
Revealing the Supramolecular Nature of Side-Chain Terpyridine-Functionalized Polymer Networks



Abstract
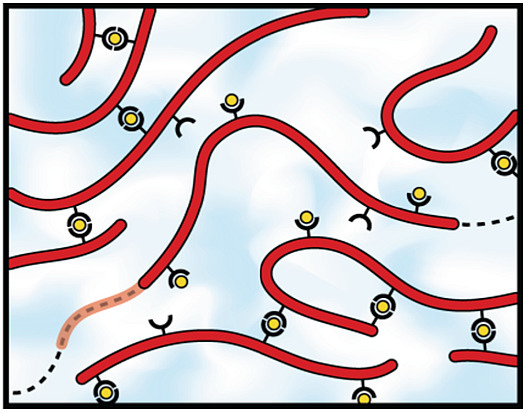
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion

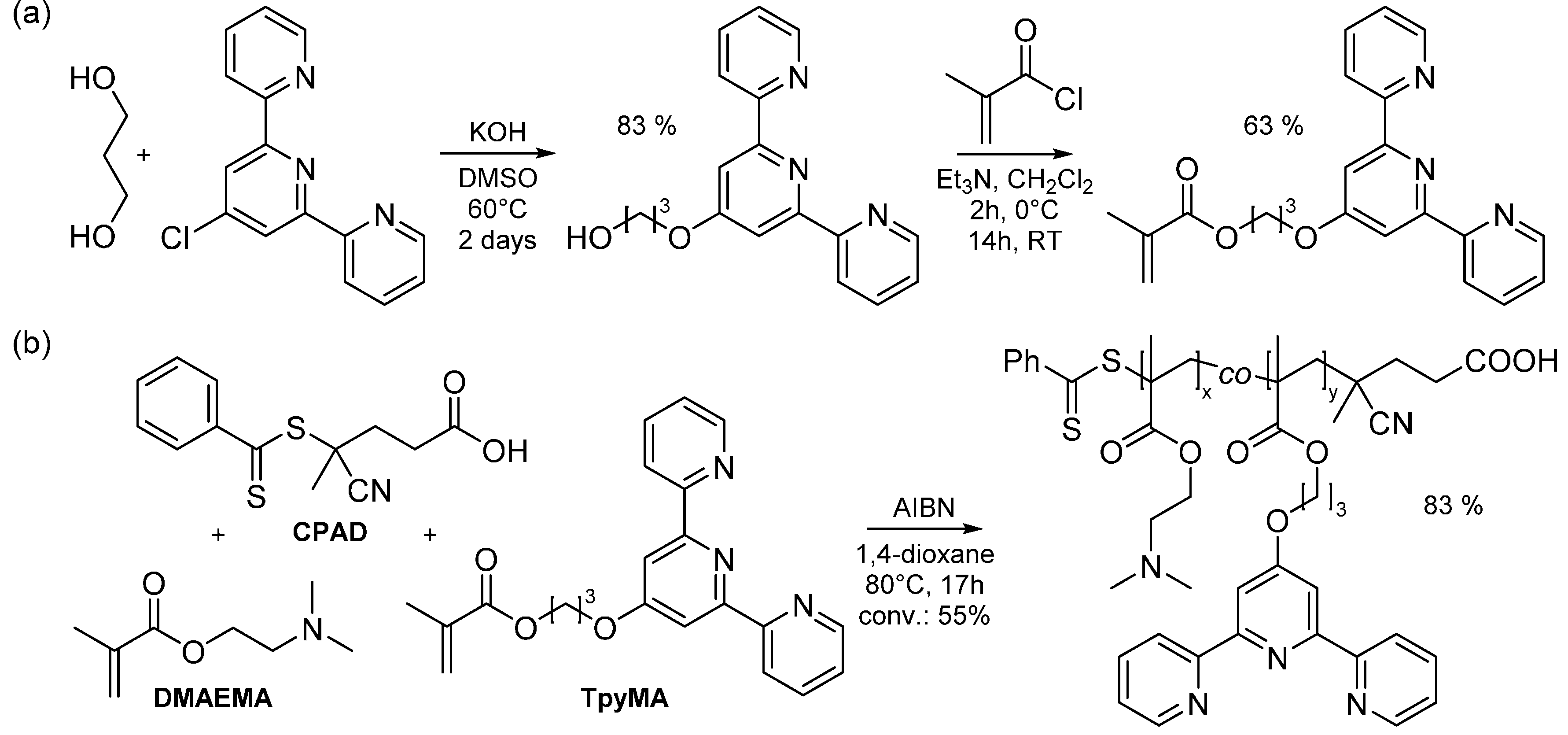
2.1. Synthesis of Side-Chain Terpyridine-Functionalized Poly(2-(dimethylamino)ethyl methacrylate)



2.2. Self-Assembly into Metallo-Supramolecular Hydrogels

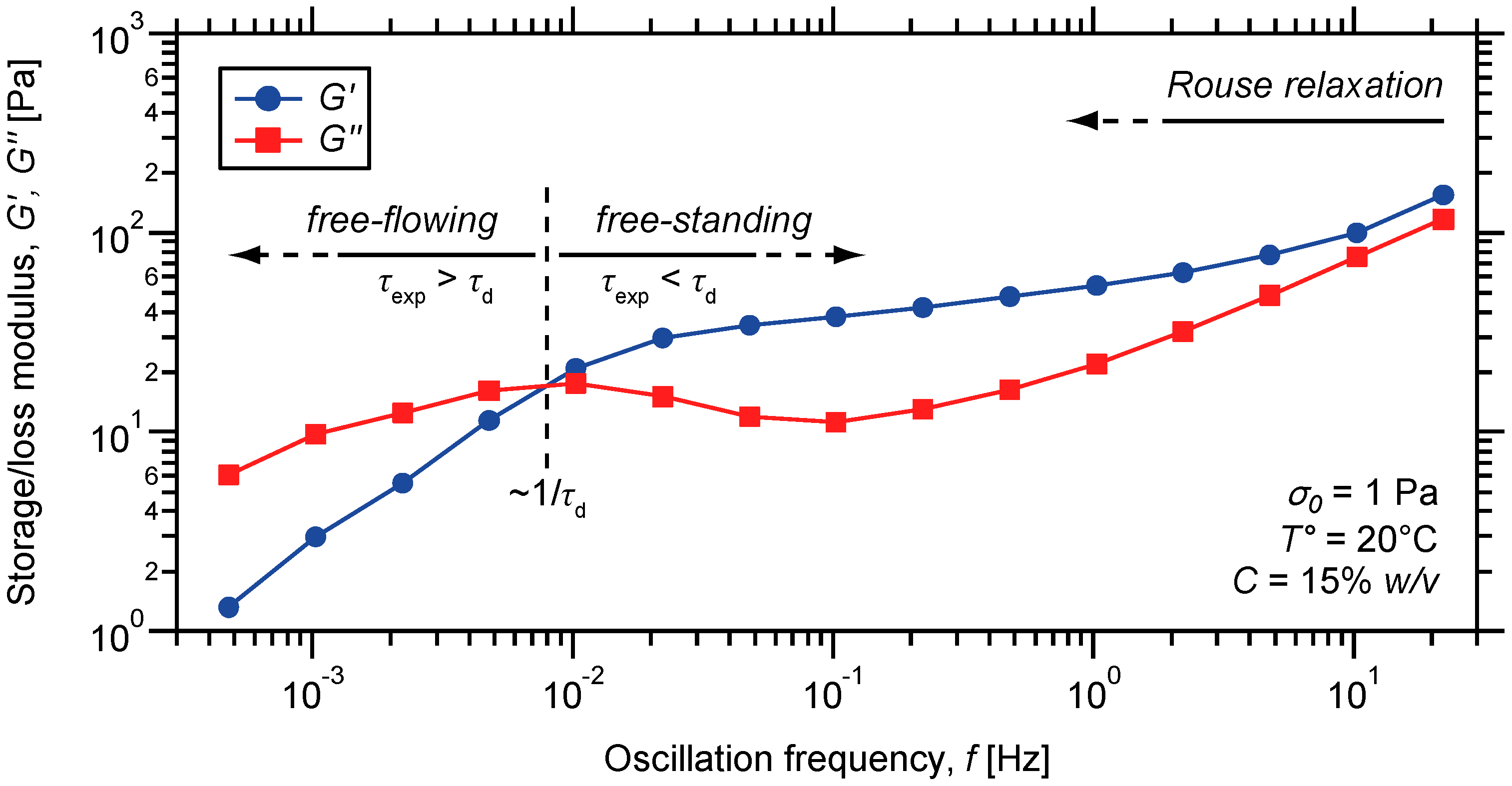
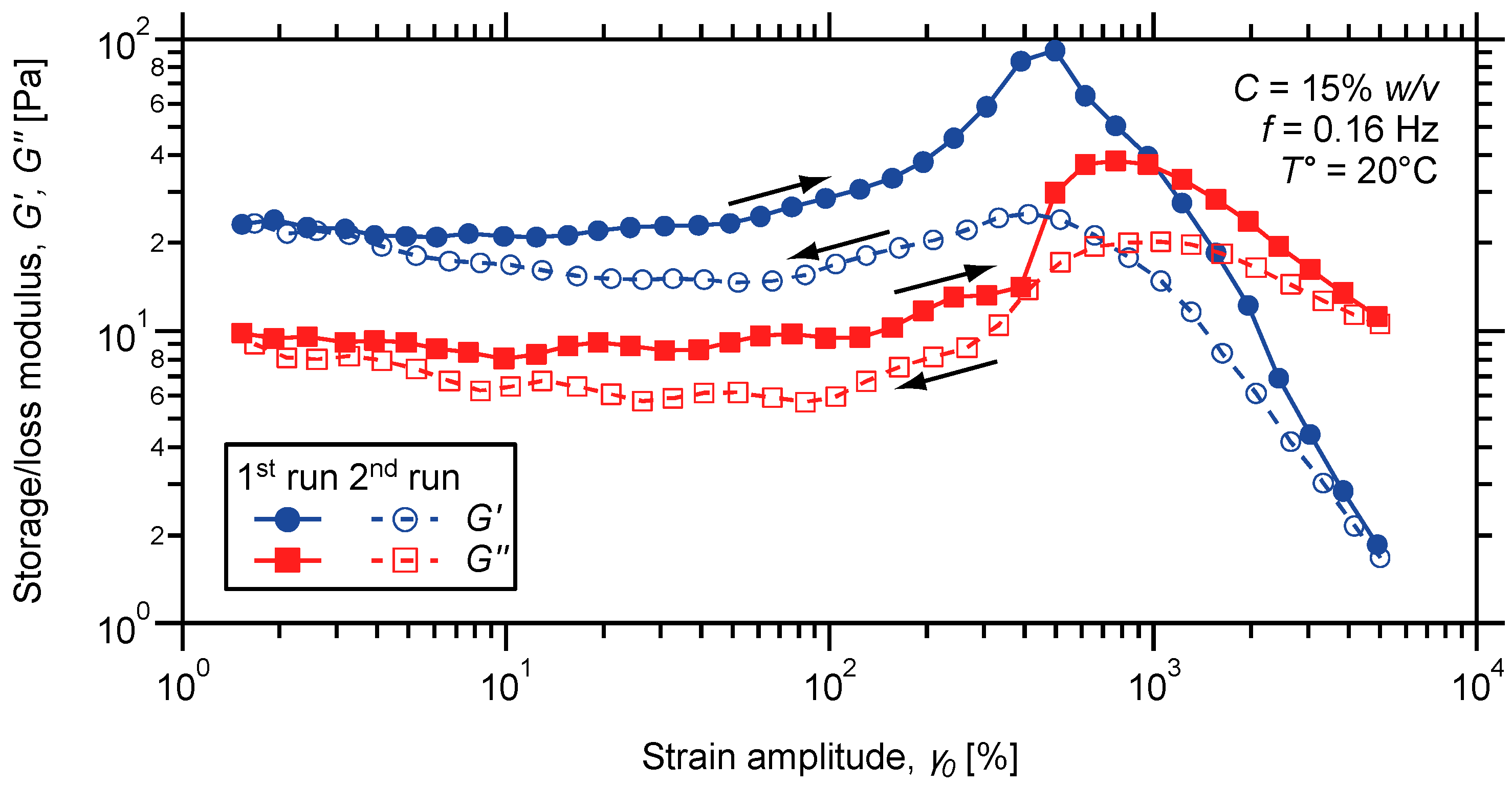
2.3. Characterization of the Dynamic Mechanical Response of the Gels

3. Experimental Section
3.1. Materials
3.2. Instrumentation
3.3. Synthesis of 4'-(3-Hydroxypropoxy)-2,2':6',2''-terpyridine
3.4. Synthesis of 2-Methacrylic acid-3-(2,2':6',2''-terpyridine-4'-yloxy)propyl ester (TpyMA)
3.5. Synthesis of Side-Chain Terpyridine-Functionalized Poly(2-(dimethylamino)ethyl methacrylate) (P(DMAMEA200-co-TpyMA4.5))
3.6. Preparation of Metallo-Supramolecular Hydrogels
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Appel, W.P.J.; Nieuwenhuizen, M.M.L.; Meijer, E.W. Multiple hydrogen-bonded supramolecular polymers. In Supramolecular Polymer Chemistry, 1st ed.; Harada, A., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 3–28. [Google Scholar]
- Yan, X.; Wang, F.; Zheng, B.; Huang, F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 2012, 41, 6042–6065. [Google Scholar] [PubMed]
- Appel, E.A.; del Barrio, J.; Loh, X.J.; Scherman, O.A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 2012, 41, 6195–6214. [Google Scholar] [PubMed]
- Noro, A.; Hayashi, M.; Matsushita, Y. Design and properties of supramolecular polymer gels. Soft Matter 2012, 8, 2416–2429. [Google Scholar] [CrossRef]
- Yount, W.C.; Loveless, D.M.; Craig, S.L. Small-molecule dynamics and mechanisms underlying the macroscopic mechanical properties of coordinatively cross-linked polymer networks. J. Am. Chem. Soc. 2005, 127, 14488–14496. [Google Scholar] [CrossRef] [PubMed]
- Loveless, D.M.; Jeon, S.L.; Craig, S.L. Rational control of viscoelastic properties in multicomponent associative polymer networks. Macromolecules 2005, 38, 10171–10177. [Google Scholar] [CrossRef]
- Brassinne, J.; Stevens, A.M.; van Ruymbeke, E.; Gohy, J.-F.; Fustin, C.-A. Hydrogels with dual relaxation and two-step gel-sol transition from heterotelechelic polymers. Macromolecules 2013, 46, 9134–9143. [Google Scholar] [CrossRef]
