iHyd-PseAAC: Predicting Hydroxyproline and Hydroxylysine in Proteins by Incorporating Dipeptide Position-Specific Propensity into Pseudo Amino Acid Composition

Abstract
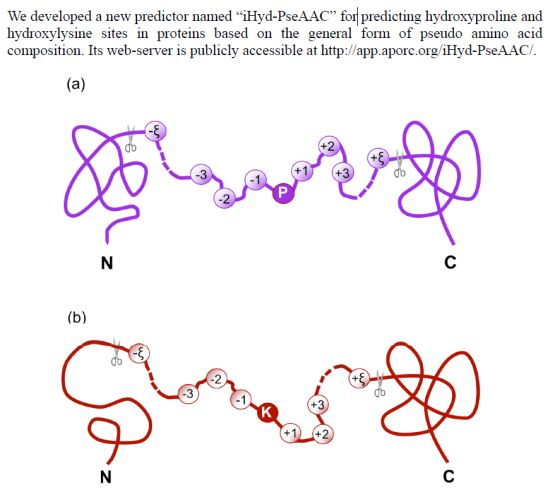
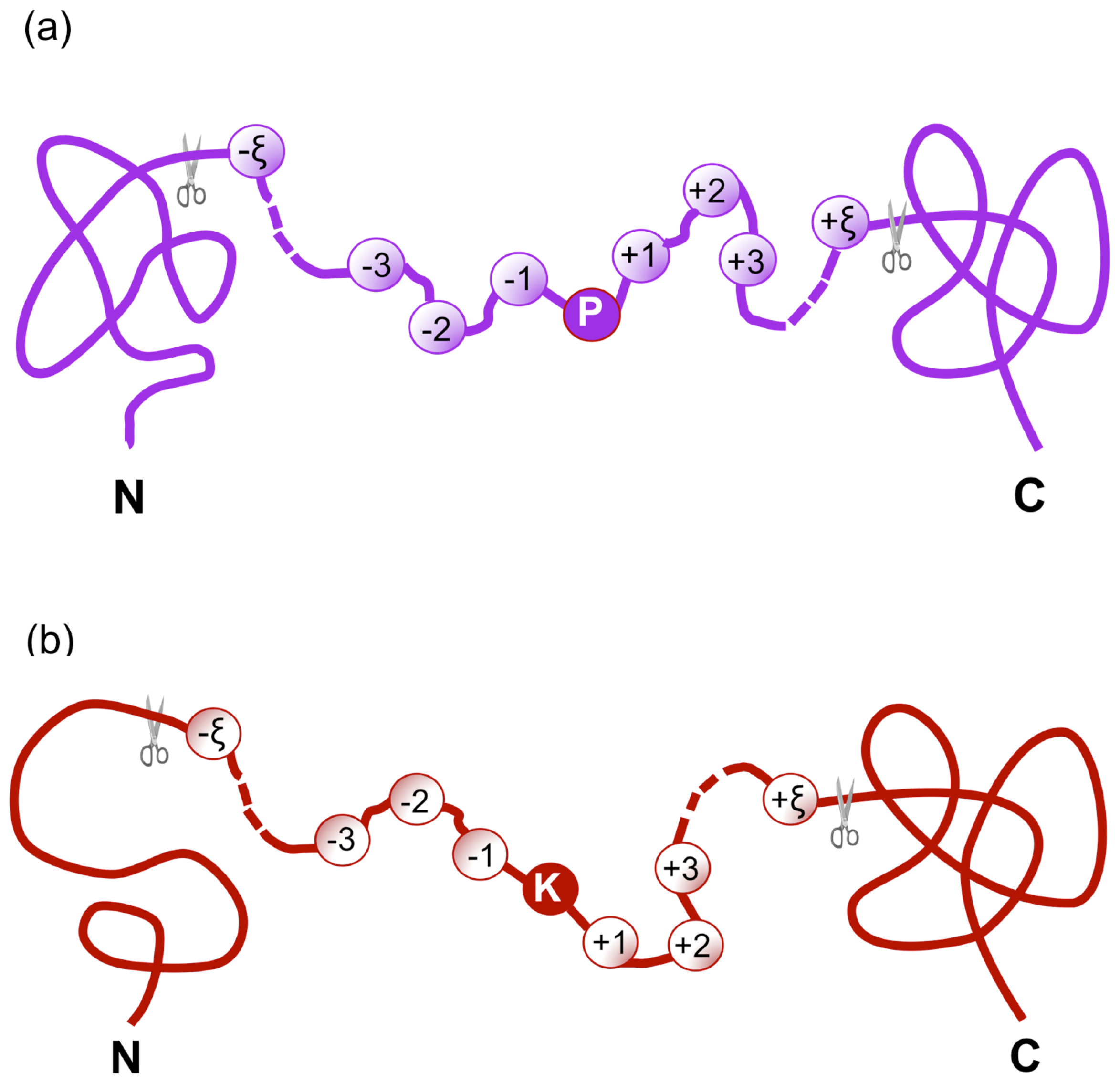
:
1. Introduction
2. Results and Discussion
2.1. Benchmark Dataset
 ) with the profile of Equation (1) can be further classified into the following categories:
) with the profile of Equation (1) can be further classified into the following categories: is the benchmark dataset for studying hydroxyproline residues,
is the benchmark dataset for studying hydroxyproline residues,
 the benchmark dataset for studying hydroxylysine residues, ⋃ the symbol for “union” in the set theory,
contains the samples for the Pro-hydroxylated peptide only,
contains the non-Pro-hydroxylated peptide only (cf. Equation (2)),
contains the samples for the Lys-hydroxylated peptide only and
contains the non-Lys-hydroxylated peptide only (cf. Equation (3))., contained 636 + 2699 = 3338 peptide samples, of which 636 were Pro-hydroxylated peptides belonging the positive subset
, and 2669 were non-Pro-hydroxylated peptides belonging to the negative subset,
; and that the benchmark dataset,
, contained 107 + 836 = 943 peptide samples, of which 107 were Lys-hydroxylated peptides belonging to the positive subset,
