brien, J.F.; Szarek, W.A.; Nakatsu, K. Selectivity of imidazole-dioxolane compounds for in vitro inhibition of microsomal haem oxygenase isoforms. Br. J. Pharmacol. 2006, 147, 307–315. [Google Scholar] [CrossRef]
- Rahman, M.N.; Vlahakis, J.Z.; Szarek, W.A.; Nakatsu, K.; Jia, Z. X-ray crystal structure of human heme oxygenase-1 in complex with 1-(adamantan-1-yl)-2-(1H-imidazol-1-yl)ethanone: A common binding mode for imidazole-based heme oxygenase-1 inhibitors. J. Med. Chem. 2008, 51, 5943–5952. [Google Scholar]
- Vlahakis, J.Z.; Kinobe, R.T.; Bowers, R.J.; Brien, J.F.; Nakatsu, K.; Szarek, W.A. Synthesis and evaluation of azalanstat analogues as heme oxygenase inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1457–1461. [Google Scholar] [CrossRef]
- Lin, H.Y.; Shen, S.C.; Lin, C.W.; Yang, L.Y.; Chen, Y.C. Baicalein inhibition of hydrogen peroxide-induced apoptosis via ROS-dependent heme oxygenase 1 gene expression. Biochim. Biophys. Acta 2007, 1773, 1073–1086. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, B.H.; Chen, L.; An, W. Overexpression of heme oxygenase-1 protects smooth muscle cells against oxidative injury and inhibits cell proliferation. Cell Res. 2002, 12, 123–132. [Google Scholar] [CrossRef]
- Fu, Y.Q.; Fang, F.; Lu, Z.Y.; Kuang, F.W.; Xu, F. N-Acetylcysteine protects alveolar epithelial cells from hydrogen peroxide-induced apoptosis through scavenging reactive oxygen species and suppressing c-Jun N-terminal kinase. Exp. Lung Res. 2010, 36, 352–361. [Google Scholar]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar]
- Fernandez-Checa, J.C.; Garcia-Ruiz, C.; Colell, A.; Morales, A.; Mari, M.; Miranda, M.; Ardite, E. Oxidative stress: Role of mitochondria and protection by glutathione. Biofactors 1998, 8, 7–11. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Mari, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 1997, 273, G7–G17. [Google Scholar]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef]
- Aronis, A.; Aharoni-Simon, M.; Madar, Z.; Tirosh, O. Triacylglycerol-induced impairment in mitochondrial biogenesis and function in J774.2 and mouse peritoneal macrophage foam cells. Arch. Biochem. Biophys. 2009, 492, 74–81. [Google Scholar] [CrossRef]
- Bruggisser, R.; von Daeniken, K.; Jundt, G.; Schaffner, W.; Tullberg-Reinert, H. Interference of plant extracts, phytoestrogens and antioxidants with the MTT tetrazolium assay. Planta Med. 2002, 68, 445–448. [Google Scholar] [CrossRef]
- Krifka, S.; Hiller, K.A.; Spagnuolo, G.; Jewett, A.; Schmalz, G.; Schweikl, H. The influence of glutathione on redox regulation by antioxidant proteins and apoptosis in macrophages exposed to 2-hydroxyethyl methacrylate (HEMA). Biomaterials 2012, 33, 5177–5186. [Google Scholar] [CrossRef]
- Xiong, L.; Sun, J.; Hirche, C.; Yang, J.; Yang, Y.; Xia, Y.; Lehnhardt, M.; Wang, R.; Fu, X. In vitro N-acetyl-l-cysteine promotes proliferation and suppresses interleukin-8 expression in adipose-derived stem cells. Aesthet. Plast. Surg. 2012, 36, 1260–1265. [Google Scholar] [CrossRef]
- Ji, H.; Liu, Y.; Zhao, X.; Zhang, M. N-acetyl-l-cysteine enhances the osteogenic differentiation and inhibits the adipogenic differentiation through up regulation of Wnt 5a and down regulation of PPARG in bone marrow stromal cells. Biomed. Pharmacother. 2011, 65, 369–374. [Google Scholar] [CrossRef]
- Park, C.; So, H.S.; Shin, C.H.; Baek, S.H.; Moon, B.S.; Shin, S.H.; Lee, H.S.; Lee, D.W.; Park, R. Quercetin protects the hydrogen peroxide-induced apoptosis via inhibition of mitochondrial dysfunction in H9c2 cardiomyoblast cells. Biochem. Pharmacol. 2003, 66, 1287–1295. [Google Scholar] [CrossRef]