- Brassinne, J.; Bourgeois, J.-P.; Fustin, C.-A.; Gohy, J.-F. Thermo-responsive properties of metallo-supramolecular block copolymer micellar hydrogels. Soft Matter 2014, 10, 3086–3092. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.N.; Feldman, K.E.; Lynd, N.A.; Deek, J.; Campos, L.M.; Spruell, J.M.; Hernandez, B.M.; Kramer, E.J.; Hawker, C.J. Tunable, high modulus hydrogels driven by ionic coacervation. Adv. Mater. 2011, 23, 2327–2331. [Google Scholar] [CrossRef] [PubMed]
- Hisamatsu, Y.; Banerjee, S.; Avinash, M.B.; Govindaraju, T.; Schmuck, C. A supramolecular gel from a quadruple zwitterion that responds to both acid and base. Angew. Chem. Int. Ed. 2013, 52, 12550–12554. [Google Scholar] [CrossRef]
- Brassinne, J.; Fustin, C.-A.; Gohy, J.-F. Polymer gels constructed through metal-ligand coordination. J. Inorg. Organomet. Polym. Mater. 2013, 23, 24–40. [Google Scholar] [CrossRef]
- Noro, A.; Hayashi, M.; Ohshika, A.; Matsushita, Y. Simple preparation of supramolecular polymer gels via hydrogen bonding by blending two liquid polymers. Soft Matter 2011, 7, 1667–1670. [Google Scholar] [CrossRef]
- Nair, K.P.; Breedveld, V.; Weck, M. Complementary hydrogen-bonded thermoreversible polymer networks with tunable properties. Macromolecules 2008, 41, 3429–3438. [Google Scholar] [CrossRef]
- Li, S.; Lu, H.-Y.; Shen, Y.; Chen, C.-F. A stimulus-response and self-healing supramolecular polymer gel based on host-guest interactions. Macromol. Chem. Phys. 2013, 214, 1596–1601. [Google Scholar] [CrossRef]
- Liu, D.; Wang, D.; Wang, M.; Zheng, Y.; Koynov, K.; Auernhammer, G.K.; Butt, H.-J.; Ikeda, T. Supramolecular organogel based on crown ether and secondary ammoniumion functionalized glycidyl triazole polymers. Macromolecules 2013, 46, 4617–4625. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, D.; Yan, X.; Chen, J.; Dong, S.; Zheng, B.; Huang, F. Self-healing supramolecular gels formed by crown ether based host-guest interactions. Angew. Chem. Int. Ed. 2012, 51, 7011–7015. [Google Scholar] [CrossRef]
- Chassenieux, C.; Nicolai, T.; Benyahia, L. Rheology of associative polymer solutions. Curr. Opin. Colloid Interface Sci. 2011, 16, 18–26. [Google Scholar] [CrossRef]
- Hietala, S.; Strandman, S.; Jarvi, P.; Torkkeli, M.; Jankova, K.; Hvilsted, S.; Tenhu, H. Rheological properties of associative star polymers in aqueous solutions: Effect of hydrophobe length and polymer topology. Macromolecules 2009, 42, 1726–1732. [Google Scholar] [CrossRef]
- Suzuki, S.; Uneyama, T.; Watanabe, H. Concentration dependence of nonlinear rheological properties of hydrophobically modified ethoxylated urethane aqueous solutions. Macromolecules 2013, 46, 3497–3504. [Google Scholar] [CrossRef]