, and 836 were non-Lys-hydroxylated peptides belonging to the negative subset,
. For the reader’s convenience, the peptide sequences, as well as their hydroxylation or non-hydroxylation sites in proteins are given in the Supplementary Information, S1 and S2, for
and
, respectively.
the benchmark dataset for studying hydroxylysine residues, ⋃ the symbol for “union” in the set theory,
contains the samples for the Pro-hydroxylated peptide only,
contains the non-Pro-hydroxylated peptide only (cf. Equation (2)),
contains the samples for the Lys-hydroxylated peptide only and
contains the non-Lys-hydroxylated peptide only (cf. Equation (3))., contained 636 + 2699 = 3338 peptide samples, of which 636 were Pro-hydroxylated peptides belonging the positive subset
, and 2669 were non-Pro-hydroxylated peptides belonging to the negative subset,
; and that the benchmark dataset,
, contained 107 + 836 = 943 peptide samples, of which 107 were Lys-hydroxylated peptides belonging to the positive subset,
, and 836 were non-Lys-hydroxylated peptides belonging to the negative subset,
. For the reader’s convenience, the peptide sequences, as well as their hydroxylation or non-hydroxylation sites in proteins are given in the Supplementary Information, S1 and S2, for
and
, respectively.2.2. Feature Vector Construction
 and
and
 , to define the peptide samples via Equation (5).