- Singh, M.; Sharma, H.; Singh, N. Hydrogen peroxide induces apoptosis in HeLa cells through mitochondrial pathway. Mitochondrion 2007, 7, 367–373. [Google Scholar] [CrossRef]
- Qian, Y.; Du, Y.H.; Tang, Y.B.; Lv, X.F.; Liu, J.; Zhou, J.G.; Guan, Y.Y. ClC-3 chloride channel prevents apoptosis induced by hydrogen peroxide in basilar artery smooth muscle cells through mitochondria dependent pathway. Apoptosis 2011, 16, 468–477. [Google Scholar] [CrossRef]
- Liu, N.S.; Du, X.; Lu, J.; He, B.P. Diva reduces cell death in response to oxidative stress and cytotoxicity. PLoS One 2012, 7, e43180. [Google Scholar] [CrossRef]
- Chen, H.Y.; Zhang, X.; Chen, S.F.; Zhang, Y.X.; Liu, Y.H.; Ma, L.L.; Wang, L.X. The protective effect of 17β-estradiol against hydrogen peroxide-induced apoptosis on mesenchymal stem cell. Biomed. Pharmacother. 2012, 66, 57–63. [Google Scholar] [CrossRef]
- Wei, H.; Li, Z.; Hu, S.; Chen, X.; Cong, X. Apoptosis of mesenchymal stem cells induced by hydrogen peroxide concerns both endoplasmic reticulum stress and mitochondrial death pathway through regulation of caspases, p38 and JNK. J. Cell. Biochem. 2010, 111, 967–978. [Google Scholar] [CrossRef]
- Lee, S.J.; Ryter, S.W.; Xu, J.F.; Nakahira, K.; Kim, H.P.; Choi, A.M.K.; Kim, Y.S. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am. J. Respir. Cell Mol. 2011, 45, 867–873. [Google Scholar] [CrossRef]
- Choi, Y.K.; Por, E.D.; Kwon, Y.G.; Kim, Y.M. Regulation of ROS production and vascular function by carbon monoxide. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef]
- Piantadosi, C.A. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic. Biol. Med. 2008, 45, 562–569. [Google Scholar] [CrossRef]
- Chi, P.L.; Liu, C.J.; Lee, I.T.; Chen, Y.W.; Hsiao, L.D.; Yang, C.M. HO-1 induction by CO-RM2 attenuates TNF-α-induced cytosolic phospholipase A2 expression via inhibition of PKCα-dependent NADPH oxidase/ROS and NF-κB. Med. Inflamm. 2014, 2014, 279171. [Google Scholar] [CrossRef]
- Kim, H.J.; Zheng, M.; Kim, S.K.; Cho, J.J.; Shin, C.H.; Joe, Y.; Chung, H.T. CO/HO-1 Induces NQO-1 Expression via Nrf2 Activation. Immune Netw. 2011, 11, 376–382. [Google Scholar] [CrossRef]
- Lee, B.S.; Heo, J.; Kim, Y.M.; Shim, S.M.; Pae, H.O.; Kim, Y.M.; Chung, H.T. Carbon monoxide mediates heme oxygenase 1 induction via Nrf2 activation in hepatoma cells. Biochem. Biophys. Res. Commun. 2006, 343, 965–972. [Google Scholar] [CrossRef]
- Kim, H.P.; Choi, A.M. A new road to induce heme oxygenase-1 expression by carbon monoxide. Circ. Res. 2007, 101, 862–864. [Google Scholar] [CrossRef]
- Ozaki, K.S.; Kimura, S.; Murase, N. Use of carbon monoxide in minimizing ischemia reperfusion injury in transplantation. Transpl. Rev. 2012, 26, 125–139. [Google Scholar] [CrossRef]
- Bilban, M.; Haschemi, A.; Wegiel, B.; Chin, B.Y.; Wagner, O.; Otterbein, L.E. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J. Mol. Med. 2008, 86, 267–279. [Google Scholar] [CrossRef]
- Heo, J.M.; Kim, H.J.; Ha, Y.M.; Park, M.K.; Kang, Y.J.; Lee, Y.S.; Seo, H.G.; Lee, J.H.; Yun-Choi, H.S.; Chang, K.C. YS 51, 1-(β-naphtylmethyl)-6,7-dihydroxy-1,2,3,4,-tetrahydroisoquinoline, protects endothelial cells against hydrogen peroxide-induced injury via carbon monoxide derived from heme oxygenase-1. Biochem. Pharmacol. 2007, 74, 1361–1370. [Google Scholar] [CrossRef]
- Wei, L.; Fraser, J.L.; Lu, Z.Y.; Hu, X.; Yu, S.P. Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and neurogenesis after cerebral ischemia in rats. Neurobiol. Dis. 2012, 46, 635–645. [Google Scholar] [CrossRef]