- Kurth, D.G.; Higuchi, M. Transition metal ions: Weak links for strong polymers. Soft Matter 2006, 2, 915–927. [Google Scholar] [CrossRef]
- Burnworth, M.; Tang, L.M.; Kumpfer, J.R.; Duncan, A.J.; Beyer, F.L.; Fiore, G.L.; Rowan, S.J.; Weder, C. Optically healable supramolecular polymers. Nature 2011, 472, 334–230. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kang, S.; Lee, J.Y.; Jung, J.H. Coordination polymer gel derived from a tetrazole ligand and Zn2+: Spectroscopic and mechanical properties of an amorphous coordination polymer gel. Soft Matter 2012, 8, 2950–2955. [Google Scholar] [CrossRef]
- Marin, V.; Holder, E.; Hoogenboom, R.; Schubert, U.S. Functional ruthenium (II)- and iridium (III)-containing polymers for potential electro-optical applications. Chem. Soc. Rev. 2007, 36, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Kokil, A.; Shiyanovskaya, I.; Singer, K.D.; Weder, C. High charge carrier mobility in conjugated organometallic polymer networks. J. Am. Chem. Soc. 2002, 124, 9978–9979. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Gao, Y.; Shi, J.; Wang, L.; Xu, B. A versatile supramolecular hydrogel of nitrilotriacetic acid (NTA) for binding metal ions and magnetorheological response. J. Mater. Chem. 2011, 21, 6804–6806. [Google Scholar] [CrossRef]
- Roubeau, O.; Colin, A.; Schmitt, V.; Clérac, R. Thermoreversible gels as magneto-optical switches. Angew. Chem. Int. Ed. 2004, 43, 3283–3286. [Google Scholar] [CrossRef]
- Xing, B.; Choi, M.F.; Xu, B. Design of coordination polymer gels as stable catalytic systems. Chem. Eur. J. 2002, 8, 5028–5032. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; He, L.; Zhang, J.; Chen, L.; Su, C.Y. Dynamic functionalised metallogel: An approach to immobilised catalysis with improved activity. J. Mol. Catal. 2010, 317, 97–103. [Google Scholar] [CrossRef]
- He, Y.; Bian, Z.; Kang, C.; Cheng, Y.; Gao, L. Chiral binaphthylbisbipyridine-based copper (I) coordination polymer gels as supramolecular catalysts. Chem. Commun. 2010, 46, 3532–3534. [Google Scholar] [CrossRef]
- Moad, G.; Chiefari, J.; Krstina, J.; Mayadunne, R.T.A.; Postma, A.; Rizzardo, E.; Thang, S.H. Living free radical polymerization with reversible addition-fragmentation chain transfer (the life of RAFT). Polym. Int. 2000, 49, 993–1001. [Google Scholar] [CrossRef]
- Perrier, S.; Takolpuckdee, P. Macromolecular design via reversible addition-fragmentation chain transfer (RAFT)/Xanthates (MADIX) polymerization. J. Polym. Sci. 2005, 43, 5347–5393. [Google Scholar] [CrossRef]
