
Medullary Thymic Epithelial Cells and Central Tolerance in Autoimmune Hepatitis Development: Novel Perspective from a New Mouse Model

Abstract
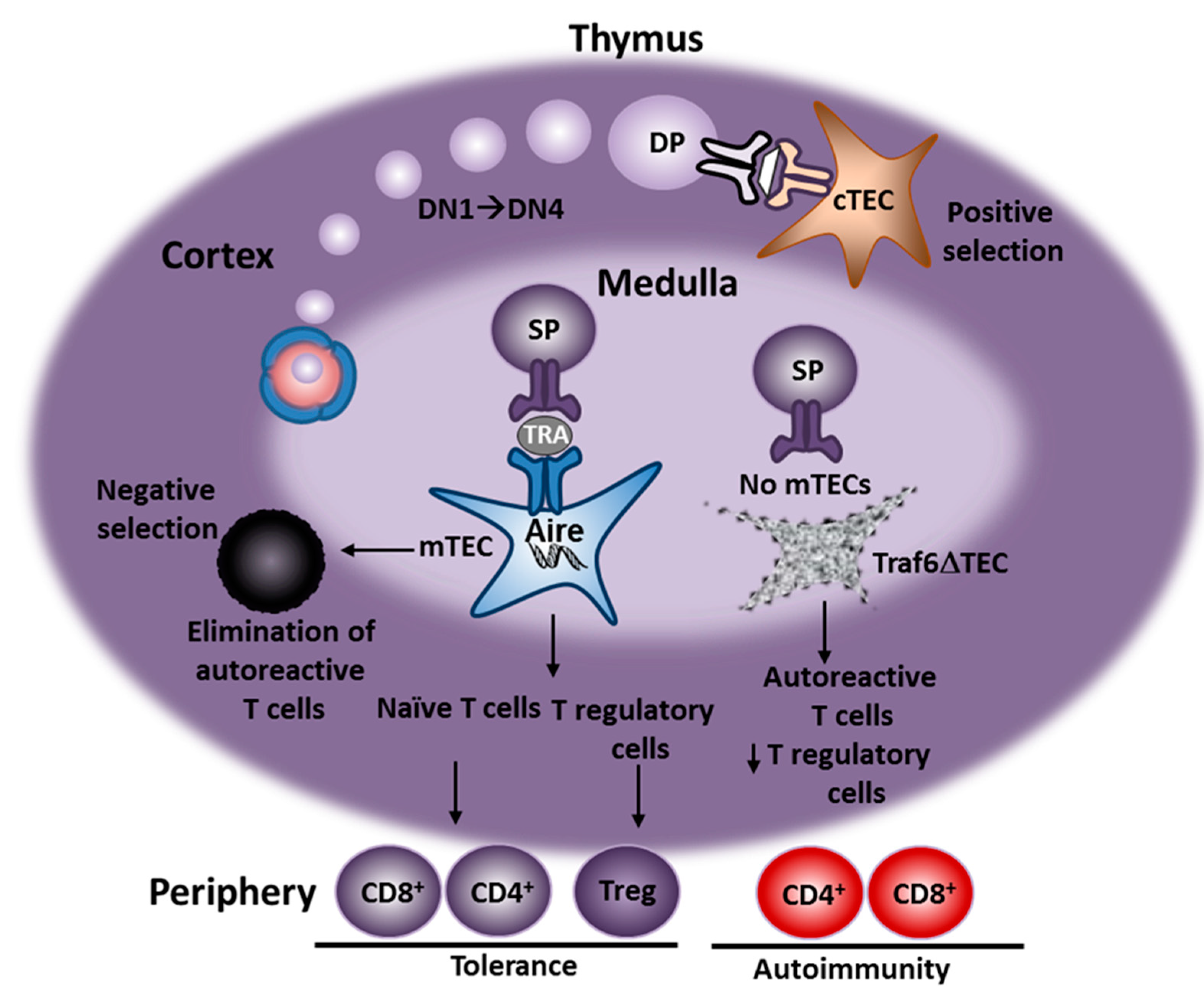
:1. Introduction

2. Animal Models of AIH
3. Generation of Traf6∆TEC Conditional Knockout Mice
4. Characterization of AIH in Traf6∆TEC Mice

{kind=link}
{kind=link}
| Mouse Model | Traf6∆TEC | Aire-∆ex2 |
|---|---|---|
| Parameters | ||
| Genetic Background | C57BL/6 | Balb/c |
| Thymus | ||
| Central tolerance | Impaired | Impaired |
| mTEC number | Depleted | Normal * |
| Treg production | Reduced (50%) | Normal * |
| Periphery | ||
| AIH | ||
| Features | Chronic/Spontaneous | Chronic/Spontaneous |
| Penetrance | 100% | 24% |
| Aminotransferases | ALT | AST |
| Portal infiltrates | CD4+ T cells, CD19+CD138+ Plasma cells | CD4+ T cells, B cells |
| Autoantibodies | Mostly ANA, anti-SLA, Polyclonal | Mostly cytoplasmic, Polyclonal |
| Intrahepatic autoantigens | Present | Present |
| Cytokines | ||
| Intrahepatic | Increased (IFN-γ, IL-4, IL-10, TGF-β) | ND |
| Serum | Normal | Increased (TNF-α, IL-2, IL-9) |
| Tregs | ||
| Hepatic | Increased | Increased |
| Splenic | Normal | Normal |
5. AIH in Traf6∆TEC Mice Is T-Cell-Dependent

6. Role of Tregs in AIH Development in Traf6∆TEC Mice
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liberal, R.; Longhi, M.S.; Mieli-Vergani, G.; Vergani, D. Pathogenesis of autoimmune hepatitis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T. Genetics in autoimmune hepatitis. Semin. Liver Dis. 2002, 22, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T. Genetics of liver disease: Immunogenetics and disease pathogenesis. Gut 2004, 53, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Vergani, D.; Choudhuri, K.; Bogdanos, D.P.; Mieli-Vergani, G. Pathogenesis of autoimmune hepatitis. Clin. Liver Dis. 2002, 6, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Carpenter, H.A.; Santrach, P.J.; Moore, S.B. Significance of HLA DR4 in type 1 autoimmune hepatitis. Gastroenterology 1993, 105, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Carpenter, H.A.; Santrach, P.J.; Moore, S.B. Genetic predispositions for the immunological features of chronic active hepatitis. Hepatology 1993, 18, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.C.; Porta, G.; Marin, M.L.; Bittencourt, P.L.; Kalil, J.; Goldberg, A.C. Autoimmune hepatitis, HLA and extended haplotypes. Autoimmun. Rev. 2011, 10, 189–193. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Williams, A.; Skavdis, G.; Harker, N.; Coles, M.; Tolaini, M.; Norton, T.; Williams, K.; Roderick, K.; Potocnik, A.J.; et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 2003, 33, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Mieli-Vergani, G.; Vergani, D. Autoimmune hepatitis. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Grant, C.R.; Mieli-Vergani, G.; Vergani, D. Autoimmune hepatitis: A comprehensive review. J. Autoimmun. 