
Potential Relationship between Inadequate Response to DNA Damage and Development of Myelodysplastic Syndrome

Abstract
:1. Introduction
2. Myelodysplastic Syndrome (MDS) Is a Disease of Genomic Instability
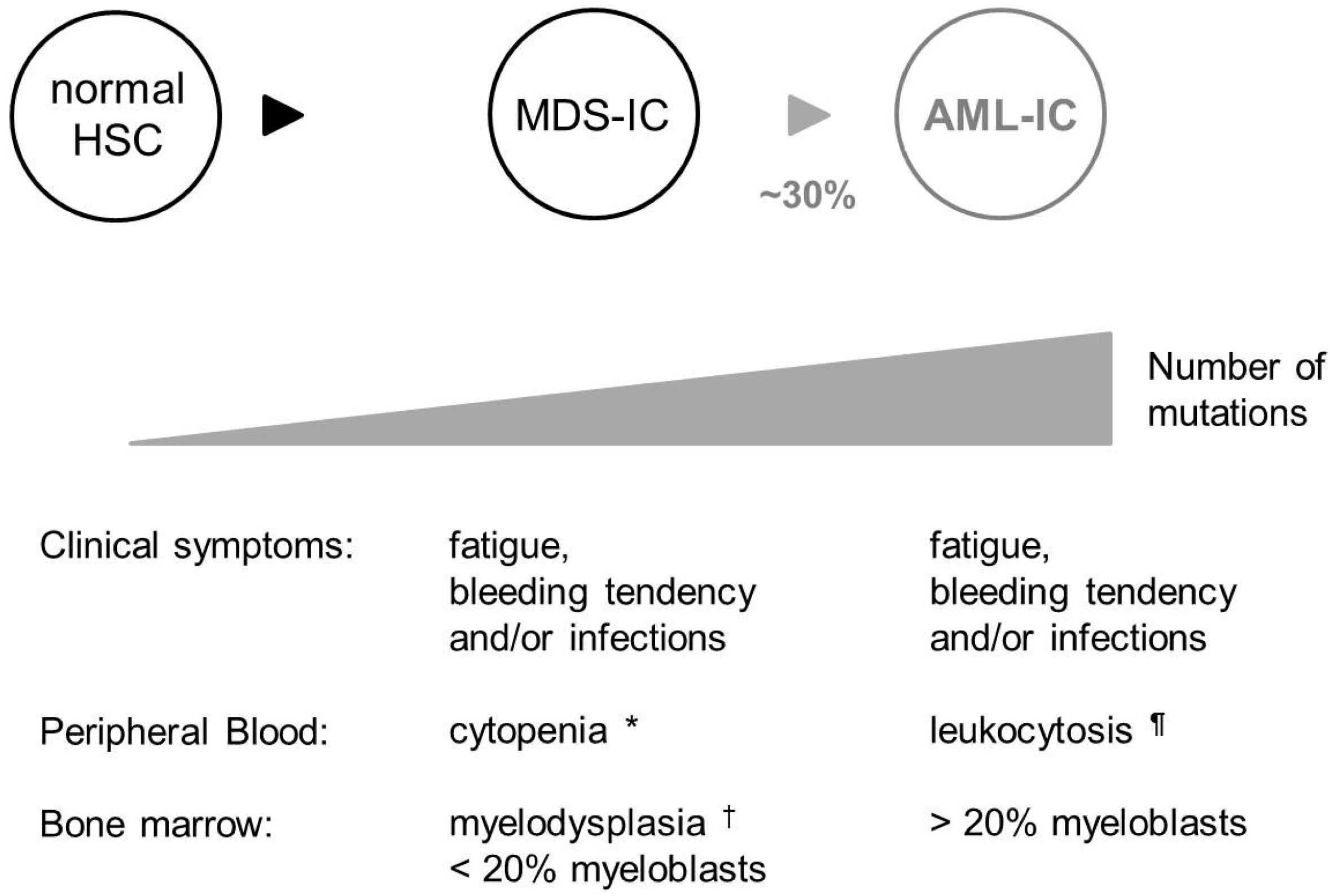
3. MDS Is Associated with Excessive DNA Damage
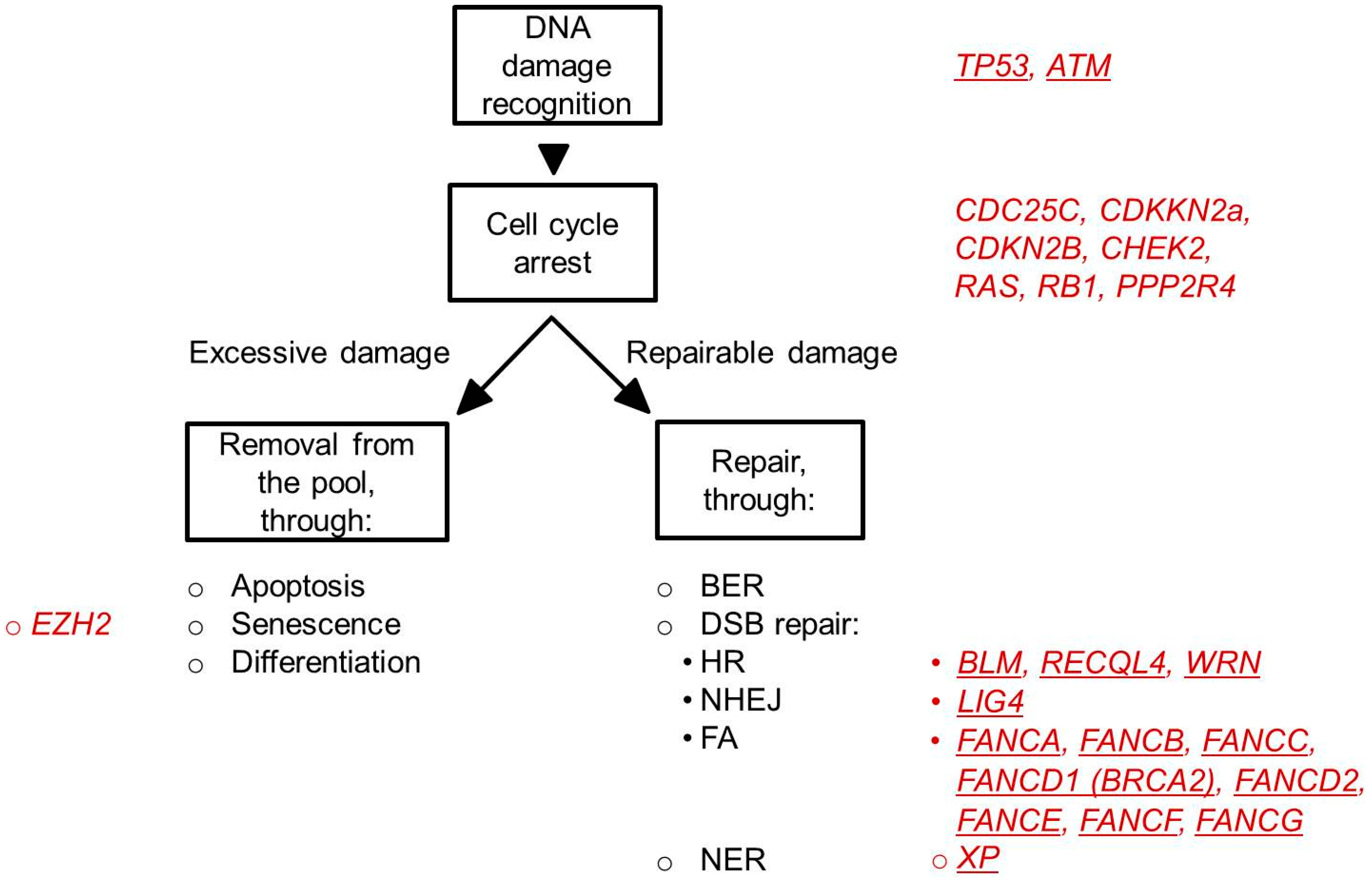
4. Cellular Mechanisms to Protect against Permanent DNA Damage

4.1. DNA Damage Checkpoints
4.2. DNA Repair Pathways
4.2.1. Base Excision Repair (BER)
4.2.2. DNA Double Strand Break Repair

{kind=link}
{kind=link}
| Process Involved | Disease | Gene(s) Involved * | Incidence of Disease | Incidence of MDS in Patients § |
|---|---|---|---|---|
| DNA damage response and cell cycle checkpoint | Li-Fraumeni syndrome [135] | TP53 or CHEK2 | 400 Cases reported | 3 Cases reported |
| DSB repair | ||||
| Global | Ataxia Telangiectasia [136] | ATM | 1/100,000 to 40,000 | Not reported |
| HR | Bloom syndrome [124] | BLM, BLAP75/RMI1 | 1/48,000 | 4 Cases reported |
| Rothmund-Thomson syndrome [125,126,127,128] | RECQL4 | 300 Cases reported | 4 Cases reported | |
| Werner syndrome [129,130] | WRN | 1300 Cases reported | 6 Cases reported | |
| FA pathway | Fanconi anemia [122] | FANC(A–G) | 1/350,000 | 20.7% |
| NHEJ | Lig4 syndrome [137] | LIG4 | Few cases reported | 1 Case reported |
| NER | Xeroderma Pigmentosa [136] | XP | 1/250,000 | Not reported |
| Other ¶ | ||||
| Unclear | Neurofibromatosis type 1 [138] | NF1 | 1/3500 | 200–500-Fold increase in children |
| Shwachman-Diamond syndrome [139,140,141] | SBDS | 1/50,000 | 8%–33% | |
| Telomere maintenance | Dyskeratosis congenital [142,143,144] | DKC1, NOP10, NHP2, TERC, TERT, TINF2 | 1/1,000,000 | 4%–5% |
| Oxidative DNA damage repair | Down syndrome [145,146,147] | Trisomy 21 | 1/1000 to 1/650 | 1/1000 to 1/500 |
4.2.3. Mismatch Repair (MMR)
4.2.4. Nucleotide Excision Repair (NER)
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cho, R.H.; Sieburg, H.B.; Muller-Sieburg, C.E. A new mechanism for the aging of hematopoietic stem cells: Aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood 2008, 111, 5553–5561. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.; Copley, M.R.; Kent, D.G.; Wohrer, S.; Cortes, A.; Aghaeepour, N.; Ma, E.; Mader, H.; Rowe, K.; Day, C.; et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 2012, 10, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; DePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar] [PubMed]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Astle, C.M.; Harrison, D.E. Genetic regulation of primitive hematopoietic stem cell senescence. Exp. Hematol. 2000, 28, 442–450. [Google Scholar] [CrossRef] [PubMed]
- De Haan, G.; Nijhof, W.; van Zant, G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: Correlation between lifespan and cycling activity. Blood 1997, 89, 1543–1550. [Google Scholar]
- De Haan, G.; van Zant, G. Intrinsic and extrinsic control of hemopoietic stem cell numbers: Mapping of a stem cell gene. J. Exp. Med. 1997, 186, 529–536. [Google Scholar]
- Harrison, D.E.; Astle, C.M. Loss of stem cell repopulating ability upon transplantation. Effects of donor age, cell number, and transplantation procedure. J. Exp. Med. 1982, 156, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Astle, C.M.; Stone, M. Numbers and functions of transplantable primitive immunohematopoietic stem cells. Effects of age. J. Immunol. 1989, 142, 3833–3840. [Google Scholar] [PubMed]
- Kamminga, L.M.; van Os, R.; Ausema, A.; Noach, E.J.; Weersing, E.; Dontje, B.; Vellenga, E.; de Haan, G. Impaired hematopoietic stem cell functioning after serial transplantation and during normal aging. Stem Cells 2005, 23, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Qian, D.; Jerabek, L.; Thiel, B.A.; Park, I.K.; Ford, P.S.; Kiel, M.J.; Schork, N.J.; Weissman, I.L.; Clarke, M.F. A genetic determinant that specifically regulates the frequency of hematopoietic stem cells. J. Immunol. 2002, 168, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Fricke, A.; Widmann, T.A.; Furst, T.; Madry, H.; Pfreundschuh, M.; Rube, C. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One 2011, 6, e17487. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Kim, T.M.; Son, M.Y.; Kim, S.A.; Holland, C.L.; Tateishi, S.; Kim, D.H.; Yew, P.R.; Montagna, C.; Dumitrache, L.C.; et al. Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes. Nature 2013, 501, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Ko, J.H.; Hu, L.; Kim, S.A.; Bishop, A.J.; Vijg, J.; Montagna, C.; Hasty, P. RAD51 mutants cause replication defects and chromosomal instability. Mol. Cell. Biol. 2012, 32, 3663–3680. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Strande, N.; Burkhalter, M.D.; Strom, C.; Havener, J.M.; Hasty, P.; Ramsden, D.A. Ku is a 5'-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 2010, 464, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.J.; Choi, C.W.; Slape, C.; Fry, T.; Aplan, P.D. Transplantation of a myelodysplastic syndrome by a long-term repopulating hematopoietic cell. Proc. Natl. Acad. Sci. USA 2008, 105, 14088–14093. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.; Eden, P.; Olsson, E.; Mansson, R.; Astrand-Grundstrom, I.; Strombeck, B.; Theilgaard-Monch, K.; Anderson, K.; Hast, R.; Hellstrom-Lindberg, E.; et al. The molecular signature of MDS stem cells supports a stem-cell origin of 5q myelodysplastic syndromes. Blood 2007, 110, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G. Myelodysplastic syndromes: 2012 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2012, 87, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Ma, X. Epidemiology of myelodysplastic syndromes. Am. J. Med. 2012, 125, S2–S5. [Google Scholar] [CrossRef] [PubMed]
- Mufti, G.J. Pathobiology, classification, and diagnosis of myelodysplastic syndrome. Best Pract. Res. Clin. Haematol. 2004, 17, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Ban, S.; Oda, K.; Tanaka, H.; Kimura, A.; Suzuki, G. Chromosomal instability and radiosensitivity in myelodysplastic syndrome cells. Leukemia 2002, 16, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Steensma, D.P. The molecular pathogenesis of myelodysplastic syndromes. Cancer Biol. Ther. 2010, 10, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L. Molecular and genetic features of myelodysplastic syndromes. Int. J. Lab. Hematol. 2012, 34, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.; Walter, M.J. Genetics of myelodysplastic syndromes: New insights. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 543–549. [Google Scholar]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstocker, M.; Nosslinger, T.; Hildebrandt, B.; Kundgen, A.; Lubbert, M.; Kunzmann, R.; Giagounidis, A.A.; et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, A.M.; Smith, A.E.; Gaken, J.; Lea, N.C.; Mian, S.A.; Westwood, N.B.; Strupp, C.; Gattermann, N.; Germing, U.; Mufti, G.J. Novel TET2 mutations associated with UPD4q24 in myelodysplastic syndrome. J. Clin. Oncol. 2009, 27, 4002–4006. [Google Scholar] [CrossRef] [PubMed]
- Secker-Walker, L.M.; Mehta, A.; Bain, B. Abnormalities of 3q21 and 3q26 in myeloid malignancy: A United Kingdom Cancer Cytogenetic Group study. Br. J. Haematol. 1995, 91, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W. The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: An overview with emphasis on the myeloid neoplasms. Chem. Biol. Interact. 2010, 184, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Malcovati, L.; Invernizzi, R. Myelodysplastic/myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 264–272. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G., Jr. Inverse radiation dose-rate effects on somatic and germ-line mutations and DNA damage rates. Proc. Natl. Acad. Sci. USA 2000, 97, 5381–5386. [Google Scholar] [CrossRef] [PubMed]
- Novotna, B.; Bagryantseva, Y.; Siskova, M.; Neuwirtova, R. Oxidative DNA damage in bone marrow cells of patients with low-risk myelodysplastic syndrome. Leuk. Res. 2009, 33, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, A.; Concia, L.; Maggio, C.; Raynaud, C.; Bergounioux, C.; Crespan, E.; Cella, R.; Maga, G. Oxidative DNA damage bypass in Arabidopsis thaliana requires DNA polymerase λ and proliferating cell nuclear antigen 2. Plant Cell 2011, 23, 806–822. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.M.; Gondek, L.P.; Szpurka, H.; Nearman, Z.P.; Tiu, R.V.; Maciejewski, J.P. Base excision repair dysfunction in a subgroup of patients with myelodysplastic syndrome. Leukemia 2008, 22, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Peddie, C.M.; Wolf, C.R.; McLellan, L.I.; Collins, A.R.; Bowen, D.T. Oxidative DNA damage in CD34+ myelodysplastic cells is associated with intracellular redox changes and elevated plasma tumour necrosis factor-α concentration. Br. J. Haematol. 1997, 99, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Bottollier-Depois, J.F.; Chau, Q.; Bouisset, P.; Kerlau, G.; Plawinski, L.; Lebaron-Jacobs, L. Assessing exposure to cosmic radiation on board aircraft. Adv. Space Res. 2003, 32, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, S.; Trott, K.; Atkinson, M.; Bink, K.; Bebeshko, V.; Bazyka, D.; Dmytrenko, I.; Abramenko, I.; Bilous, N.; Misurin, A.; et al. Aml1 gene rearrangements and mutations in radiation-associated acute myeloid leukemia and myelodysplastic syndromes. J. Radiat. Res. 2005, 46, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Dyagil, I.; Adam, M.; Beebe, G.W.; Burch, J.D.; Gaidukova, S.N.; Gluzman, D.; Gudzenko, N.; Klimenko, V.; Peterson, L.; Reiss, R.F.; et al. Histologic verification of leukemia, myelodysplasia, and multiple myeloma diagnoses in patients in Ukraine, 1987–1998. Int. J. Hematol. 2002, 76, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, H.; Iwanaga, M.; Miyazaki, Y. Late effect of atomic bomb radiation on myeloid disorders: Leukemia and myelodysplastic syndromes. Int. J. Hematol. 2012, 95, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, M.; Hsu, W.L.; Soda, M.; Takasaki, Y.; Tawara, M.; Joh, T.; Amenomori, T.; Yamamura, M.; Yoshida, Y.; Koba, T.; et al. Risk of myelodysplastic syndromes in people exposed to ionizing radiation: A retrospective cohort study of Nagasaki atomic bomb survivors. J. Clin. Oncol. 2011, 29, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, A. The role of DNA repair in benzene-induced carcinogenesis. Chem. Biol. Interact. 2010, 184, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Aul, C.; Bowen, D.T.; Yoshida, Y. Pathogenesis, etiology and epidemiology of myelodysplastic syndromes. Haematologica 1998, 83, 71–86. [Google Scholar] [PubMed]
- Ren, X.; Lim, S.; Smith, M.T.; Zhang, L. Werner syndrome protein, WRN, protects cells from DNA damage induced by the benzene metabolite hydroquinone. Toxicol. Sci. 2009, 107, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Razin, S.V. Chemotherapy-related secondary leukemias: A role for DNA repair by error-prone non-homologous end joining in topoisomerase II-Induced chromosomal rearrangements. Gene 2007, 391, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Baehring, J.M.; Marks, P.W. Treatment-related myelodysplasia in patients with primary brain tumors. Neuro-Oncol. 2012, 14, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.E. Risk for the development of treatment-related acute myelocytic leukemia and myelodysplastic syndrome among patients with breast cancer: Review of the literature and the National Surgical Adjuvant Breast and Bowel Project experience. Clin. Breast Cancer 2003, 4, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Au, W.Y.; Ma, S.K.; Wan, T.S.; Man, C.; Kwong, Y.L. Pentasomy 8q in therapy-related myelodysplastic syndrome due to cyclophosphamide therapy for fibrosing alveolitis. Cancer Genet. Cytogenet. 2003, 141, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, F.E. Risk of acute myelogenous leukaemia and myelodysplasia following cancer treatment. Baillière’s Clin. Haematol. 1996, 9, 57–85. [Google Scholar] [CrossRef]
- Winick, N.J.; McKenna, R.W.; Shuster, J.J.; Schneider, N.R.; Borowitz, M.J.; Bowman, W.P.; Jacaruso, D.; Kamen, B.A.; Buchanan, G.R. Secondary acute myeloid leukemia in children with acute lymphoblastic leukemia treated with etoposide. J. Clin. Oncol. 1993, 11, 209–217. [Google Scholar] [PubMed]
- Zhang, L.; Wang, S.A. A focused review of hematopoietic neoplasms occurring in the therapy-related setting. Int. J. Clin. Exp. Pathol. 2014, 7, 3512–3523. [Google Scholar] [PubMed]
- Marchesi, F.; Turriziani, M.; Tortorelli, G.; Avvisati, G.; Torino, F.; de Vecchis, L. Triazene compounds: Mechanism of action and related DNA repair systems. Pharmacol. Res. 2007, 56, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Heisig, P. Type II topoisomerases—Inhibitors, repair mechanisms and mutations. Mutagenesis 2009, 24, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W. DNA Repair and Mutagenesis, 2nd ed.; AMC Press: Washington, DC, USA, 1995. [Google Scholar]