- Schmuck, C.; Wienand, W. Self-complementary quadruple hydrogen-bonding motifs as a functional principle: From dimeric supramolecules to supramolecular polymers. Angew. Chem. Int. Ed. 2001, 40, 4363–4369. [Google Scholar] [CrossRef]
- Baruah, P.K.; Khan, S. Self-complementary quadruple hydrogen bonding motifs: From design to function. RSC Adv. 2013, 3, 21202–21217. [Google Scholar] [CrossRef]
- Hu, J.; Liu, S. Engineering responsive polymer building blocks with host-guest molecular recognition for functional applications. Acc. Chem. Res. 2014, 47, 2084–2095. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zheng, B.; Wang, F.; Huang, F. Supramolecular polymers constructed from macrocycle-based host-guest molecular recognition motifs. Acc. Chem. Res. 2014, 47, 1982–1994. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.V.K.J.; Hetzer, M.; Ritter, H.; Barner-Kowollik, C. Complex macromolecular architecture design via cyclodextrin host/guest complexes. Prog. Polym. Sci. 2014, 39, 235–249. [Google Scholar] [CrossRef]
- Armstrong, G.; Buggy, M. Hydrogen-bonded supramolecular polymers: A literature review. J. Mater. Sci. 2005, 40, 547–559. [Google Scholar] [CrossRef]
- Ten Brinke, G.; Ruokolainen, J.; Ikkala, O. Supramolecular materials based on hydrogen-bonded polymers. Adv. Polym. Sci. 2007, 207, 113–177. [Google Scholar]
- Dobrawa, R.; Wurthner, F. Metallosupramolecular approach toward functional coordination polymers. J. Polym. Sci. 2005, 43, 4981–4995. [Google Scholar] [CrossRef]
- Whittell, G.R.; Manners, I. Metallopolymers: New multifunctional materials. Adv. Mater. 2007, 19, 3439–3468. [Google Scholar] [CrossRef]
- Williams, K.A.; Boydston, A.J.; Bielawski, C.W. Main-chain organometallic polymers: Synthetic strategies, applications, and perspectives. Chem. Soc. Rev. 2007, 36, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, F.J.M.; Jonkheijm, P.; Meijer, E.W.; Schenning, A.P.H.J. About supramolecular assemblies of π-conjugated systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef] [PubMed]
- Uraneck, C.; Hsieh, H.; Buck, O. Telechelic polymers. J. Polym. Sci. 1960, 46, 535–539. [Google Scholar] [CrossRef]
- Tezuka, Y. Telechelic polymers. Prog. Polym. Sci. 1992, 17, 471–514. [Google Scholar] [CrossRef]
- Jerome, R.; Henrioulle-Granville, M.; Boutevin, B.; Robin, J.J. Telechelic polymers: Synthesis, characterization and applications. Prog. Polym. Sci. 1991, 16, 837–906. [Google Scholar] [CrossRef]