2013, 41, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Heneghan, M.A.; Yeoman, A.D.; Verma, S.; Smith, A.D.; Longhi, M.S. Autoimmune hepatitis. Lancet 2013, 382, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, H.J.; Wands, J.R.; Isselbacher, K.J. Alteration in suppressor cell activity in chronic active hepatitis. Proc. Natl. Acad. Sci. USA 1978, 75, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Nouri-Aria, K.T.; Hegarty, J.E.; Alexander, G.J.; Eddleston, A.L.; Williams, R. Effect of corticosteroids on suppressor-cell activity in “autoimmune” and viral chronic active hepatitis. N. Engl. J. Med. 1982, 307, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Vento, S.; Hegarty, J.E.; Bottazzo, G.; Macchia, E.; Williams, R.; Eddleston, A.L. Antigen specific suppressor cell function in autoimmune chronic active hepatitis. Lancet 1984, 1, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Ferri, S.; Longhi, M.S.; de Molo, C.; Lalanne, C.; Muratori, P.; Granito, A.; Hussain, M.J.; Ma, Y.; Lenzi, M.; Mieli-Vergani, G.; et al. A multifaceted imbalance of T cells with regulatory function characterizes type 1 autoimmune hepatitis. Hepatology 2010, 52, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Ma, Y.; Bogdanos, D.P.; Cheeseman, P.; Mieli-Vergani, G.; Vergani, D. Impairment of CD4+CD25+ regulatory T-cells in autoimmune liver disease. J. Hepatol. 2004, 41, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Ma, Y.; Mitry, R.R.; Bogdanos, D.P.; Heneghan, M.; Cheeseman, P.; Mieli-Vergani, G.; Vergani, D. Effect of CD4+CD25+ regulatory T-cells on CD8 T-cell function in patients with autoimmune hepatitis. J. Autoimmun. 2005, 25, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Calne, R.Y.; Sells, R.A.; Pena, J.R.; Davis, D.R.; Millard, P.R.; Herbertson, B.M.; Binns, R.M.; Davies, D.A. Induction of immunological tolerance by porcine liver allografts. Nature 1969, 223, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Demetris, A.J.; Murase, N.; Rao, A.S.; Fung, J.J.; Starzl, T.E. Murine liver allograft transplantation: Tolerance and donor cell chimerism. Hepatology 1994, 19, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, N. The immunology of experimental liver transplantation in the rat. Immunology 1985, 55, 369–389. [Google Scholar] [PubMed]
- Crispe, I.N. Hepatic T cells and liver tolerance. Nat. Rev. Immunol. 2003, 3, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Jaeckel, E. Mouse models for experimental autoimmune hepatitis: Limits and chances. Dig. Dis. 2010, 28, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Zlotoff, D.A.; Bhandoola, A. Hematopoietic progenitor migration to the adult thymus. Ann. N. Y. Acad. Sci. 2011, 1217, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jeremiah Bell, J.; Bhandoola, A. T-cell lineage determination. Immunol. Rev. 2010, 238, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Alexandropoulos, K.; Danzl, N.M. Thymic epithelial cells: Antigen presenting cells that regulate T cell repertoire and tolerance development. Immunol. Res. 2012, 54, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mouchess, M.L.; Anderson, M. Central tolerance induction. Curr. Top. Microbiol. Immunol. 2014, 373, 69–86. [Google Scholar] [PubMed]
- Perry, J.S.; Lio, C.W.; Kau, A.L.; Nutsch, K.; Yang, Z.; Gordon, J.I.; Murphy, K.M.; Hsieh, C.S. Distinct contributions of Aire and antigen-presenting-cell subsets to the generation of self-tolerance in the thymus. Immunity 2014, 41, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Laan, M.; Peterson, P. The many faces of aire in central tolerance. Front. Immunol. 2013, 4, 326. [Google Scholar] [CrossRef] [PubMed]