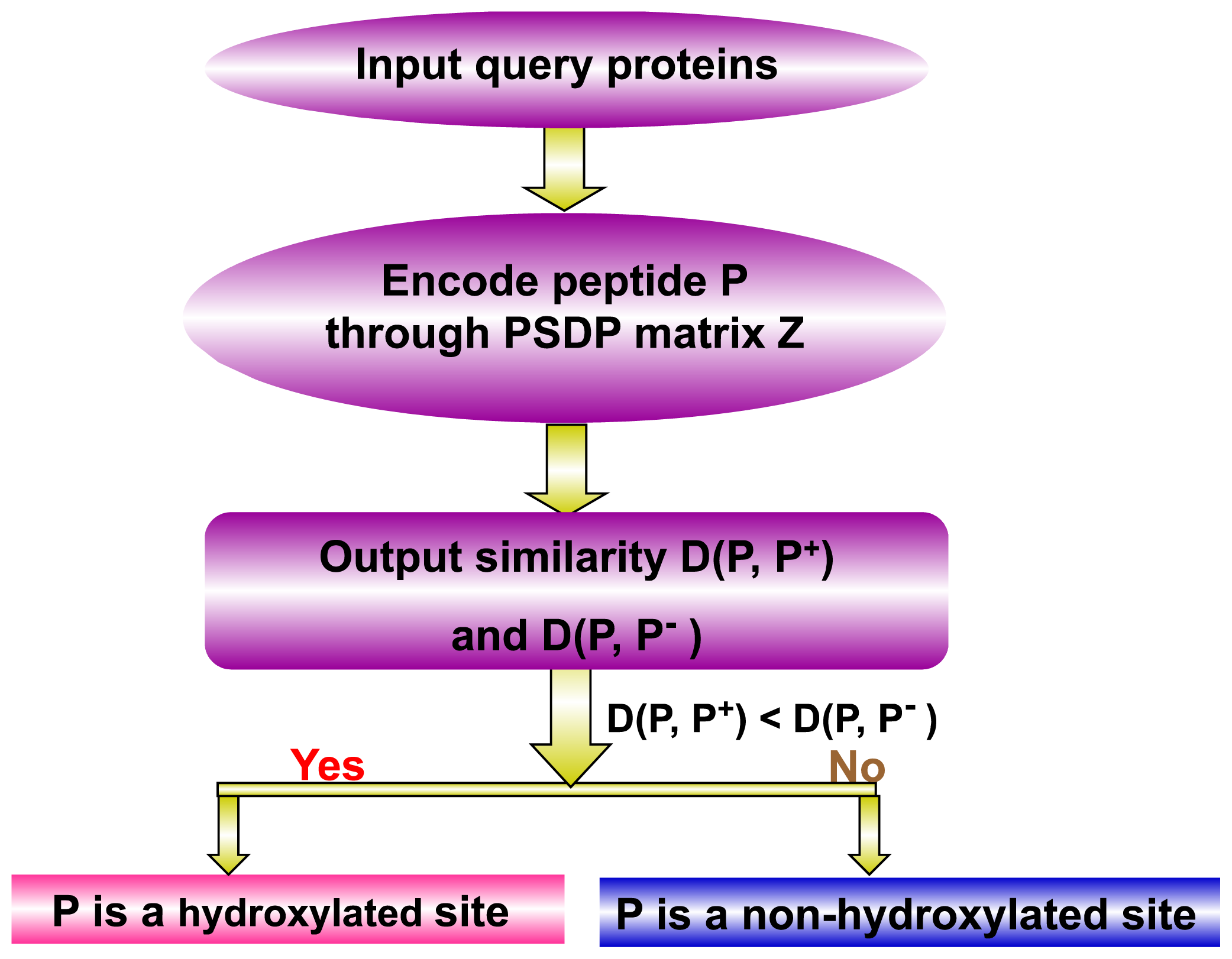
, to define the peptide samples via Equation (5).2.3. Prediction Algorithm
 (P, ℙ+) is its similarity to the norm of hydroxylated peptides and
(P, ℙ−) its similarity to the norm of non-hydroxylated peptides, as formulated by:(P, ℙ+) and
(P, ℙ−), the query peptide would be randomly assigned between the hydroxylated peptide and non-hydroxylated peptide categories. However, this kind of tie case rarely happened and actually never happened in our study.