- Pollino, J.M.; Weck, M. Non-covalent side-chain polymers: Design principles, functionalization strategies, and perspectives. Chem. Soc. Rev. 2005, 34, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Dobrynin, A.V. Solutions of associative polymers. Trends Polym. Sci. 1997, 5, 181–186. [Google Scholar]
- Brassinne, J.; Mugemana, C.; Guillet, P.; Bertrand, O.; Auhl, D.; Bailly, C.; Fustin, C.-A.; Gohy, J.-F. Tuning micellar morphology and rheological behavior of metallo-supramolecular micellar gels. Soft Matter 2012, 8, 4499–4506. [Google Scholar] [CrossRef]
- Rossow, T.; Seiffert, S. Supramolecular polymer gels with potential model-network structure. Polym. Chem. 2014, 5, 3018–3029. [Google Scholar] [CrossRef]
- Rossow, T.; Habicht, A.; Seiffert, S. Relaxation and dynamics in transient polymer model networks. Macromolecules 2014, 47, 6473–6482. [Google Scholar] [CrossRef]
- Ueki, T.; Takasaki, Y.; Bundo, K.; Ueno, T.; Sakai, T.; Akagi, Y.; Yoshida, R. Autonomous viscosity oscillation via metallo-supramolecular terpyridine chemistry of branched poly(ethylene glycol) driven by the Belousov–Zhabotinsky reaction. Soft Matter 2014, 10, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Asoh, T.-A.; Yoshitake, H.; Takano, Y.; Kikuchi, A. Fabrication of self-healable hydrogels through sol-gel transition in metallo-supramolecular aqueous solution by aeration. Macromol. Chem. Phys. 2013, 214, 2534–2539. [Google Scholar] [CrossRef]
- Nair, K.P.; Breedveld, V.; Weck, M. Multiresponsive reversible polymer networks based on hydrogen bonding and metal coordination. Macromolecules 2011, 44, 3346–3357. [Google Scholar] [CrossRef]
- Pollino, J.M.; Nair, K.P.; Stubbs, L.P.; Adams, J.; Weck, M. Cross-linked and functionalized “universal polymer backbones” via simple, rapid, and orthogonal multi-site self-assembly. Tetrahedron 2004, 60, 7205–7215. [Google Scholar] [CrossRef]
- Tew, G.N.; Aamer, K.A.; Shunmugam, R. Incorporation of terpyridine into the side chain of copolymers to create multi-functional materials. Polymer 2005, 46, 8440–8447. [Google Scholar] [CrossRef]
- Bode, S.; Zedler, L.; Schacher, F.H.; Dietzek, B.; Schmitt, M.; Popp, J.; Hager, M.D.; Schubert, U.S. Self-healing polymer coatings based on crosslinked metallosupramolecular copolymers. Adv. Mater. 2013, 25, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C.; Beyer, F.L.; Price, S.C.; Rinderspacher, B.C.; Lambeth, R.H. Role of metal-ligand bond strength and phase separation on the mechanical properties of metallopolymer films. Macromolecules 2013, 46, 5416–5422. [Google Scholar] [CrossRef]
