
Progress and Challenges in Developing Aptamer-Functionalized Targeted Drug Delivery Systems

 ,
, 
Abstract
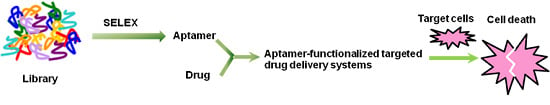
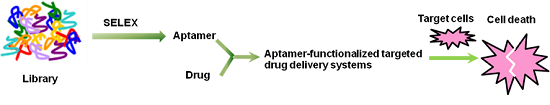
:
1. Introduction
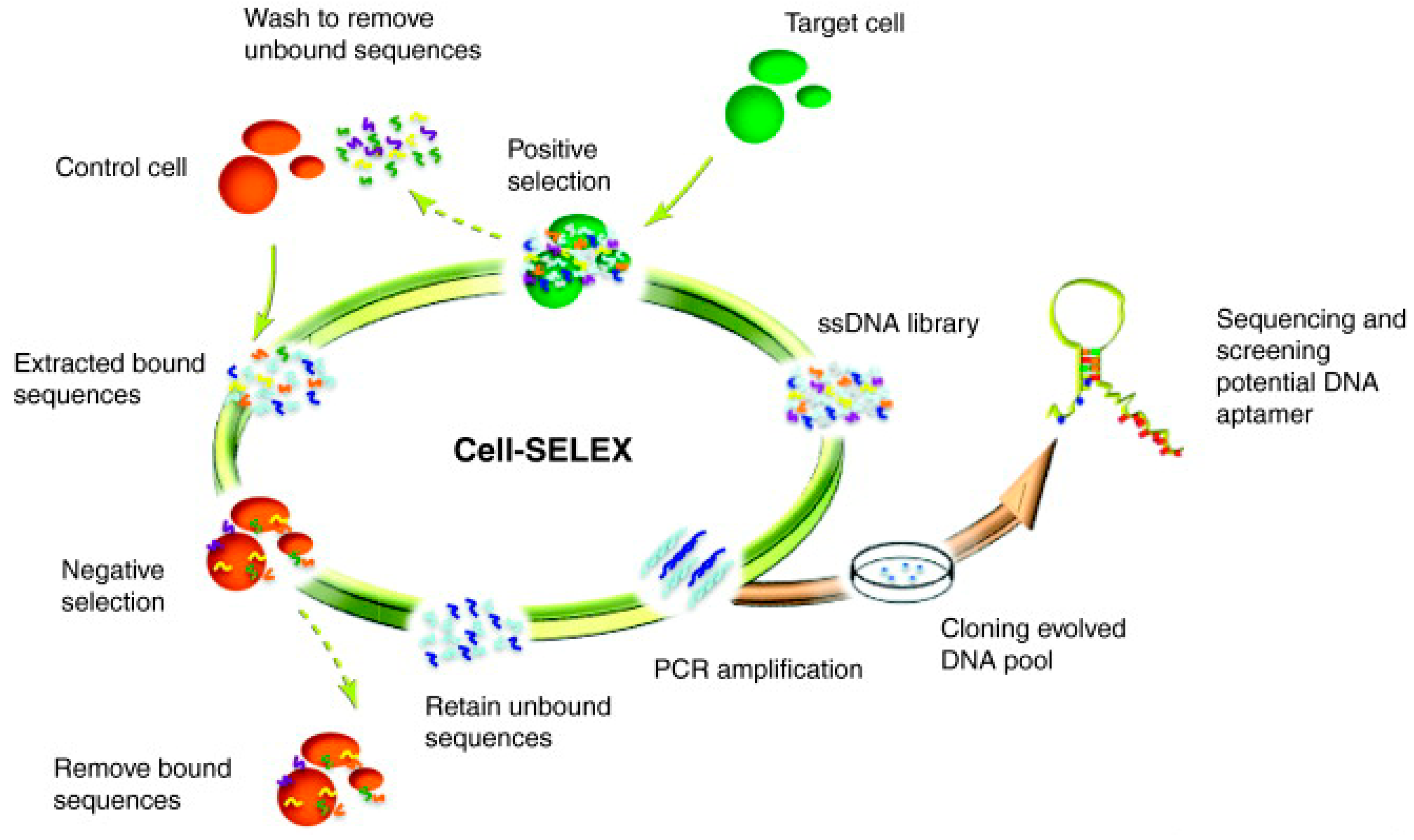
2. Aptamer SELEX