- Peterson, P.; Org, T.; Rebane, A. Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat. Rev. Immunol. 2008, 8, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Bonito, A.J.; Aloman, C.; Fiel, M.I.; Danzl, N.M.; Cha, S.; Weinstein, E.G.; Jeong, S.; Choi, Y.; Walsh, M.C.; Alexandropoulos, K. Medullary thymic epithelial cell depletion leads to autoimmune hepatitis. J. Clin. Investig. 2013, 123, 3510–3524. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T.; Doherty, D.G.; Hayllar, K.M.; McFarlane, I.G.; Johnson, P.J.; Williams, R. Susceptibility to autoimmune chronic active hepatitis: Human leukocyte antigens DR4 and A1-B8-DR3 are independent risk factors. Hepatology 1991, 13, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Strettell, M.D.; Donaldson, P.T.; Thomson, L.J.; Santrach, P.J.; Moore, S.B.; Czaja, A.J.; Williams, R. Allelic basis for HLA-encoded susceptibility to type 1 autoimmune hepatitis. Gastroenterology 1997, 112, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Hintermann, E.; Jaeckel, E. New animal models for autoimmune hepatitis. Semin. Liver Dis. 2009, 29, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Jaeckel, E.; Hardtke-Wolenski, M.; Fischer, K. The benefit of animal models for autoimmune hepatitis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Noyan, F.; Jaeckel, E. Requirements and challenges of a preclinical autoimmune hepatitis mouse model. Dig. Dis. 2011, 29, 402–410. [Google Scholar] [PubMed]
- Czaja, A.J. Animal models of autoimmune hepatitis. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Scheiffarth, F.; Warnatz, H.; Mayer, K. Studies concerning the importance of mononuclear cells in the development of experimental hepatitis. J. Immunol. 1967, 98, 396–401. [Google Scholar] [PubMed]
- Kuriki, J.; Murakami, H.; Kakumu, S.; Sakamoto, N.; Yokochi, T.; Nakashima, I.; Kato, N. Experimental autoimmune hepatitis in mice after immunization with syngeneic liver proteins together with the polysaccharide of Klebsiella pneumoniae. Gastroenterology 1983, 84, 596–603. [Google Scholar] [PubMed]
- Mori, Y.; Mori, T.; Yoshida, H.; Ueda, S.; Iesato, K.; Wakashin, Y.; Wakashin, M.; Okuda, K. Study of cellular immunity in experimental autoimmune hepatitis in mice. Clin. Exp. Immunol. 1984, 57, 85–92. [Google Scholar] [PubMed]
- Watanabe, Y.; Kawakami, H.; Kawamoto, H.; Ikemoto, Y.; Masuda, K.; Takezaki, E.; Nakanishi, T.; Kajiyama, G.; Takeno, H. Effect of neonatal thymectomy on experimental autoimmune hepatitis in mice. Clin. Exp. Immunol. 1987, 67, 105–113. [Google Scholar] [PubMed]
- Lohse, A.W.; Manns, M.; Dienes, H.P.; Meyer zum Buschenfelde, K.H.; Cohen, I.R. Experimental autoimmune hepatitis: Disease induction, time course and T-cell reactivity. Hepatology 1990, 11, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Toyonaga, T.; Hino, O.; Sugai, S.; Wakasugi, S.; Abe, K.; Shichiri, M.; Yamamura, K. Chronic active hepatitis in transgenic mice expressing interferon-γ in the liver. Proc. Natl. Acad. Sci. USA 1994, 91, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G. Experimental hepatitis and role of cytokines. Acta Gastroenterol. Belg. 1997, 60, 176–179. [Google Scholar] [PubMed]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Kusters, S.; Gantner, F.; Kunstle, G.; Tiegs, G. Interferon γ plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996, 111, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Gantner, F.; Leist, M.; Lohse, A.W.; Germann, P.G.; Tiegs, G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: The role of tumor necrosis factor. Hepatology 1995, 21, 190–198. [Google Scholar] [PubMed]