- Fan, J.R.; Peng, A.L.; Chen, H.C.; Lo, S.C.; Huang, T.H.; Li, T.K. Cellular processing pathways contribute to the activation of etoposide-induced DNA damage responses. DNA Repair 2008, 7, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Essers, J.; Vermeulen, W.; Houtsmuller, A.B. DNA damage repair: Anytime, anywhere? Curr. Opin. Cell Biol. 2006, 18, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Cyclin-dependent protein kinases: Key regulators of the eukaryotic cell cycle. Bioessays 1995, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Callegari, A.J.; Kelly, T.J. UV irradiation induces a postreplication DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 2006, 103, 15877–15882. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Teng, X.; Liu, Y.; Tseng, M.; Spardy, N.; Duensing, S. A role of the mitotic spindle checkpoint in the cellular response to DNA replication stress. J. Cell. Biochem. 2006, 99, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, D.H.; Andersen, M.K.; Pedersen-Bjergaard, J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J. Clin. Oncol. 2001, 19, 1405–1413. [Google Scholar] [PubMed]
- Constantinidou, M.; Chalevelakis, G.; Economopoulos, T.; Koffa, M.; Liloglou, T.; Anastassiou, C.; Yalouris, A.; Spandidos, D.A.; Raptis, S. Codon 12 ras mutations in patients with myelodysplastic syndrome: Incidence and prognostic value. Ann. Hematol. 1997, 74, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Chen, X.; Rocha, K.; Epling-Burnette, P.K.; Djeu, J.Y.; Liu, Q.; Byrd, J.; Sokol, L.; Lawrence, N.; Pireddu, R.; et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc. Natl. Acad. Sci. USA 2009, 106, 12974–12979. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Wei, S.; List, A. PP2A: The achilles heal in MDS with 5q deletion. Front. Oncol. 2014, 4, 264. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Kinoshita, T.; Nagai, H.; Nakahara, Y.; Saito, H.; Hotta, T.; Murate, T. Hypermethylation of the p15INK4B gene in myelodysplastic syndromes. Blood 1997, 90, 1403–1409. [Google Scholar] [PubMed]
- Rodrigues, E.F.; Santos-Reboucas, C.B.; Goncalves Pimentel, M.M.; Mencalha, A.L.; Dobbin, J.; da Costa, E.S.; Fernandez Cde, S.; Bouzas, L.F.; Abdelhay, E.; de Souza Fernandez, T. Epigenetic alterations of p15(INK4B) and p16(INK4A) genes in pediatric primary myelodysplastic syndrome. Leuk. Lymphoma 2010, 51, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.K.; Miller, C.W.; Tsukasaki, K.; Tavor, S.; Ikezoe, T.; Hoelzer, D.; Takeuchi, S.; Koeffler, H.P. Mutation analysis of the DNA-damage checkpoint gene CHK2 in myelodysplastic syndromes and acute myeloid leukemias. Leuk. Res. 2001, 25, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Preudhomme, C.; Vachee, A.; Lepelley, P.; Vanrumbeke, M.; Zandecki, M.; Quesnel, B.; Cosson, A.; Fenaux, P. Inactivation of the retinoblastoma gene appears to be very uncommon in myelodysplastic syndromes. Br. J. Haematol. 1994, 87, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Bally, C.; Ades, L.; Renneville, A.; Sebert, M.; Eclache, V.; Preudhomme, C.; Mozziconacci, M.J.; de The, H.; Lehmann-Che, J.; Fenaux, P. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk. Res. 2014, 38, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Perez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Saft, L.; Karimi, M.; Ghaderi, M.; Matolcsy, A.; Mufti, G.J.; Kulasekararaj, A.; Gohring, G.; Giagounidis, A.; Selleslag, D.; Muus, P.; et al. p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica 2014, 99, 1041–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkert, S.; Kohlmann, A.; Schnittger, S.; Kern, W.; Haferlach, T.; Haferlach, C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer 2014, 53, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Kulasekararaj, A.G.; Smith, A.E.; Mian, S.A.; Mohamedali, A.M.; Krishnamurthy, P.; Lea, N.C.; Gaken, J.; Pennaneach, C.; Ireland, R.; Czepulkowski, B.; et al. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br. J. Haematol. 2013, 160, 660–672. [Google Scholar] [CrossRef] [PubMed]