- Rossow, T.; Hackelbusch, S.; van Assenbergh, P.; Seiffert, S. A modular construction kit for supramolecular polymer gels. Polym. Chem. 2013, 4, 2515–2527. [Google Scholar] [CrossRef]
- Aamer, K.A.; de Jeu, W.H.; Tew, G.N. Diblock copolymers containing metal complexes in the side chain of one block. Macromolecules 2008, 41, 2022–2029. [Google Scholar] [CrossRef]
- Hackelbusch, S.; Rossow, T.; Becker, H.; Seiffert, S. Multiresponsive polymer hydrogels by orthogonal supramolecular chain cross-linking. Macromolecules 2014, 47, 4028–4036. [Google Scholar] [CrossRef]
- Schubert, U.; Hofmeier, H.; Newkome, G.R. Modern Terpyridine Chemistry; Wiley-VCH: Weinheim, Germany, 2006; p. 229. [Google Scholar]
- Holyer, R.H.; Hubbard, C.D.; Kettle, S.F.A.; Wilkins, R.G. The kinetics of replacement reactions of complexes of the transition metals with 2,2',2''-terpyridine. Inorg. Chem. 1966, 5, 622–625. [Google Scholar] [CrossRef]
- Hogg, R.; Wilkins, R. Exchange studies of certain chelate compounds of the transitional metals. Part VIII. 2,2',2''-terpyridine complexes. J. Chem. Soc. 1962, 341–350. [Google Scholar]
- Kotova, O.; Daly, R.; dos Santos, C.M.G.; Boese, M.; Kruger, P.E.; Boland, J.J.; Gunnlaugsson, T. Europium-directed self-assembly of a luminescent supramolecular gel from a tripodal terpyridine-based ligand. Angew. Chem. Int. Ed. 2012, 51, 7208–7212. [Google Scholar] [CrossRef]
- Han, F.S.; Higuchi, M.; Ikeda, T.; Negishi, Y.; Tsukuda, T.; Kurth, D.G. Luminescence properties of metallo-supramolecular coordination polymers assembled from pyridine ring functionalized ditopic bis-terpyridines and Ru (II) ion. J. Mater. Chem. 2008, 18, 4555–4560. [Google Scholar] [CrossRef]
- El-ghayoury, A.; Hofmeier, H.; de Ruiter, B.; Schubert, U.S. Combining covalent and noncovalent cross-linking: A novel terpolymer for two-step curing applications. Macromolecules 2003, 36, 3955–3959. [Google Scholar] [CrossRef]
- Calzia, K.J.; Tew, G.N. Methacrylate polymers containing metal binding ligands for use in supramolecular materials: Random copolymers containing terpyridines. Macromolecules 2002, 35, 6090–6093. [Google Scholar] [CrossRef]
- Shunmugam, R.; Tew, G.N. Unique emission from polymer based lanthanide alloys. J. Am. Chem. Soc. 2005, 127, 13567–13572. [Google Scholar] [CrossRef] [PubMed]
- Hofmeier, H.; Schubert, U.S. Supramolecular branching and crosslinking of terpyridine-modified copolymers: Complexation and decomplexation studies in diluted solution. Macromol. Chem. Phys. 2003, 204, 1391–1397. [Google Scholar] [CrossRef]