- Gorham, J.D.; Lin, J.T.; Sung, J.L.; Rudner, L.A.; French, M.A. Genetic regulation of autoimmune disease: BALB/c background TGF-β 1-deficient mice develop necroinflammatory IFN- γ-dependent hepatitis. J. Immunol. 2001, 166, 6413–6422. [Google Scholar] [CrossRef] [PubMed]
- Jones-Youngblood, S.L.; Wieties, K.; Forman, J.; Hammer, R.E. Effect of the expression of a hepatocyte-specific MHC molecule in transgenic mice on T cell tolerance. J. Immunol. 1990, 144, 1187–1195. [Google Scholar] [PubMed]
- Morahan, G.; Brennan, F.E.; Bhathal, P.S.; Allison, J.; Cox, K.O.; Miller, J.F. Expression in transgenic mice of class I histocompatibility antigens controlled by the metallothionein promoter. Proc. Natl. Acad. Sci. USA 1989, 86, 3782–3786. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Guilhot, S.; Klopchin, K.; Moss, B.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L.; Kanagawa, O.; Chisari, F.V. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science 1990, 248, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F.V.; Ferrari, C. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 1995, 13, 29–60. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F.V. Hepatitis B virus transgenic mice: Models of viral immunobiology and pathogenesis. Curr. Top. Microbiol. Immunol. 1996, 206, 149–173. [Google Scholar] [PubMed]
- Christen, U.; Holdener, M.; Hintermann, E. Cytochrome P450 2D6 as a model antigen. Dig. Dis. 2010, 28, 80–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zierden, M.; Kuhnen, E.; Odenthal, M.; Dienes, H.P. Effects and regulation of autoreactive CD8+ T cells in a transgenic mouse model of autoimmune hepatitis. Gastroenterology 2010, 139, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Ferber, I.; Schonrich, G.; Schenkel, J.; Mellor, A.L.; Hammerling, G.J.; Arnold, B. Levels of peripheral T cell tolerance induced by different doses of tolerogen. Science 1994, 263, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Djilali-Saiah, I.; Lapierre, P.; Vittozi, S.; Alvarez, F. DNA vaccination breaks tolerance for a neo-self antigen in liver: A transgenic murine model of autoimmune hepatitis. J. Immunol. 2002, 169, 4889–4896. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, P.; Djilali-Saiah, I.; Vitozzi, S.; Alvarez, F. A murine model of type 2 autoimmune hepatitis: Xenoimmunization with human antigens. Hepatology 2004, 39, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Marceau, G.; Yang, R.; Lapierre, P.; Beland, K.; Alvarez, F. Low-dose anti-CD3 antibody induces remission of active autoimmune hepatitis in xenoimmunized mice. Liver Int. 2014. [Google Scholar] [CrossRef]
- Lapierre, P.; Beland, K.; Yang, R.; Alvarez, F. Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology 2013, 57, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Holdener, M.; Hintermann, E.; Bayer, M.; Rhode, A.; Rodrigo, E.; Hintereder, G.; Johnson, E.F.; Gonzalez, F.J.; Pfeilschifter, J.; Manns, M.P.; et al. Breaking tolerance to the natural human liver autoantigen cytochrome P450 2D6 by virus infection. J. Exp. Med. 2008, 205, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Fischer, K.; Noyan, F.; Schlue, J.; Falk, C.S.; Stahlhut, M.; Woller, N.; Kuehnel, F.; Taubert, R.; Manns, M.P.; et al. Genetic predisposition and environmental danger signals initiate chronic autoimmune hepatitis driven by CD4+ T cells. Hepatology 2013, 58, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.G. Animal models of autoimmune liver disease. Immunol. Cell Biol. 2002, 80, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Irla, M.; Hollander, G.; Reith, W. Control of central self-tolerance induction by autoreactive CD4+ thymocytes. Trends Immunol. 