(P, ℙ+) is its similarity to the norm of hydroxylated peptides and
(P, ℙ−) its similarity to the norm of non-hydroxylated peptides, as formulated by:(P, ℙ+) and
(P, ℙ−), the query peptide would be randomly assigned between the hydroxylated peptide and non-hydroxylated peptide categories. However, this kind of tie case rarely happened and actually never happened in our study.3. Experimental Section
3.1. A Set of Metrics for Measuring Prediction Quality
3.2. Jackknife Cross-Validation
3.3. Test by Public Database
4. Conclusions
5. The User Guide for the Web-Server iHyd-PseAAC
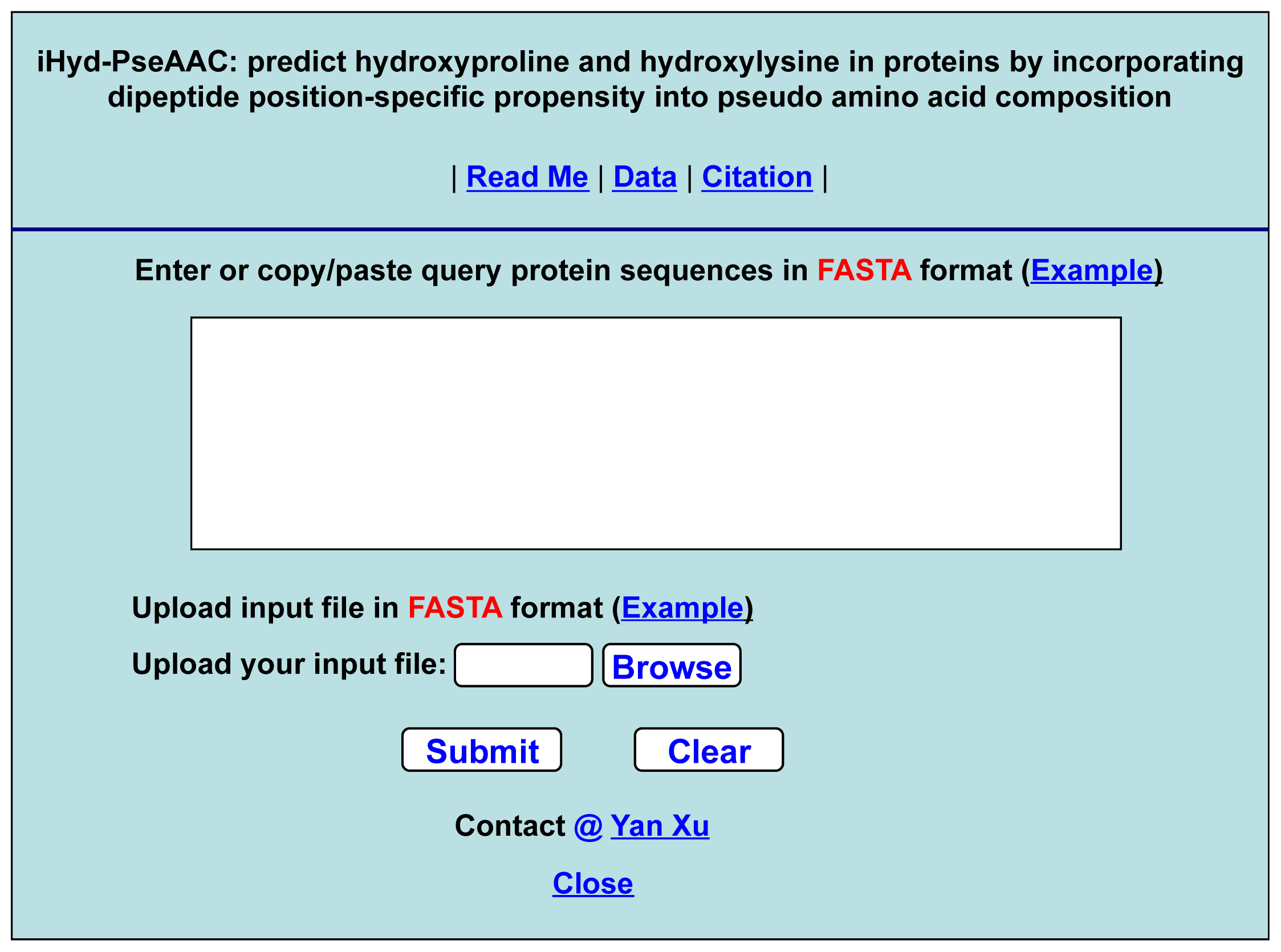
- Step 1. Open the web-server at the site at http://app.aporc.org/iHyd-PseAAC/, and you will see the top page of the predictor on your computer screen, as shown in Figure 4. Click on the “Read Me” button to see a brief introduction about the iHyd-PseAAC predictor and the caveat when using it.