- Bode, S.; Bose, R.K.; Matthes, S.; Ehrhardt, M.; Seifert, A.; Schacher, F.H.; Paulus, R.M.; Stumpf, S.; Sandmann, B.; Vitz, J.; et al. Self-healing metallopolymers based on cadmium bis(terpyridine) complex containing polymer networks. Polym. Chem. 2013, 4, 4966–4973. [Google Scholar] [CrossRef]
- Aamer, K.A.; Tew, G.N. Supramolecular polymers containing terpyridine-metal complexes in the side chain. Macromolecules 2007, 40, 2737–2744. [Google Scholar] [CrossRef]
- Hackelbusch, S.; Rossow, T.; van Assenbergh, P.; Seiffert, S. Chain dynamics in supramolecular polymer networks. Macromolecules 2013, 46, 6273–6286. [Google Scholar] [CrossRef]
- Heller, M.; Schubert, U.S. Polystyrene with pendant mixed functional ruthenium (II)-terpyridine complexes. Macromol. Rapid Commun. 2002, 23, 411–415. [Google Scholar] [CrossRef]
- Shunmugam, R.; Tew, G.N. White-light emission from mixing blue and red-emitting metal complexes. Polym. Adv. Technol. 2008, 19, 596–601. [Google Scholar] [CrossRef]
- Ott, C.; Ulbricht, C.; Hoogenboom, R.; Schubert, U.S. Metallo-supramolecular materials based on amine-grafting onto polypentafluorostyrene. Macromol. Rapid Commun. 2012, 33, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 2012, 41, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Plamper, F.A.; Ruppel, M.; Schmalz, A.; Borisov, O.; Ballauff, M.; Muller, A.H.E. Tuning the thermoresponsive properties of weak polyelectrolytes: Aqueous solutions of star-shaped and linear poly(N,N-dimethylaminoethyl methacrylate). Macromolecules 2007, 40, 8361–8366. [Google Scholar] [CrossRef]
- Li, F.-M.; Chen, S.-J.; Du, F.-S.; Wu, Z.-Q.; Li, Z.-C. Stimuli-responsive behavior of N,N-dimethylaminoethyl methacrylate polymers and their hydrogels. ACS Symp. Ser. 1999, 726, 266–276. [Google Scholar]
- Bütün, V.; Armes, S.; Billingham, N. Synthesis and aqueous solution properties of near-monodisperse tertiary amine methacrylate homopolymers and diblock copolymers. Polymer 2001, 42, 5993–6008. [Google Scholar] [CrossRef]
- Schubert, U.S.; Hofmeier, H. Metallo-supramolecular graft copolymers: A novel approach toward polymer-analogous reactions. Macromol. Rapid Commun. 2002, 23, 561–566. [Google Scholar] [CrossRef]
- Jochum, F.D.; Brassinne, J.; Fustin, C.-A.; Gohy, J.-F. Metallo-supramolecular hydrogels based on copolymers bearing terpyridine side-chain ligands. Soft Matter 2013, 9, 2314–2320. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Brassinne, J.; Gohy, J.-F.; Fustin, C.-A. Controlling the cross-linking density of supramolecular hydrogels formed by heterotelechelic associating copolymers. Macromolecules 2014, 47, 4514–4524. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon: Oxford, UK, 1986. [Google Scholar]