2010, 31, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Walsh, P.T.; Walsh, M.C.; Speirs, K.M.; Chiffoleau, E.; King, C.G.; Hancock, W.W.; Caamano, J.H.; Hunter, C.A.; Scott, P.; et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity 2003, 19, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.; Xiao, S.; Hughes, B., III; Su, D.M.; Navarre, S.P.; Condie, B.G.; Manley, N.R. Specific expression of lacZ and cre recombinase in fetal thymic epithelial cells by multiplex gene targeting at the Foxn1 locus. BMC Dev. Biol. 2007, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Corbeaux, T.; Hess, I.; Swann, J.B.; Kanzler, B.; Haas-Assenbaum, A.; Boehm, T. Thymopoiesis in mice depends on a Foxn1-positive thymic epithelial cell lineage. Proc. Natl. Acad. Sci. USA 2010, 107, 16613–16618. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, K.; D’Cruz, L.M.; Vollmann, E.H.; Hinterberger, M.; Emmerich, J.; Swee, L.K.; Rolink, A.; Klein, L. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat. Immunol. 2007, 8, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, I.; Sarukhan, A.; Klein, L.; von Boehmer, H. Origin of regulatory T cells with known specificity for antigen. Nat. Immunol. 2002, 3, 756–763. [Google Scholar]
- Jordan, M.S.; Boesteanu, A.; Reed, A.J.; Petrone, A.L.; Holenbeck, A.E.; Lerman, M.A.; Naji, A.; Caton, A.J. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001, 2, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D.; Benoist, C. Aire. Annu. Rev. Immunol. 2009, 27, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Akirav, E.M.; Ruddle, N.H.; Herold, K.C. The role of AIRE in human autoimmune disease. Nat. Rev. Endocrinol. 2011, 7, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lankisch, T.O.; Jaeckel, E.; Strassburg, C.P. The autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy or autoimmune polyglandular syndrome type 1. Semin. Liver Dis. 2009, 29, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Strassburg, C.P. Autoimmune hepatitis. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.G.; Meloni, A.; Obermayer-Straub, P.; Frau, F.; Manns, M.P.; de Virgiliis, S. Two cytochromes P450 are major hepatocellular autoantigens in autoimmune polyglandular syndrome type 1. Gastroenterology 1998, 114, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.G.; Obermayer-Straub, P.; Meloni, A.; Strassburg, C.P.; Arangino, V.; Tukey, R.H.; de Virgiliis, S.; Manns, M.P. Cytochrome P450 1A2 is a hepatic autoantigen in autoimmune polyglandular syndrome type 1. J. Clin. Endocrinol. Metab. 1997, 82, 1353–1361. [Google Scholar] [PubMed]
- Obermayer-Straub, P.; Perheentupa, J.; Braun, S.; Kayser, A.; Barut, A.; Loges, S.; Harms, A.; Dalekos, G.; Strassburg, C.P.; Manns, M.P. Hepatic autoantigens in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Gastroenterology 2001, 121, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Ma, Y.; Mieli-Vergani, G.; Vergani, D. Aetiopathogenesis of autoimmune hepatitis. J. Autoimmun. 2010, 34, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Autoantibodies as prognostic markers in autoimmune liver disease. Dig. Dis. Sci. 2010, 55, 2144–2161. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Sebode, M.; Franke, B.; Wortmann, F.; Schwinge, D.; Quaas, A.; Baron, U.; Olek, S.; Wiegard, C.; Lohse, A.W.; et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J. Hepatol. 2012, 57, 125–132. [Google Scholar]