- Step 2. Either type or copy/paste the query protein sequences into the input box at the center of Figure 4. The protein sequences should be in FASTA format. The input examples can be seen by clicking on the “Example” button right above the input box.
- Step 4. Click on the “Citation” button to find the relevant paper that documents the detailed development and algorithm of iHyd-PseAAC.
- Step 5. Click on the “Data” button to download the benchmark dataset used to train and test the iHyd-PseAAC predictor.
Supplementary Information
Acknowledgments
Conflicts of Interest
- Author ContributionsXu and Chou conceived and designed the experiments. Wen and Shao processed and analyzed data. Xu and Deng wrote the manuscript.
References
- Cockman, M.E.; Webb, J.D.; Kramer, H.B.; Kessler, B.M.; Ratcliffe, P.J. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell Proteomics 2009, 8, 535–546. [Google Scholar]
- Yamauchi, M.; Shiiba, M. Lysine hydroxylation and cross-linking of collagen. Methods Mol. Biol 2008, 446, 95–108. [Google Scholar]
- Chopra, R.K.; Ananthanarayanan, V.S. Conformational implications of enzymatic proline hydroxylation in collagen. Proc. Natl. Acad. Sci. USA 1982, 79, 7180–7184. [Google Scholar]
- Krane, S.M. The importance of proline residues in the structure, stability and susceptibility to proteolytic degradation of collagens. Amino Acids 2008, 35, 703–710. [Google Scholar]
- Palfi, V.K.; Perczel, A. How stable is a collagen triple helix? An ab initio study on various collagen and beta-sheet forming sequences. J. Comput. Chem 2008, 29, 1374–1386. [Google Scholar]
- Guszczyn, T.; Sobolewski, K. Deregulation of collagen metabolism in human stomach cancer. Pathobiology 2004, 71, 308–313. [Google Scholar]
- Sunila, E.S.; Kuttan, G. A preliminary study on antimetastatic activity of Thuja occidentalis L. in mice model. Immunopharmacol. Immunotoxicol 2006, 28, 269–280. [Google Scholar]
- Guruvayoorappan, C.; Kuttan, G. Anti-metastatic effect of Biophytum sensitivum is exerted through its cytokine and immunomodulatory activity and its regulatory effect on the activation and nuclear translocation of transcription factors in B16F-10 melanoma cells. J. Exp. Ther. Oncol 2008, 7, 49–63. [Google Scholar]
- Richards, A.A.; Stephens, T.; Charlton, H.K.; Jones, A.; Macdonald, G.A.; Prins, J.B.; Whitehead, J.P. Adiponectin multimerization is dependent on conserved lysines in the collagenous domain: Evidence for regulation of multimerization by alterations in posttranslational modifications. Mol. Endocrinol 2006, 20, 1673–1687. [Google Scholar]
- Yang, Z.R. Predict collagen hydroxyproline sites using support vector machines. J. Comput. Biol 2009, 16, 691–702. [Google Scholar]
- Hu, L.L.; Niu, S.; Huang, T.; Wang, K.; Shi, X.H.; Cai, Y.D. Prediction and analysis of protein hydroxyproline and hydroxylysine. PLoS One 2010, 5, e15917. [Google Scholar]
- Garnier, J.; Osguthorpe, D.J.; Robson, B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol 1978, 120, 97–120. [Google Scholar]
- Guo, S.H.; Deng, E.Z.; Xu, L.Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar]
- Fan, Y.N.; Xiao, X.; Min, J.L.; Chou, K.C. iNR-Drug: Predicting the interaction of drugs with nuclear receptors in cellular networking. Int. J. Mol. Sci 2014, 15, 4915–4937. [Google Scholar]