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamer | Molecular Target | Associated Disease | Aptamer Structure | Ref. |
|---|---|---|---|---|
| Macugen | Vascular endothelial growth factor (VEGF) | Age-related macular degeneration | 5′-CGGAAUCAGUGAAUG CUUAUACAUCCG-3′ | [37] |
| AS1411 | Nucleoin | Cancer | 5′-d(GGTGGTGGTGGT TGTGGTGGTGGTGG)-3′ | [38] |
| Sgc8 | Protein tyrosine kinase 7 (PTK-7) | Cancer | 5′-ATCTAACTGCTGC GCCGCCGGGAAAATA CTGTACGGTTAGA-3′ | [39] |
| TD05 | Immunoglobulin μ heavy chains (IGHM) | Lymphoma | 5′-AACACCGGGAGGAT AGTTCGGTGGCTGTTCA GGGTCTCCTCCCGGTG-3′ | [40] |
| ARC1779 | A1 Domain of von Willebrand factor (vWF) | Thrombotic microangiopathies and carotid artery disease | 5′-GCGUGCAGUGCCU UCGGCCGTGCGGT GCCUCCGUCACGC-3′ | [41] |
| TBA | α-Thrombin | Thrombosis | 5′-GGTTGGTGTGGTTGG-3′ | [42] |
2.1. Cell-SELEX

2.2. Complex-Target SELEX
3. Aptamer-Small Molecule Conjugated Systems
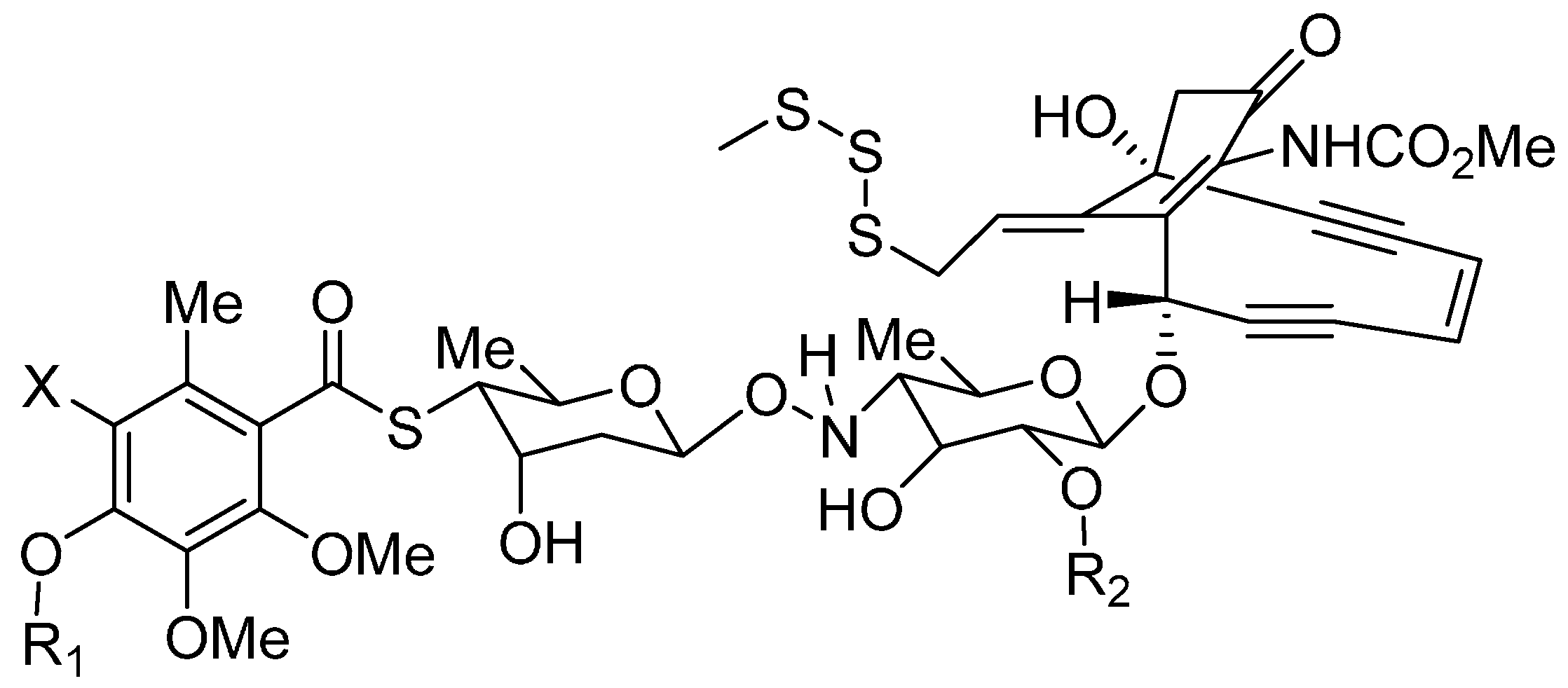
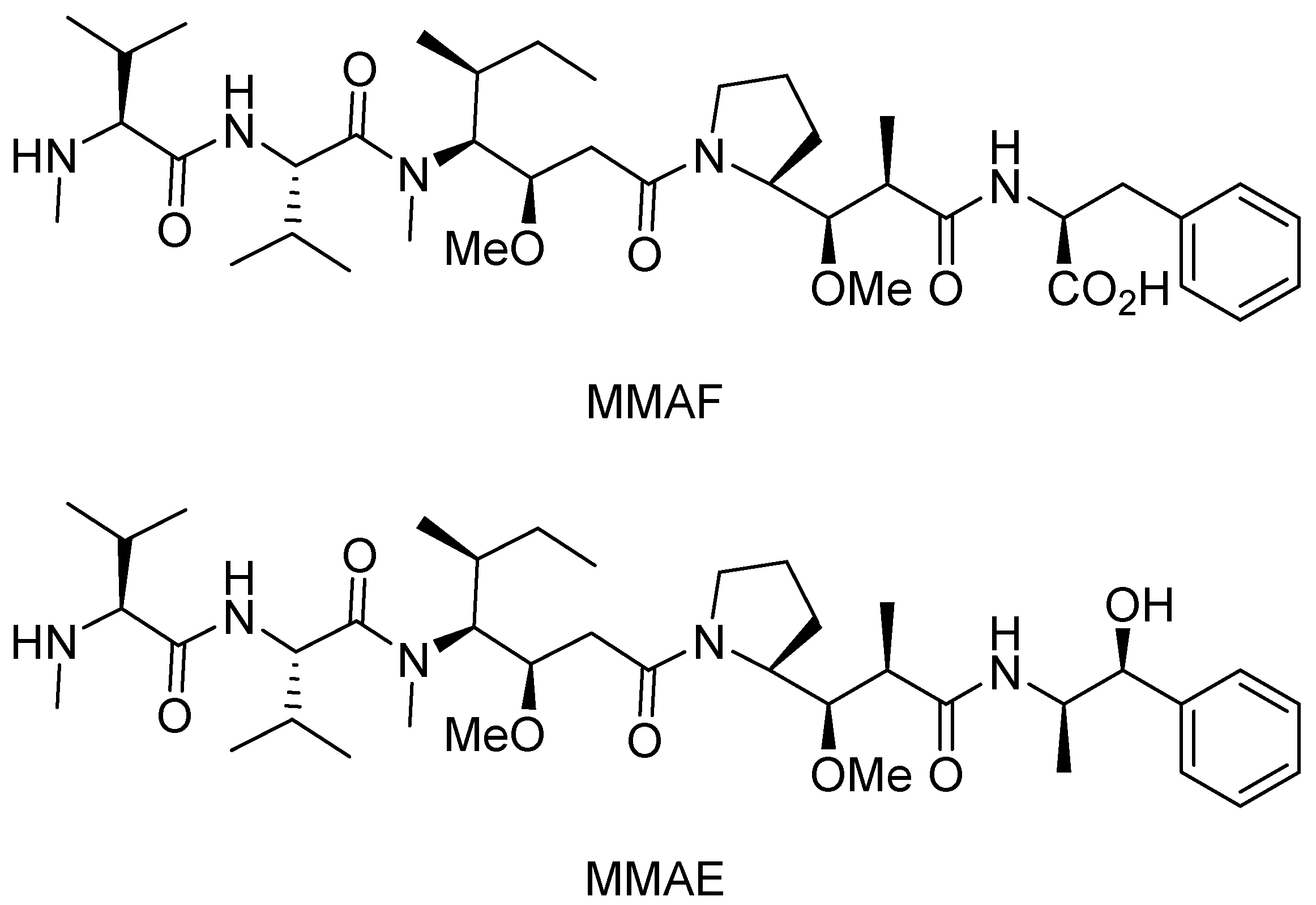
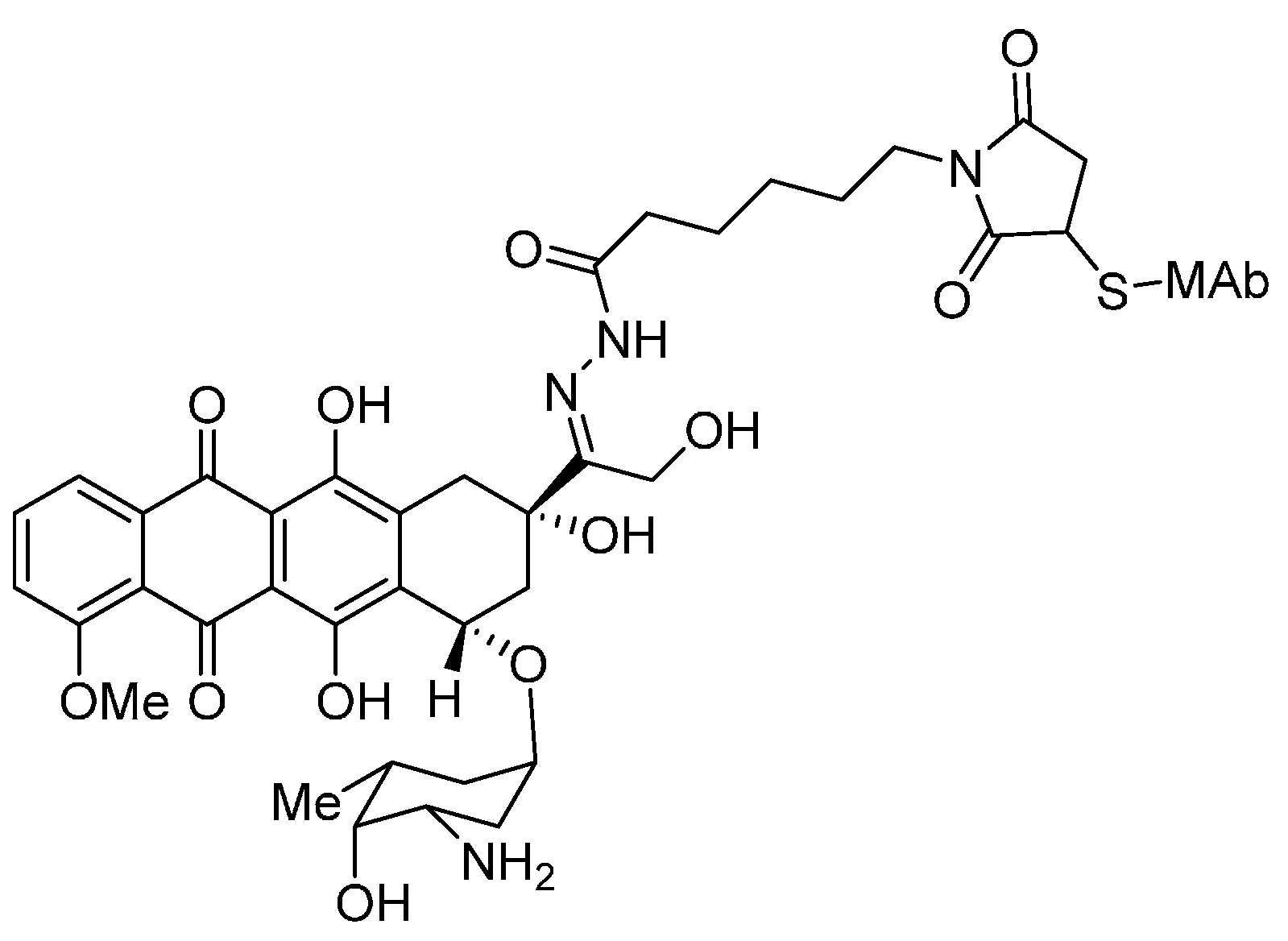