- Rubinstein, M.; Semenov, A.N. Dynamics of entangled solutions of associating polymers. Macromolecules 2001, 34, 1058–1068. [Google Scholar] [CrossRef]
- Jongschaap, R.; Wientjes, R.; Duits, M.; Mellema, J. A generalized transient network model for associative polymer networks. Macromolecules 2001, 34, 1031–1038. [Google Scholar] [CrossRef]
- Rubinstein, M.; Dobrynin, A.V. Associations leading to formation of reversible networks and gels. Curr. Opin. Colloid Interface Sci. 1999, 4, 83–87. [Google Scholar] [CrossRef]
- Tanaka, F.; Edwards, S.F. Viscoelastic properties of physically crosslinked networks: Part 1. Non-linear stationary viscoelasticity. J. Non-Newton. Fluid Mech. 1992, 43, 247–271. [Google Scholar] [CrossRef]
- Tirtaatmadja, V.; Tam, K.; Jenkins, R. Superposition of oscillations on steady shear flow as a technique for investigating the structure of associative polymers. Macromolecules 1997, 30, 1426–1433. [Google Scholar] [CrossRef]
- Kim, S.H.; Sim, H.G.; Ahn, K.H.; Lee, S.J. Large amplitude oscillatory shear behavior of the network model for associating polymeric systems. Korea-Aust. Rheol. J. 2002, 14, 49–55. [Google Scholar]
- Sim, H.G.; Ahn, K.H.; Lee, S.J. Large amplitude oscillatory shear behavior of complex fluids investigated by a network model: A guideline for classification. J. Non-Newton. Fluid Mech. 2003, 112, 237–250. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brassinne, J.; Jochum, F.D.; Fustin, C.-A.; Gohy, J.-F. Revealing the Supramolecular Nature of Side-Chain Terpyridine-Functionalized Polymer Networks. Int. J. Mol. Sci. 2015, 16, 990-1007. https://doi.org/10.3390/ijms16010990
Brassinne J, Jochum FD, Fustin C-A, Gohy J-F. Revealing the Supramolecular Nature of Side-Chain Terpyridine-Functionalized Polymer Networks. International Journal of Molecular Sciences. 2015; 16(1):990-1007. https://doi.org/10.3390/ijms16010990
Chicago/Turabian StyleBrassinne, Jérémy, Florian D. Jochum, Charles-André Fustin, and Jean-François Gohy. 2015. "Revealing the Supramolecular Nature of Side-Chain Terpyridine-Functionalized Polymer Networks" International Journal of Molecular Sciences 16, no. 1: 990-1007. https://doi.org/10.3390/ijms16010990
APA StyleBrassinne, J., Jochum, F. D., Fustin, C.-A., & Gohy, J.-F. (2015). Revealing the Supramolecular Nature of Side-Chain Terpyridine-Functionalized Polymer Networks. International Journal of Molecular Sciences, 16(1), 990-1007. https://doi.org/10.3390/ijms16010990