- Qiu, W.R.; Xiao, X.; Chou, K.C. iRSpot-TNCPseAAC: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int. J. Mol. Sci 2014, 15, 1746–1766. [Google Scholar]
- Min, J.L.; Xiao, X.; Chou, K.C. iEzy-Drug: A web server for identifying the interaction between enzymes and drugs in cellular networking. BioMed Res. Int 2013, 2013, 701317. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P. M.; Lin, H.; Chou, K.C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res 2013, 41, e69. [Google Scholar]
- Feng, P.M.; Chen, W.; Lin, H.; Chou, K.C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem 2013, 442, 118–125. [Google Scholar]
- Xiao, X.; Min, J.L.; Wang, P.; Chou, K.C. iCDI-PseFpt: Identify the channel-drug interaction in cellular networking with PseAAC and molecular fingerprints. J. Theor. Biol 2013, 337C, 71–79. [Google Scholar]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J. Theor. Biol 2011, 273, 236–247. [Google Scholar]
- Lu, C.T.; Huang, K.Y.; Su, M.G.; Lee, T.Y.; Bretana, N.A.; Chang, W.C.; Chen, Y.J.; Huang, H.D. DbPTM 3.0: An informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res 2013, 41, D295–D305. [Google Scholar]
- Chou, K.C. A vectorized sequence-coupling model for predicting HIV protease cleavage sites in proteins. J. Biol. Chem 1993, 268, 16938–16948. [Google Scholar]
- Chou, K.C. Review: Prediction of human immunodeficiency virus protease cleavage sites in proteins. Anal. Biochem 1996, 233, 1–14. [Google Scholar]
- Chou, K.C. A sequence-coupled vector-projection model for predicting the specificity of GalNAc-transferase. Protein Sci 1995, 4, 1365–1383. [Google Scholar]
- Chou, K.C. Prediction of protein signal sequences and their cleavage sites. Proteins: Struct. Funct. Genet 2001, 42, 136–139. [Google Scholar]
- Chou, K.C. Using subsite coupling to predict signal peptides. Protein Eng 2001, 14, 75–79. [Google Scholar]
- Chou, K.C. Prediction of signal peptides using scaled window. Peptides 2001, 22, 1973–1979. [Google Scholar]
- Chou, K.C. Review: Prediction of protein signal sequences. Curr. Protein Peptide Sci 2002, 3, 615–622. [Google Scholar]
- Chou, K.C.; Shen, H.B. Review: Recent progresses in protein subcellular location prediction. Anal. Biochem 2007, 370, 1–16. [Google Scholar]
- Hajisharifi, Z.; Piryaiee, M.; Mohammad Beigi, M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou’s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol 2014, 341, 34–40. [Google Scholar]
- Chen, Y.K.; Li, K.B. Predicting membrane protein types by incorporating protein topology, domains, signal peptides, and physicochemical properties into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol 2013, 318, 1–12. [Google Scholar]
- Xu, Y.; Ding, J.; Wu, L.Y.; Chou, K.C. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One 2013, 8, e55844. [Google Scholar]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem 2013, 436, 168–177. [Google Scholar]
- Mohabatkar, H.; Beigi, M.M.; Abdolahi, K.; Mohsenzadeh, S. Prediction of allergenic proteins by means of the concept of chou’s pseudo amino acid composition and a machine learning approach. Med. Chem 2013, 9, 133–137. [Google Scholar]
- Xu, Y.; Shao, X.J.; Wu, L.Y.; Deng, N.Y.; Chou, K.C. iSNO-AAPair: Incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ 2013, 1, e171. [Google Scholar]