- D’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Lobrich, M. Contribution of DNA repair and cell cycle checkpoint arrest to the maintenance of genomic stability. DNA Repair 2006, 5, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Q.; Morita, Y.; Jiang, H.; Gross, A.; Lechel, A.; Hildner, K.; Guachalla, L.M.; Gompf, A.; Hartmann, D.; et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012, 148, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Tehranchi, R.; Invernizzi, R.; Grandien, A.; Zhivotovsky, B.; Fadeel, B.; Forsblom, A.M.; Travaglino, E.; Samuelsson, J.; Hast, R.; et al. Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes. Blood 2005, 106, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Mufti, G.J.; Rasool, F.; Mijovic, A.; Devereux, S.; Pagliuca, A. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000, 96, 3932–3938. [Google Scholar] [PubMed]
- Davis, R.E.; Greenberg, P.L. Bcl-2 expression by myeloid precursors in myelodysplastic syndromes: Relation to disease progression. Leuk. Res. 1998, 22, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Neiger, J.D.; Porcher, J.C.; Keats, J.J.; Bergsagel, P.L.; Dennis, T.R.; Knudson, R.A.; Jenkins, R.B.; Santana-Davila, R.; Kumar, R.; et al. Rearrangements and amplification of IER3 (IEX-1) represent a novel and recurrent molecular abnormality in myelodysplastic syndromes. Cancer Res. 2009, 69, 7518–7523. [Google Scholar] [CrossRef] [PubMed]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Fishlock, K.L.; Mijovic, A.; Czepulkowski, B.; Pagliuca, A.; Mufti, G.J. “Low-risk” myelodysplastic syndrome is associated with excessive apoptosis and an increased ratio of pro- vs. anti-apoptotic Bcl-2-related proteins. Br. J. Haematol. 1998, 103, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Monch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ai, X.; Gale, R.P.; Xu, Z.; Qin, T.; Fang, L.; Zhang, H.; Pan, L.; Hu, N.; Zhang, Y.; et al. TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk. Res. 2013, 37, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Souza-Pinto, N.; Nyaga, S.G.; Thybo, T.; Stevnsner, T.; Bohr, V.A. Base excision repair in nuclear and mitochondrial DNA. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 285–297. [Google Scholar] [PubMed]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, K.; Iwai, S.; Tainer, J.A. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair (Amst.) 2007, 6, 410–428. [Google Scholar] [CrossRef]
- Liu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007, 282, 13532–13541. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Kasparek, T.R.; Humphrey, T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol. 2011, 22, 886–897. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Alt, F.W. The cellular response to general and programmed DNA double strand breaks. DNA Repair 2004, 3, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, L.; Small, D.; Rassool, F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood 2010, 116, 5298–5305. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E.; Rassool, F.V. Up-regulation of WRN and DNA ligase IIIα in chronic myeloid leukemia: Consequences for the repair of DNA double-strand breaks. Blood 2008, 112, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.F.; Wiemels, J. Origins of chromosome translocations in childhood leukaemia. Nat. Rev. Cancer 2003, 3, 639–649. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Kabotyanski, E.B.; Gomelsky, L.; Han, J.O.; Stamato, T.D.; Roth, D.B. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 1998, 26, 5333–5342. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Hasty, P.; Walter, C.A.; Bishop, A.J.; Scott, L.M.; Rebel, V.I. Myelodysplastic syndrome: An inability to appropriately respond to damaged DNA? Exp. Hematol. 2013, 41, 665–674. [Google Scholar] [CrossRef] [PubMed]
- De Laval, B.; Pawlikowska, P.; Barbieri, D.; Besnard-Guerin, C.; Cico, A.; Kumar, R.; Gaudry, M.; Baud, V.; Porteu, F. Thrombopoietin promotes NHEJ DNA repair in hematopoietic stem cells through specific activation of Erk and NF-κB pathways and their target, IEX-1. Blood 2014, 123, 509–519. [Google Scholar] [CrossRef] [PubMed]