3.1. Cytotoxic Drugs



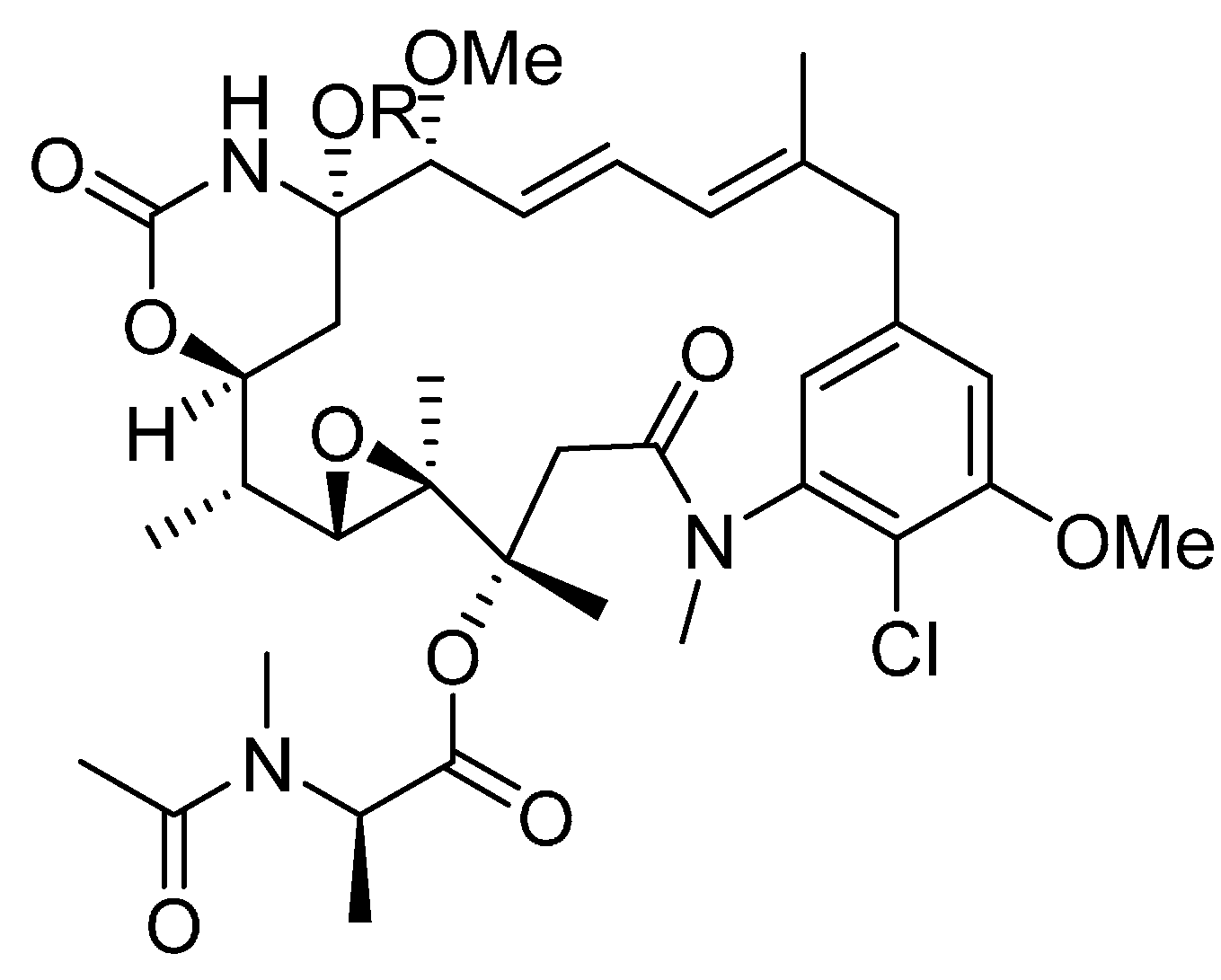
3.2. Linkers
3.2.1. Chemically Labile Linkers
Acid-Cleavable Linkers

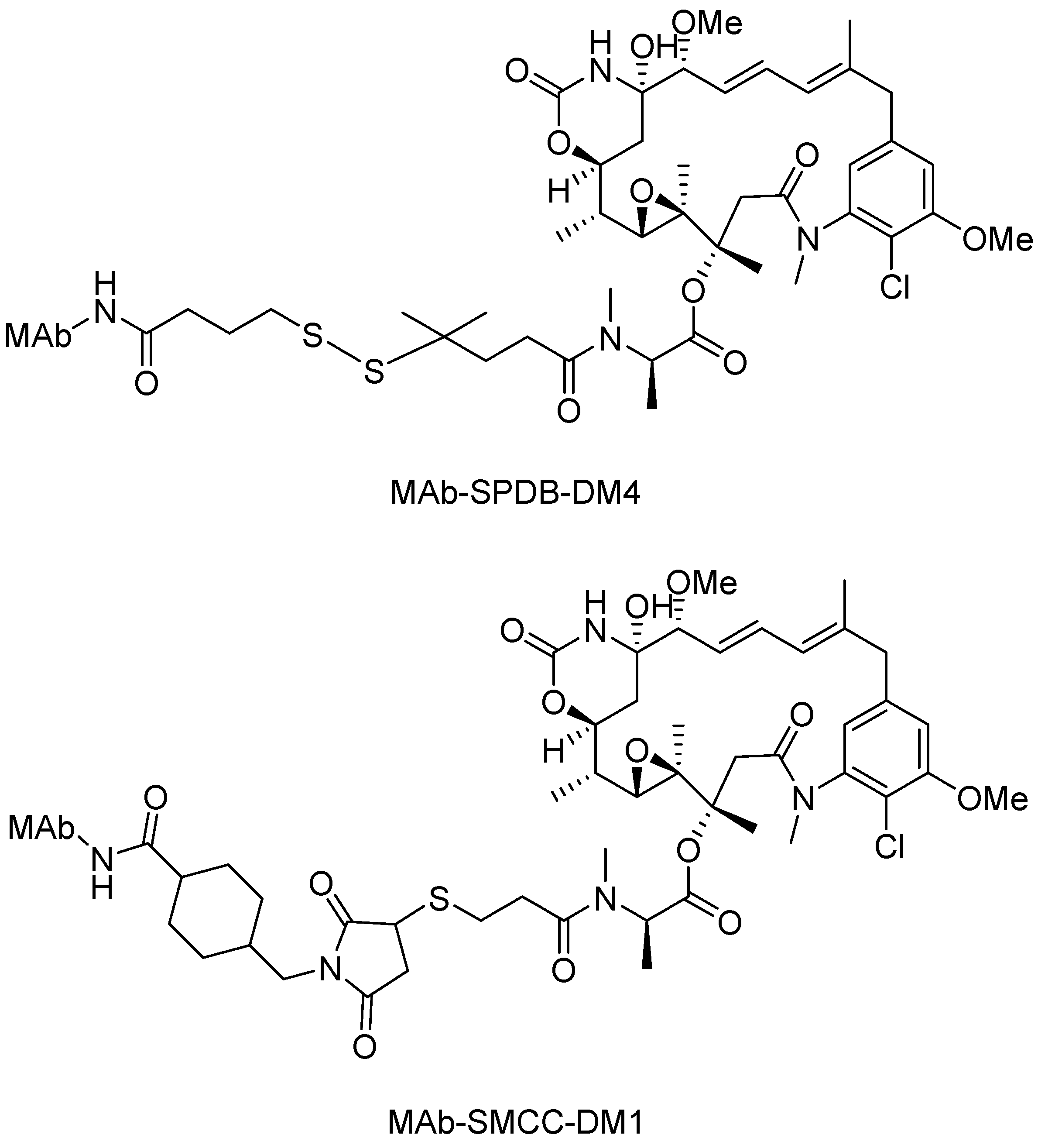
Disulfide Linkers

3.2.2. Enzyme-Labile Linkers
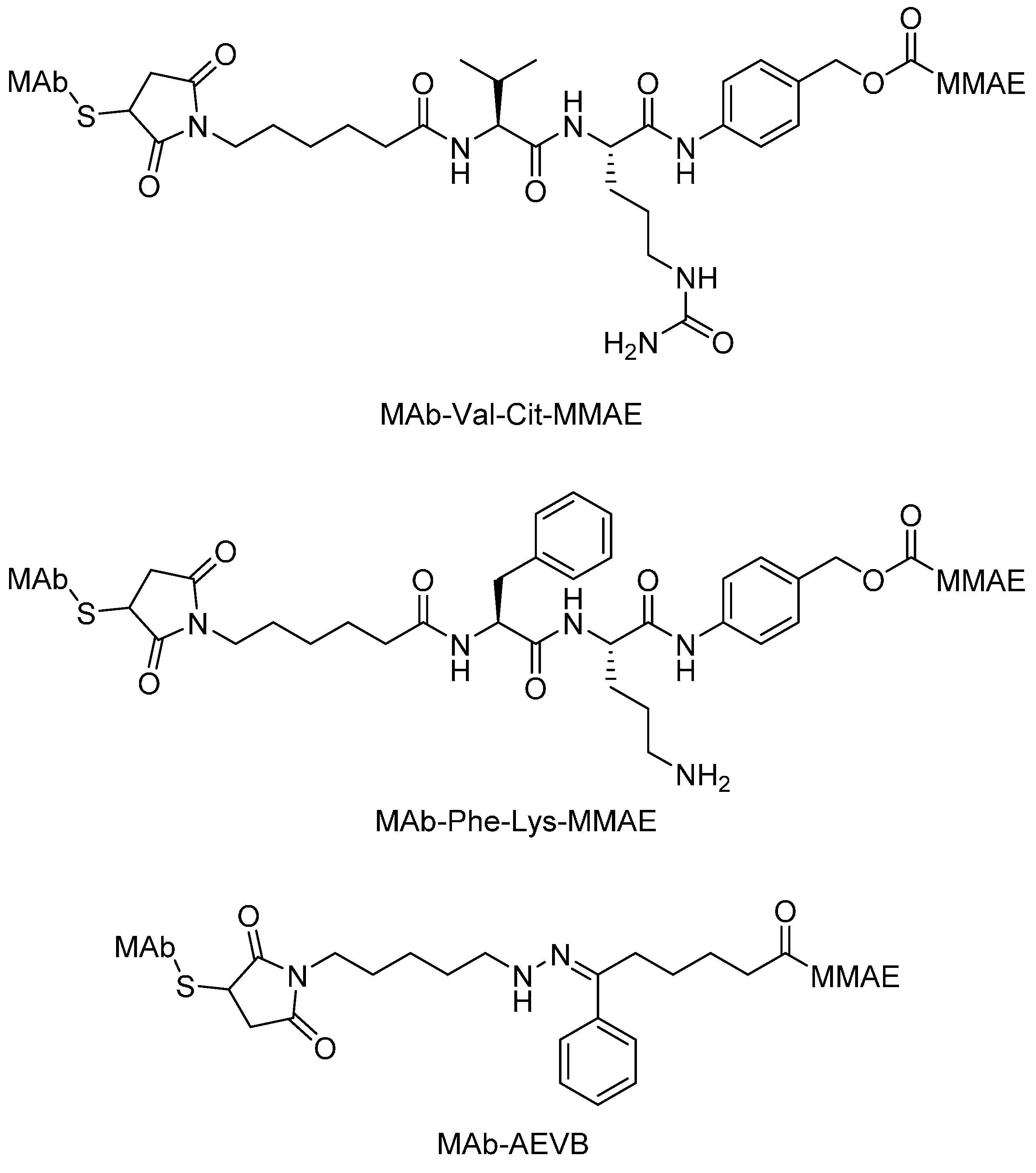