- Chou, K.C. Prediction of protein cellular attributes using pseudo amino acid composition. Proteins: Struct. Funct. Genet 2001, 43, 246–255. [Google Scholar]
- Chou, K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar]
- Du, P.; Wang, X.; Xu, C.; Gao, Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Anal. Biochem 2012, 425, 117–119. [Google Scholar]
- Georgiou, D.N.; Karakasidis, T.E.; Megaritis, A.C. A short survey on genetic sequences, Chou’s pseudo amino acid composition and its combination with fuzzy set theory. Open Bioinforma. J 2013, 7, 41–48. [Google Scholar]
- Liu, B.; Wang, X.; Zou, Q.; Dong, Q.; Chen, Q. Protein remote homology detection by combining Chou’s pseudo amino acid composition and profile-based protein representation. Mol. Inform 2013, 32, 775–782. [Google Scholar]
- Lin, S.X.; Lapointe, J. Theoretical and experimental biology in one. J. Biomed. Sci. Eng 2013, 6, 435–442. [Google Scholar]
- Cao, D.S.; Xu, Q.S.; Liang, Y.Z. Propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar]
- Chou, K.C.; Tomasselli, A.L.; Reardon, I.M.; Heinrikson, R.L. Predicting HIV protease cleavage sites in proteins by a discriminant function method. Proteins: Struct. Funct. Genet 1996, 24, 51–72. [Google Scholar]
- Chen, W.; Lin, H.; Feng, P.M.; Ding, C.; Zuo, Y.C.; Chou, K.C. iNuc-PhysChem: A sequence-based predictor for identifying nucleosomes via physicochemical properties. PLoS One 2012, 7, e47843. [Google Scholar]
- Chou, K.C.; Shen, H.B. Euk-mPLoc: A fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J. Proteome Res 2007, 6, 1728–1734. [Google Scholar]
- Chou, K.C.; Wu, Z.C.; Xiao, X. iLoc-Hum: Using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol. Biosyst 2012, 8, 629–641. [Google Scholar]
- Shen, H.B.; Chou, K.C. Hum-mPLoc: An ensemble classifier for large-scale human protein subcellular location prediction by incorporating samples with multiple sites. Biochem. Biophys. Res. Commun 2007, 355, 1006–1011. [Google Scholar]
- Chou, K.C. Some Remarks on Predicting Multi-Label Attributes in Molecular Biosystems. Mol. Biosyst 2013, 9, 1092–1100. [Google Scholar]
- Chou, K.C.; Zhang, C.T. Review: Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol 1995, 30, 275–349. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc 2008, 3, 153–162. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Sci 2010, 2, 1090–1103. [Google Scholar]
- Fan, G.L.; Li, Q.Z. Predicting protein submitochondria locations by combining different descriptors into the general form of Chou’s pseudo amino acid composition. Amino Acids 2012, 43, 545–555. [Google Scholar]
- Fan, G.L.; Li, Q.Z. Discriminating bioluminescent proteins by incorporating average chemical shift and evolutionary information into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol 2013, 334, 45–51. [Google Scholar]
- Huang, C.; Yuan, J.Q. Predicting protein subchloroplast locations with both single and multiple sites via three different modes of Chou’s pseudo amino acid compositions. J. Theor. Biol 2013, 335, 205–212. [Google Scholar]
- Lin, H. The modified Mahalanobis discriminant for predicting outer membrane proteins by using Chou’s pseudo amino acid composition. J. Theor. Biol 2008, 252, 350–356. [Google Scholar]