- Taubert, R.; Hardtke-Wolenski, M.; Noyan, F.; Wilms, A.; Baumann, A.K.; Schlue, J.; Olek, S.; Falk, C.S.; Manns, M.P.; Jaeckel, E. Intrahepatic regulatory T cells in autoimmune hepatitis are associated with treatment response and depleted with current therapies. J. Hepatol. 2014, 61, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Homberg, J.C.; Abuaf, N.; Bernard, O.; Islam, S.; Alvarez, F.; Khalil, S.H.; Poupon, R.; Darnis, F.; Levy, V.G.; Grippon, P.; et al. Chronic active hepatitis associated with antiliver/kidney microsome antibody type 1: A second type of “autoimmune” hepatitis. Hepatology 1987, 7, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Abuaf, N.; Cavalli, F.; Durand, V.; Johanet, C.; Homberg, J.C. Antibody to liver cytosol (anti-LC1) in patients with autoimmune chronic active hepatitis type 2. Hepatology 1988, 8, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, S.; Weidemann, C.; Gerken, G.; Lohr, H.F.; Galle, P.R.; Meyer zum Buschenfelde, K.H.; Lohse, A.W. Clinical significance of autoantibodies to soluble liver antigen in autoimmune hepatitis. J. Hepatol. 1999, 31, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Taubert, R.; Noyan, F.; Sievers, M.; Dywicki, J.; Schlue, J.; Falk, C.S.; Lundgren, B.A.; Scott, H.S.; Pich, A.; et al. Autoimmune hepatitis in a murine APS-1 model is directed against multiple autoantigens. Hepatology 2014. [Google Scholar] [CrossRef]
- Anderson, M.S.; Venanzi, E.S.; Chen, Z.; Berzins, S.P.; Benoist, C.; Mathis, D. The cellular mechanism of Aire control of T cell tolerance. Immunity 2005, 23, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Senaldi, G.; Portmann, B.; Mowat, A.P.; Mieli-Vergani, G.; Vergani, D. Immunohistochemical features of the portal tract mononuclear cell infiltrate in chronic aggressive hepatitis. Arch. Dis. Child. 1992, 67, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T. Genetics of autoimmune and viral liver diseases: Understanding the issues. J. Hepatol. 2004, 41, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Parel, Y.; Chizzolini, C. CD4+CD8+ double positive (DP) T cells in health and disease. Autoimmun. Rev. 2004, 3, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Parel, Y.; Aurrand-Lions, M.; Scheja, A.; Dayer, J.M.; Roosnek, E.; Chizzolini, C. Presence of CD4+CD8+ double-positive T cells with very high interleukin-4 production potential in lesional skin of patients with systemic sclerosis. Arthritis Rheumatol. 2007, 56, 3459–3467. [Google Scholar] [CrossRef]
- Liberal, R.; Longhi, M.S.; Grant, C.R.; Mieli-Vergani, G.; Vergani, D. Autoimmune hepatitis after liver transplantation. Clin. Gastroenterol. Hepatol. 2012, 10, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimkhani, M.R.; Mohar, I.; Crispe, I.N. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 2011, 54, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tang, Y.; You, Z.; Wang, Q.; Liang, S.; Han, X.; Qiu, D.; Wei, J.; Liu, Y.; Shen, L.; et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One 2011, 6, e18909. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wang, L.; Liu, N.; Wang, Y.; Chu, Y. Critical role of interleukin-17/interleukin-17 receptor axis in mediating Con A-induced hepatitis. Immunol. Cell Biol. 2012, 90, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Lafdil, F.; Wang, H.; Park, O.; Zhang, W.; Moritoki, Y.; Yin, S.; Fu, X.Y.; Gershwin, M.E.; Lian, Z.X.; Gao, B. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology 2009, 137, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Karow, M.; Flavell, R.A. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007, 27, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. IL-17 in liver injury: An inflammatory issue? Immunol. Cell Biol. 2012, 90, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Sebode, M.; Lohse, A.W. Future perspective: Immunomodulatory therapy for autoimmune hepatitis. Dig. Dis. 2014, 32, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Oo, Y.H.; Adams, D.H. Regulatory T cells and autoimmune hepatitis: What happens in the liver stays in the liver. J. Hepatol. 2014, 61, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Grant, C.R.; Holder, B.S.; Ma, Y.; Mieli-Vergani, G.; Vergani, D.; Longhi, M.S. The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin-9/tim-3 pathway. Hepatology 2012, 56, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Schmitt, E.; Jin, S.; Germann, T.; Duchmann, R.; Hegenbarth, S.; Gerken, G.; Lohse, A.W. Induction of cytokine production in naive CD4+ T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology 1999, 116, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Lohse, A.W.; Knolle, P.A.; Bilo, K.; Uhrig, A.; Waldmann, C.; Ibe, M.; Schmitt, E.; Gerken, G.; Meyer Zum Buschenfelde, K.H. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology 1996, 110, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Carambia, A.; Frenzel, C.; Bruns, O.T.; Schwinge, D.; Reimer, R.; Hohenberg, H.; Huber, S.; Tiegs, G.; Schramm, C.; Lohse, A.W.; et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J. Hepatol. 2013, 58, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Kruse, N.; Neumann, K.; Schrage, A.; Derkow, K.; Schott, E.; Erben, U.; Kuhl, A.; Loddenkemper, C.; Zeitz, M.; Hamann, A.; et al. Priming of CD4+ T cells by liver sinusoidal endothelial cells induces CD25low forkhead box protein 3− regulatory T cells suppressing autoimmune hepatitis. Hepatology 2009, 50, 1904–1913. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandropoulos, K.; Bonito, A.J.; Weinstein, E.G.; Herbin, O. Medullary Thymic Epithelial Cells and Central Tolerance in Autoimmune Hepatitis Development: Novel Perspective from a New Mouse Model. Int. J. Mol. Sci. 2015, 16, 1980-2000. https://doi.org/10.3390/ijms16011980
Alexandropoulos K, Bonito AJ, Weinstein EG, Herbin O. Medullary Thymic Epithelial Cells and Central Tolerance in Autoimmune Hepatitis Development: Novel Perspective from a New Mouse Model. International Journal of Molecular Sciences. 2015; 16(1):1980-2000. https://doi.org/10.3390/ijms16011980
Chicago/Turabian StyleAlexandropoulos, Konstantina, Anthony J. Bonito, Erica G. Weinstein, and Olivier Herbin. 2015. "Medullary Thymic Epithelial Cells and Central Tolerance in Autoimmune Hepatitis Development: Novel Perspective from a New Mouse Model" International Journal of Molecular Sciences 16, no. 1: 1980-2000. https://doi.org/10.3390/ijms16011980
APA StyleAlexandropoulos, K., Bonito, A. J., Weinstein, E. G., & Herbin, O. (2015). Medullary Thymic Epithelial Cells and Central Tolerance in Autoimmune Hepatitis Development: Novel Perspective from a New Mouse Model. International Journal of Molecular Sciences, 16(1), 1980-2000. https://doi.org/10.3390/ijms16011980