- De Laval, B.; Pawlikowska, P.; Petit-Cocault, L.; Bilhou-Nabera, C.; Aubin-Houzelstein, G.; Souyri, M.; Pouzoulet, F.; Gaudry, M.; Porteu, F. Thrombopoietin-increased DNA-PK-dependent DNA repair limits hematopoietic stem and progenitor cell mutagenesis in response to DNA damage. Cell Stem Cell 2013, 12, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Prall, W.C.; Czibere, A.; Grall, F.; Spentzos, D.; Steidl, U.; Giagounidis, A.A.; Kuendgen, A.; Otu, H.; Rong, A.; Libermann, T.A.; et al. Differential gene expression of bone marrow-derived CD34+ cells is associated with survival of patients suffering from myelodysplastic syndrome. Int. J. Hematol. 2009, 89, 173–187. [Google Scholar] [CrossRef]
- Ramsey, H.; Zhang, Q.; Brown, D.E.; Steensma, D.P.; Lin, C.P.; Wu, M.X. Stress-induced hematopoietic failure in the absence of immediate early response gene X-1 (IEX-1, IER3). Haematologica 2014, 99, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Crossan, G.P.; Patel, K.J. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J. Pathol. 2012, 226, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Pichierri, P.; Franchitto, A.; Rosselli, F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 2004, 23, 3154–3163. [Google Scholar] [CrossRef] [PubMed]
- Cioc, A.M.; Wagner, J.E.; MacMillan, M.L.; DeFor, T.; Hirsch, B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: Morphologic and cytogenetic characteristics. Am. J. Clin. Pathol. 2010, 133, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Quentin, S.; Cuccuini, W.; Ceccaldi, R.; Nibourel, O.; Pondarre, C.; Pages, M.P.; Vasquez, N.; Dubois d’Enghien, C.; Larghero, J.; Peffault de Latour, R.; et al. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 2011, 117, e161–e170. [Google Scholar] [CrossRef] [PubMed]
- Poppe, B.; van Limbergen, H.; van Roy, N.; Vandecruys, E.; de Paepe, A.; Benoit, Y.; Speleman, F. Chromosomal aberrations in Bloom syndrome patients with myeloid malignancies. Cancer Genet. Cytogenet. 2001, 128, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, I.; Arikan, U.; Buyukpamukcu, M. Myelodysplastic syndromes and RTS. Pediatr. Hematol. Oncol. 1996, 13, 197. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Fleming, C.; Trainer, A.H.; Craig, J.A. Rothmund-Thomson syndrome with myelodysplasia. Pediatr. Dermatol. 2001, 18, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Pianigiani, E.; de Aloe, G.; Andreassi, A.; Rubegni, P.; Fimiani, M. Rothmund-Thomson syndrome (Thomson-type) and myelodysplasia. Pediatr. Dermatol. 2001, 18, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Rizzari, C.; Bacchiocchi, D.; Rovelli, A.; Biondi, A.; Cantu’-Rajnoldi, A.; Uderzo, C.; Masera, G. Myelodysplastic syndrome in a child with Rothmund-Thomson syndrome: A case report. J. Pediatr. Hematol. Oncol. 1996, 18, 96–97. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol. Biomark. Prev. 1996, 5, 239–246. [Google Scholar]
- Yamamoto, K.; Imakiire, A.; Miyagawa, N.; Kasahara, T. A report of two cases of Werner’s syndrome and review of the literature. J. Orthop. Surg. 2003, 11, 224–233. [Google Scholar]
- Broberg, K.; Hoglund, M.; Gustafsson, C.; Bjork, J.; Ingvar, C.; Albin, M.; Olsson, H. Genetic variant of the human homologous recombination-associated gene RMI1 (S455N) impacts the risk of AML/MDS and malignant melanoma. Cancer Lett. 2007, 258, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar] [PubMed]
- Jacoby, M.A.; de Jesus Pizarro, R.E.; Shao, J.; Koboldt, D.C.; Fulton, R.S.; Zhou, G.; Wilson, R.K.; Walter, M.J. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia 2014, 28, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Seedhouse, C.; Faulkner, R.; Ashraf, N.; Das-Gupta, E.; Russell, N. Polymorphisms in genes involved in homologous recombination repair interact to increase the risk of developing acute myeloid leukemia. Clin. Cancer Res. 2004, 10, 2675–2680. [Google Scholar] [CrossRef] [PubMed]
- Talwalkar, S.S.; Yin, C.C.; Naeem, R.C.; Hicks, M.J.; Strong, L.C.; Abruzzo, L.V. Myelodysplastic syndromes arising in patients with germline TP53 mutation and Li-Fraumeni syndrome. Arch. Pathol. Lab. Med. 2010, 134, 1010–1015. [Google Scholar] [PubMed]
- Gadner, H.; Haas, O.A. Experience in pediatric myelodysplastic syndromes. Hematol. Oncol. Clin. N. Am. 1992, 6, 655–672. [Google Scholar]