- Lin, H.; Wang, H.; Ding, H.; Chen, Y.L.; Li, Q.Z. Prediction of Subcellular Localization of Apoptosis Protein Using Chou’s Pseudo Amino Acid Composition. Acta Biotheor 2009, 57, 321–330. [Google Scholar]
- Qiu, J.D.; Huang, J.H.; Liang, R.P.; Lu, X.Q. Prediction of G-protein-coupled receptor classes based on the concept of Chou’s pseudo amino acid composition: An approach from discrete wavelet transform. Anal. Biochem 2009, 390, 68–73. [Google Scholar]
- Sahu, S.S.; Panda, G. A novel feature representation method based on Chou’s pseudo amino acid composition for protein structural class prediction. Comput. Biol. Chem 2010, 34, 320–327. [Google Scholar]
- Sun, X.Y.; Shi, S.P.; Qiu, J.D.; Suo, S.B.; Huang, S.Y.; Liang, R.P. Identifying protein quaternary structural attributes by incorporating physicochemical properties into the general form of Chou’s PseAAC via discrete wavelet transform. Mol. BioSyst 2012, 8, 3178–3184. [Google Scholar]
- Zeng, Y.H.; Guo, Y.Z.; Xiao, R.Q.; Yang, L.; Yu, L.Z.; Li, M.L. Using the augmented Chou’s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach. J. Theor. Biol 2009, 259, 366–372. [Google Scholar]
- Zhou, X.B.; Chen, C.; Li, Z.C.; Zou, X.Y. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol 2007, 248, 546–551. [Google Scholar]
- Chou, K.C.; Shen, H.B. Review: Recent advances in developing web-servers for predicting protein attributes. Nat. Sci 2009, 2, 63–92. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Benchmark dataset a | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| Supplementary Information S1 for HyP | 80.66 | 80.54 | 80.57 | 0.51 |
| Supplementary Information S2 for HyL | 87.85 | 83.01 | 83.56 | 0.50 |
| Benchmark dataset a | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| Supplementary Information S1 for HyP | 70.68 | 89.03 | 78.42 | 0.52 |
| Supplementary Information S2 for HyL | 79.04 | 86.37 | 83.12 | 0.51 |
| Hydroxylated type | Sn (%) | Sp (%) | Acc (%) |
|---|---|---|---|
| Proline | 71.2 | 79.3 | 75.3 |
| Lysine | 72.7 | 80.6 | 76.8 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, Y.; Wen, X.; Shao, X.-J.; Deng, N.-Y.; Chou, K.-C. iHyd-PseAAC: Predicting Hydroxyproline and Hydroxylysine in Proteins by Incorporating Dipeptide Position-Specific Propensity into Pseudo Amino Acid Composition. Int. J. Mol. Sci. 2014, 15, 7594-7610. https://doi.org/10.3390/ijms15057594
Xu Y, Wen X, Shao X-J, Deng N-Y, Chou K-C. iHyd-PseAAC: Predicting Hydroxyproline and Hydroxylysine in Proteins by Incorporating Dipeptide Position-Specific Propensity into Pseudo Amino Acid Composition. International Journal of Molecular Sciences. 2014; 15(5):7594-7610. https://doi.org/10.3390/ijms15057594
Chicago/Turabian StyleXu, Yan, Xin Wen, Xiao-Jian Shao, Nai-Yang Deng, and Kuo-Chen Chou. 2014. "iHyd-PseAAC: Predicting Hydroxyproline and Hydroxylysine in Proteins by Incorporating Dipeptide Position-Specific Propensity into Pseudo Amino Acid Composition" International Journal of Molecular Sciences 15, no. 5: 7594-7610. https://doi.org/10.3390/ijms15057594
APA StyleXu, Y., Wen, X., Shao, X.-J., Deng, N.-Y., & Chou, K.-C. (2014). iHyd-PseAAC: Predicting Hydroxyproline and Hydroxylysine in Proteins by Incorporating Dipeptide Position-Specific Propensity into Pseudo Amino Acid Composition. International Journal of Molecular Sciences, 15(5), 7594-7610. https://doi.org/10.3390/ijms15057594