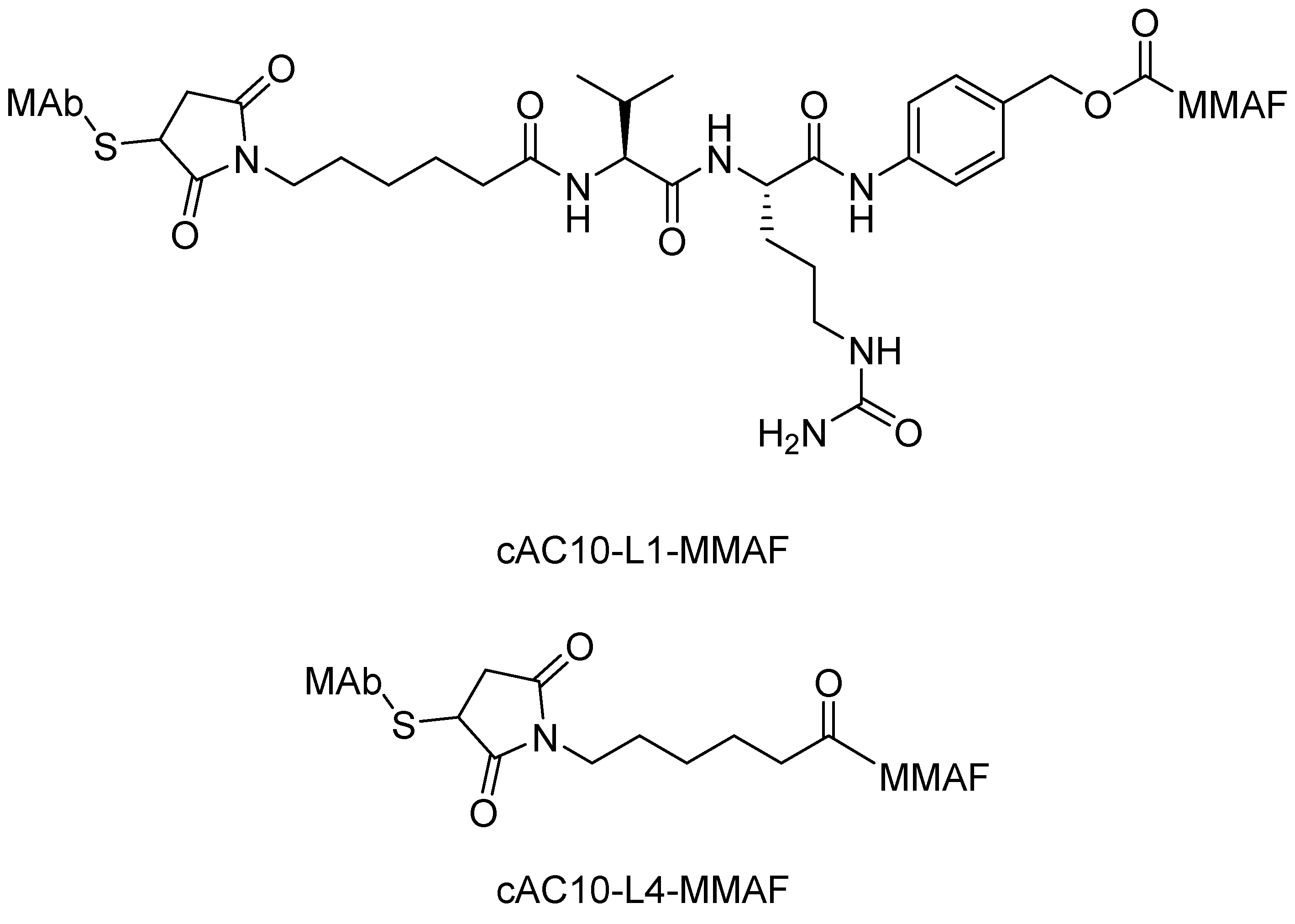
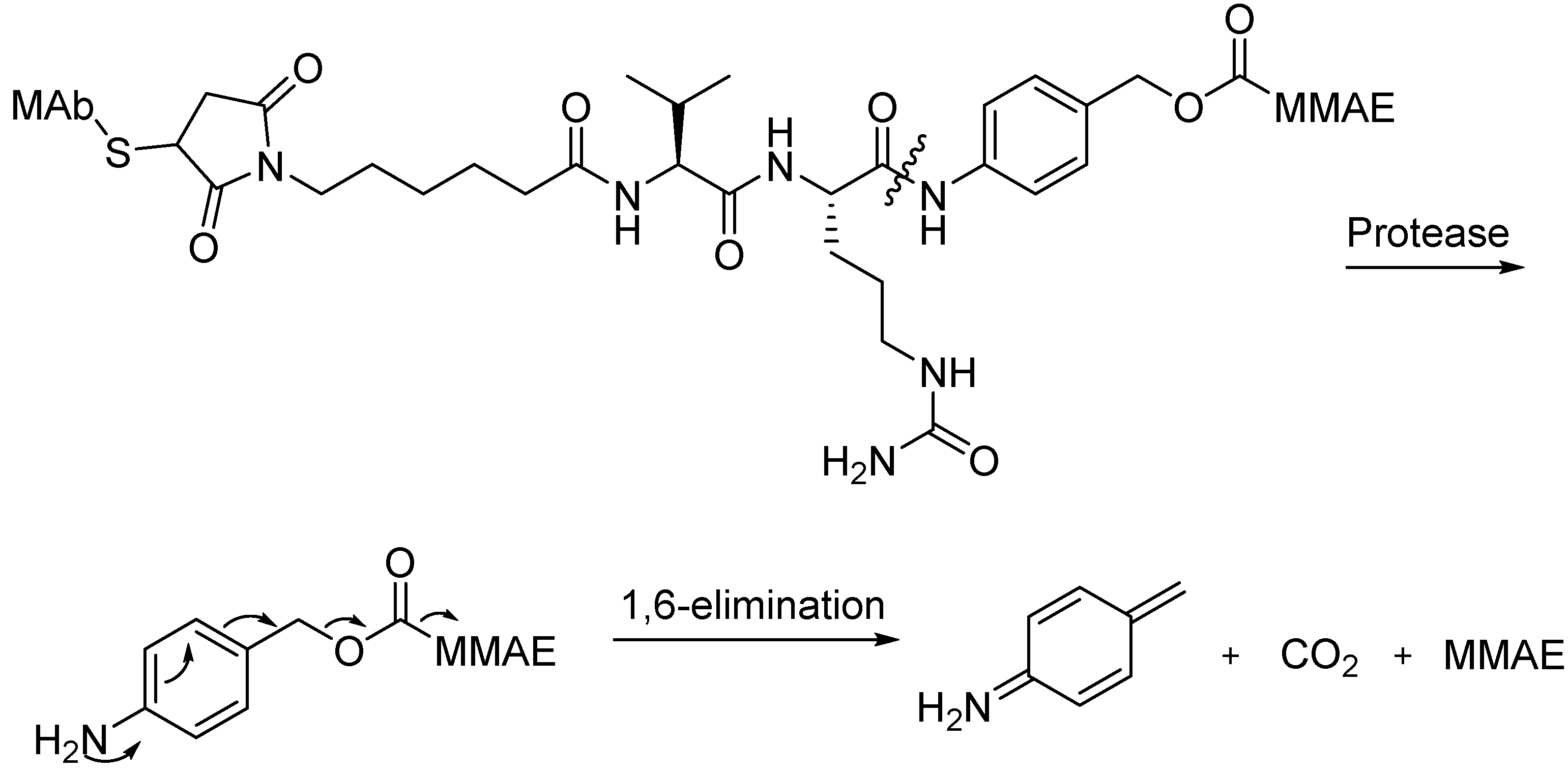
Peptide Linkers


β-Glucuronide Linkers
3.2.3. Non-Cleavable Linkers

4. Aptamer-Nanomaterial Conjugated Systems
4.1. Inorganic Nanomaterials
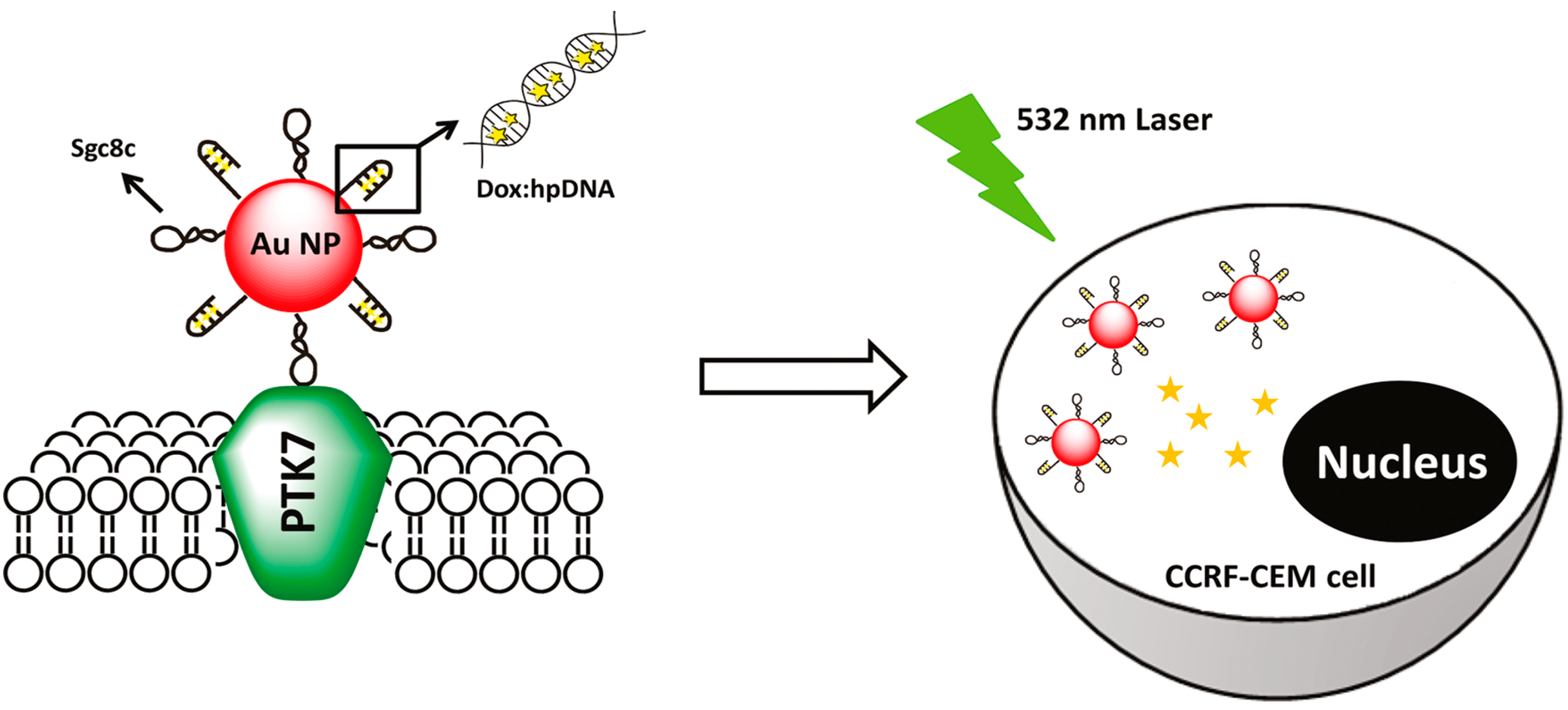