- Zhang, M.Y.; Keel, S.B.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Watts, A.C.; Pritchard, C.C.; Salipante, S.J.; Jeng, M.R.; Hofmann, I.; et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica 2015, 100, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C.A.; Chessells, J.M.; Fitchett, M. Neurofibromatosis and childhood leukaemia/lymphoma: A population-based UKCCSG study. Br. J. Cancer 1994, 70, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.L.; Zhang, B.; Riches, J.J.; Gandhi, R.; Li, J.; Rommens, J.M.; Myers, J.S. Shwachman-Bodian Diamond syndrome is a multi-functional protein implicated in cellular stress responses. Hum. Mol. Genet. 2009, 18, 3684–3695. [Google Scholar] [CrossRef] [PubMed]
- Ginzberg, H.; Shin, J.; Ellis, L.; Morrison, J.; Ip, W.; Dror, Y.; Freedman, M.; Heitlinger, L.A.; Belt, M.A.; Corey, M.; et al. Shwachman syndrome: Phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J. Pediatr. 1999, 135, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Smith, O.P. Shwachman-Diamond syndrome. Semin. Hematol. 2002, 39, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gramatges, M.M.; Bertuch, A.A. Short telomeres: From dyskeratosis congenita to sporadic aplastic anemia and malignancy. Transl. Res. 2013, 162, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Kojima, S. Recent progress in dyskeratosis congenita. Int. J. Hematol. 2010, 92, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Dokal, I. Dyskeratosis congenita in all its forms. Br. J. Haematol. 2000, 110, 768–779. [Google Scholar] [CrossRef] [PubMed]
- McLean, S.; McHale, C.; Enright, H. Hematological abnormalities in adult patients with Down’s syndrome. Ir. J. Med. Sci. 2009, 178, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Morawiec, Z.; Janik, K.; Kowalski, M.; Stetkiewicz, T.; Szaflik, J.; Morawiec-Bajda, A.; Sobczuk, A.; Blasiak, J. DNA damage and repair in children with Down’s syndrome. Mutat. Res. 2008, 637, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K. Hematopoietic disorders in Down syndrome. Int. J. Clin. Exp. Pathol. 2008, 1, 387–395. [Google Scholar] [PubMed]
- Glaubach, T.; Robinson, L.J.; Corey, S.J. Pediatric myelodysplastic syndromes: They do exist! J. Pediatr. Hematol. Oncol. 2014, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pemov, A.; Park, C.; Reilly, K.M.; Stewart, D.R. Evidence of perturbations of cell cycle and DNA repair pathways as a consequence of human and murine NF1-haploinsufficiency. BMC Genomics 2010, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Vulliamy, T.J.; Knight, S.W.; Mason, P.J.; Dokal, I. Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita. Blood Cells Mol. Dis. 2001, 27, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Pluciennik, A.; Dzantiev, L.; Iyer, R.R.; Constantin, N.; Kadyrov, F.A.; Modrich, P. PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc. Natl. Acad. Sci. USA 2010, 107, 16066–16071. [Google Scholar] [CrossRef] [PubMed]
- Buermeyer, A.B.; Deschenes, S.M.; Baker, S.M.; Liskay, R.M. Mammalian DNA mismatch repair. Annu. Rev. Genet. 1999, 33, 533–564. [Google Scholar] [CrossRef] [PubMed]
- Alemayehu, A.; Fridrichova, I. The MRE11/RAD50/NBS1 complex destabilization in Lynch-syndrome patients. Eur. J. Hum. Genet. 2007, 15, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yehuda, D.; Krichevsky, S.; Caspi, O.; Rund, D.; Polliack, A.; Abeliovich, D.; Zelig, O.; Yahalom, V.; Paltiel, O.; Or, R.; et al. Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Blood 1996, 88, 4296–4303. [Google Scholar] [PubMed]
- Casorelli, I.; Offman, J.; Mele, L.; Pagano, L.; Sica, S.; D’Errico, M.; Giannini, G.; Leone, G.; Bignami, M.; Karran, P. Drug treatment in the development of mismatch repair defective acute leukemia and myelodysplastic syndrome. DNA Repair 2003, 2, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Olipitz, W.; Hopfinger, G.; Aguiar, R.C.; Gunsilius, E.; Girschikofsky, M.; Bodner, C.; Hiden, K.; Linkesch, W.; Hoefler, G.; Sill, H. Defective DNA-mismatch repair: A potential mediator of leukemogenic susceptibility in therapy-related myelodysplasia and leukemia. Genes Chromosomes Cancer 2002, 34, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.S.; Liu, L.; Gerson, S.L. Repopulating defect of mismatch repair-deficient hematopoietic stem cells. Blood 2003, 102, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; D’Andrea, A.D. DNA repair pathways in clinical practice: Lessons from pediatric cancer susceptibility syndromes. J. Clin. Oncol. 2006, 24, 3799–3808. [Google Scholar] [CrossRef] [PubMed]