- Malgieri, A.; Kantzari, E.; Patrizi, M.P.; Gambardella, S. Bone marrow and umbilical cord blood human mesenchymal stem cells: State of the art. Int. J. Clin. Exp. Med. 2010, 3, 248–269. [Google Scholar]
- Tse, W.T.; Pendleton, J.D.; Beyer, W.M.; Egalka, M.C.; Guinan, E.C. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: Implications in transplantation. Transplantation 2003, 75, 389–397. [Google Scholar] [CrossRef]
- Shin, L.; Peterson, D.A. Human mesenchymal stem cell grafts enhance normal and impaired wound healing by recruiting existing endogenous tissue stem/progenitor cells. Stem Cells Transl. Med. 2013, 2, 33–42. [Google Scholar] [CrossRef]
- Tang, Y.L.; Tang, Y.; Zhang, Y.C.; Qian, K.; Shen, L.; Phillips, M.I. Improved graft mesenchymal stem cell survival in ischemic heart with a hypoxia-regulated heme oxygenase-1 vector. J. Am. Coll. Cardiol. 2005, 46, 1339–1350. [Google Scholar] [CrossRef]
- Caplan, A.I.; Correa, D. The MSC: An injury drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef]
- Herrmann, J.L.; Abarbanell, A.M.; Weil, B.R.; Manukyan, M.C.; Poynter, J.A.; Brewster, B.J.; Wang, Y.; Meldrum, D.R. Optimizing stem cell function for the treatment of ischemic heart disease. J. Surg. Res. 2011, 166, 138–145. [Google Scholar] [CrossRef]
- Di Francesco, L.; Totani, L.; Dovizio, M.; Piccoli, A.; di Francesco, A.; Salvatore, T.; Pandolfi, A.; Evangelista, V.; Dercho, R.A.; Seta, F.; et al. Induction of prostacyclin by steady laminar shear stress suppresses tumor necrosis factor-α biosynthesis via heme oxygenase-1 in human endothelial cells. Circ. Res. 2009, 104, 506–513. [Google Scholar] [CrossRef]
- Bellner, L.; Martinelli, L.; Halilovic, A.; Patil, K.; Puri, N.; Dunn, M.W.; Regan, R.F.; Schwartzman, M.L. Heme oxygenase-2 deletion causes endothelial cell activation marked by oxidative stress, inflammation, and angiogenesis. J. Pharmacol. Exp. Ther. 2009, 331, 925–932. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cremers, N.A.J.; Lundvig, D.M.S.; Van Dalen, S.C.M.; Schelbergen, R.F.; Van Lent, P.L.E.M.; Szarek, W.A.; Regan, R.F.; Carels, C.E.; Wagener, F.A.D.T.G. Curcumin-Induced Heme Oxygenase-1 Expression Prevents H2O2-Induced Cell Death in Wild Type and Heme Oxygenase-2 Knockout Adipose-Derived Mesenchymal Stem Cells. Int. J. Mol. Sci. 2014, 15, 17974-17999. https://doi.org/10.3390/ijms151017974
Cremers NAJ, Lundvig DMS, Van Dalen SCM, Schelbergen RF, Van Lent PLEM, Szarek WA, Regan RF, Carels CE, Wagener FADTG. Curcumin-Induced Heme Oxygenase-1 Expression Prevents H2O2-Induced Cell Death in Wild Type and Heme Oxygenase-2 Knockout Adipose-Derived Mesenchymal Stem Cells. International Journal of Molecular Sciences. 2014; 15(10):17974-17999. https://doi.org/10.3390/ijms151017974
Chicago/Turabian StyleCremers, Niels A. J., Ditte M. S. Lundvig, Stephanie C. M. Van Dalen, Rik F. Schelbergen, Peter L. E. M. Van Lent, Walter A. Szarek, Raymond F. Regan, Carine E. Carels, and Frank A. D. T. G. Wagener. 2014. "Curcumin-Induced Heme Oxygenase-1 Expression Prevents H2O2-Induced Cell Death in Wild Type and Heme Oxygenase-2 Knockout Adipose-Derived Mesenchymal Stem Cells" International Journal of Molecular Sciences 15, no. 10: 17974-17999. https://doi.org/10.3390/ijms151017974
APA StyleCremers, N. A. J., Lundvig, D. M. S., Van Dalen, S. C. M., Schelbergen, R. F., Van Lent, P. L. E. M., Szarek, W. A., Regan, R. F., Carels, C. E., & Wagener, F. A. D. T. G. (2014). Curcumin-Induced Heme Oxygenase-1 Expression Prevents H2O2-Induced Cell Death in Wild Type and Heme Oxygenase-2 Knockout Adipose-Derived Mesenchymal Stem Cells. International Journal of Molecular Sciences, 15(10), 17974-17999. https://doi.org/10.3390/ijms151017974