- Riedl, T.; Hanaoka, F.; Egly, J.M. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003, 22, 5293–5303. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C. Subpathways of nucleotide excision repair and their regulation. Oncogene 2002, 21, 8949–8956. [Google Scholar] [CrossRef] [PubMed]
- Dip, R.; Camenisch, U.; Naegeli, H. Mechanisms of DNA damage recognition and strand discrimination in human nucleotide excision repair. DNA Repair (Amst.) 2004, 3, 1409–1423. [Google Scholar] [CrossRef]
- Christians, F.C.; Hanawalt, P.C. Lack of transcription-coupled repair in mammalian ribosomal RNA genes. Biochemistry 1993, 32, 10512–10518. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Pfeifer, G.P. Lack of gene- and strand-specific DNA repair in RNA polymerase III-transcribed human tRNA genes. Mol. Cell. Biol. 1997, 17, 219–229. [Google Scholar] [PubMed]
- Fuss, J.O.; Cooper, P.K. DNA repair: Dynamic defenders against cancer and aging. PLoS Biol. 2006, 4, e203. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.K.; Nouspikel, T.; Clarkson, S.G.; Leadon, S.A. Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science 1997, 275, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Webb, S. Xeroderma pigmentosum. BMJ 2008, 336, 444–446. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, T. Pediatric Oncology, 2nd ed.; Imbach, P., Kuehne, T., Arceci, R.J., Eds.; Springer: Berlin, Germany, 2006; pp. 39–40. [Google Scholar]
- Carney, D.A.; Westerman, D.A.; Tam, C.S.; Milner, A.; Prince, H.M.; Kenealy, M.; Wolf, M.; Januszewicz, E.H.; Ritchie, D.; Came, N.; et al. Therapy-related myelodysplastic syndrome and acute myeloid leukemia following fludarabine combination chemotherapy. Leukemia 2010, 24, 2056–2062. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Seymour, J.F.; Prince, H.M.; Kenealy, M.; Wolf, M.; Januszewicz, E.H.; Westerman, D. Treatment-related myelodysplasia following fludarabine combination chemotherapy. Haematologica 2006, 91, 1546–1550. [Google Scholar] [PubMed]
- Issa, J.P. The myelodysplastic syndrome as a prototypical epigenetic disease. Blood 2013, 121, 3811–3817. [Google Scholar] [CrossRef] [PubMed]
- Vasilatou, D.; Papageorgiou, S.G.; Dimitriadis, G.; Pappa, V. Epigenetic alterations and microRNAs: New players in the pathogenesis of myelodysplastic syndromes. Epigenetics 2013, 8, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.J.; Zair, Z.; Johnson, G.E.; Doak, S.H. Genotoxic thresholds, DNA repair, and susceptibility in human populations. Toxicology 2010, 278, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Rossi, D.J. Epigenetic regulation of hematopoietic stem cell aging. Exp. Cell Res. 2014, 329, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Guillem, V.; Tormo, M. Influence of DNA damage and repair upon the risk of treatment related leukemia. Leuk. Lymphoma 2008, 49, 204–217. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Chen, P.; Gu, J.; Bishop, A.J.R.; Scott, L.M.; Hasty, P.; Rebel, V.I. Potential Relationship between Inadequate Response to DNA Damage and Development of Myelodysplastic Syndrome. Int. J. Mol. Sci. 2015, 16, 966-989. https://doi.org/10.3390/ijms16010966
Zhou T, Chen P, Gu J, Bishop AJR, Scott LM, Hasty P, Rebel VI. Potential Relationship between Inadequate Response to DNA Damage and Development of Myelodysplastic Syndrome. International Journal of Molecular Sciences. 2015; 16(1):966-989. https://doi.org/10.3390/ijms16010966
Chicago/Turabian StyleZhou, Ting, Peishuai Chen, Jian Gu, Alexander J. R. Bishop, Linda M. Scott, Paul Hasty, and Vivienne I. Rebel. 2015. "Potential Relationship between Inadequate Response to DNA Damage and Development of Myelodysplastic Syndrome" International Journal of Molecular Sciences 16, no. 1: 966-989. https://doi.org/10.3390/ijms16010966
APA StyleZhou, T., Chen, P., Gu, J., Bishop, A. J. R., Scott, L. M., Hasty, P., & Rebel, V. I. (2015). Potential Relationship between Inadequate Response to DNA Damage and Development of Myelodysplastic Syndrome. International Journal of Molecular Sciences, 16(1), 966-989. https://doi.org/10.3390/ijms16010966