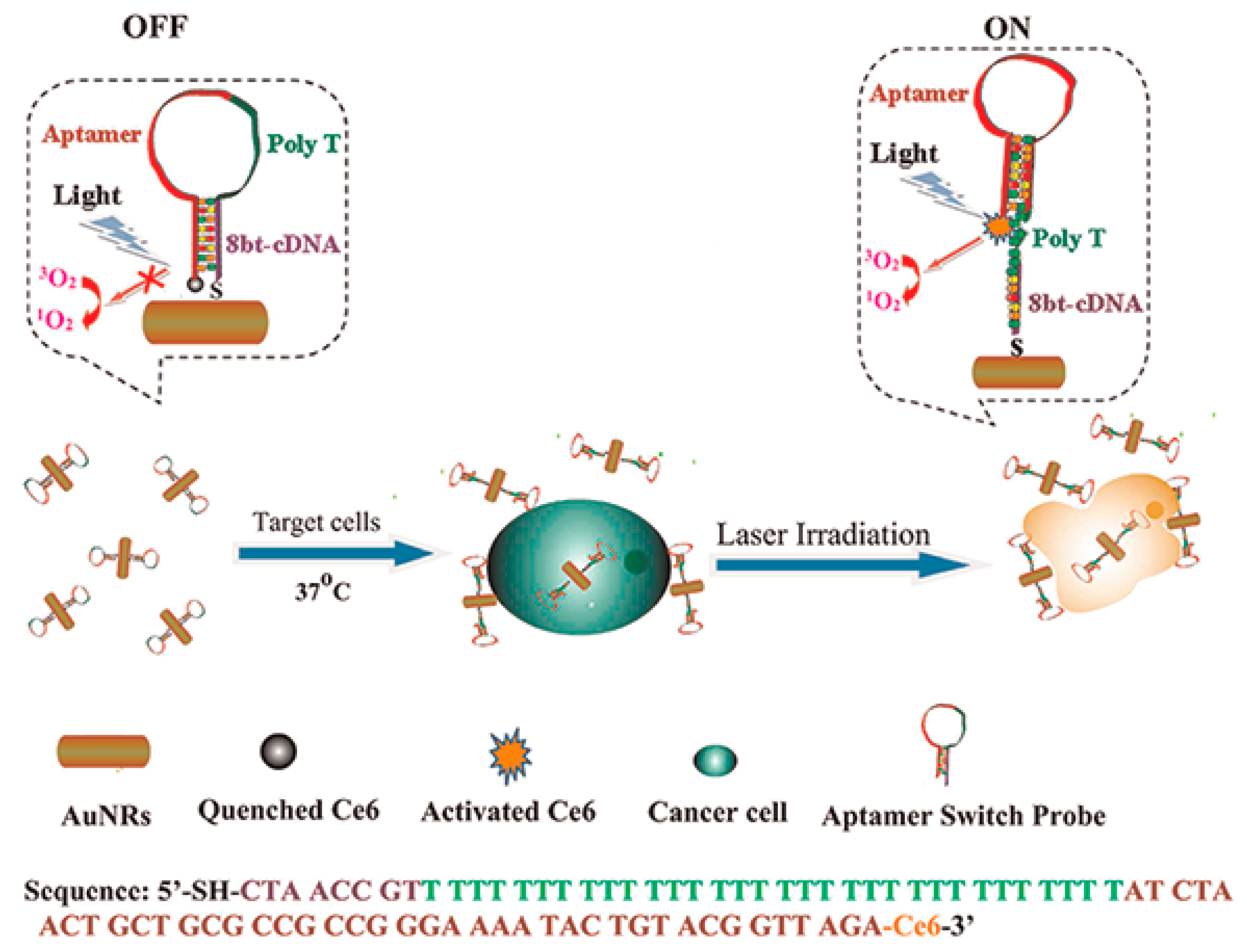
4.1.1. Gold Nanoparticles


4.1.2. Nano-Scale Iron Oxides
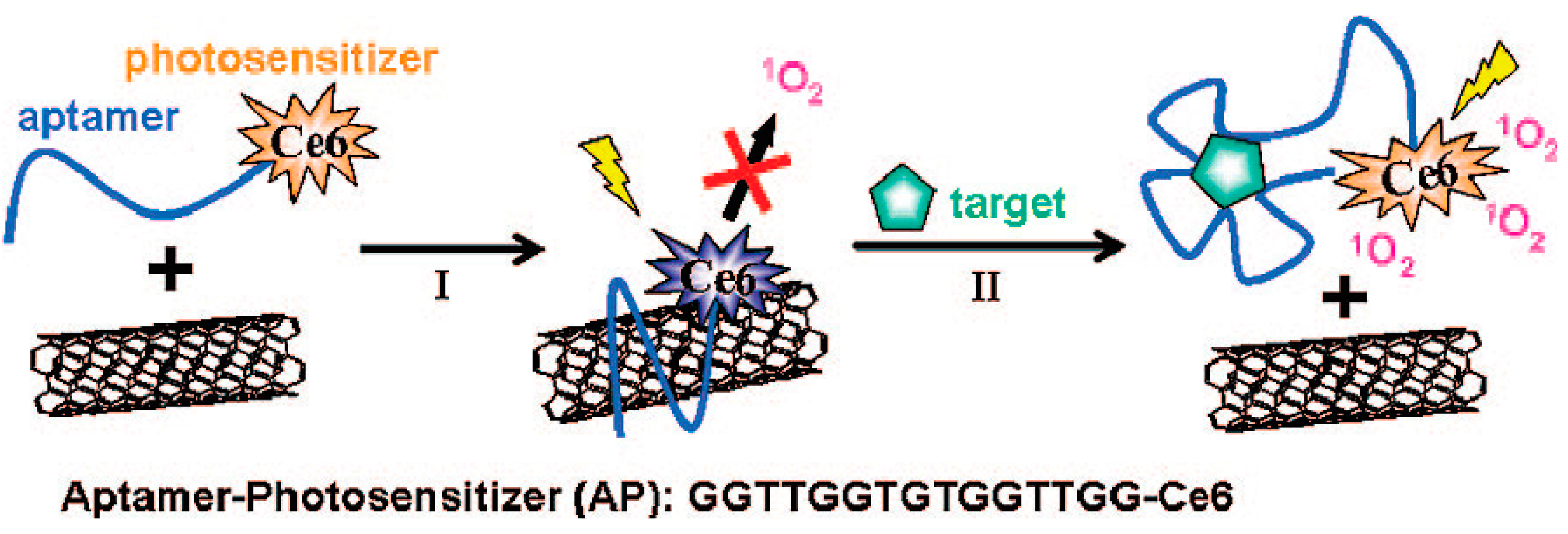
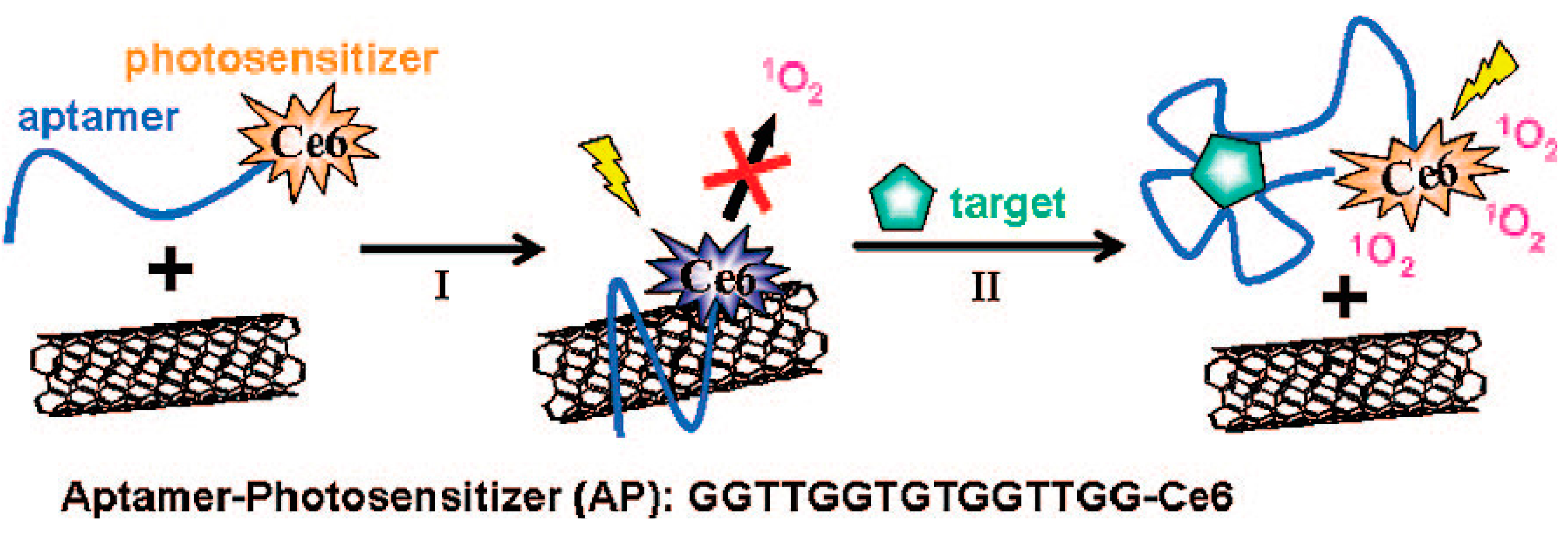
4.1.3. Carbon Nanomaterials
4.1.4. Mesoporous Silica Nanoparticles
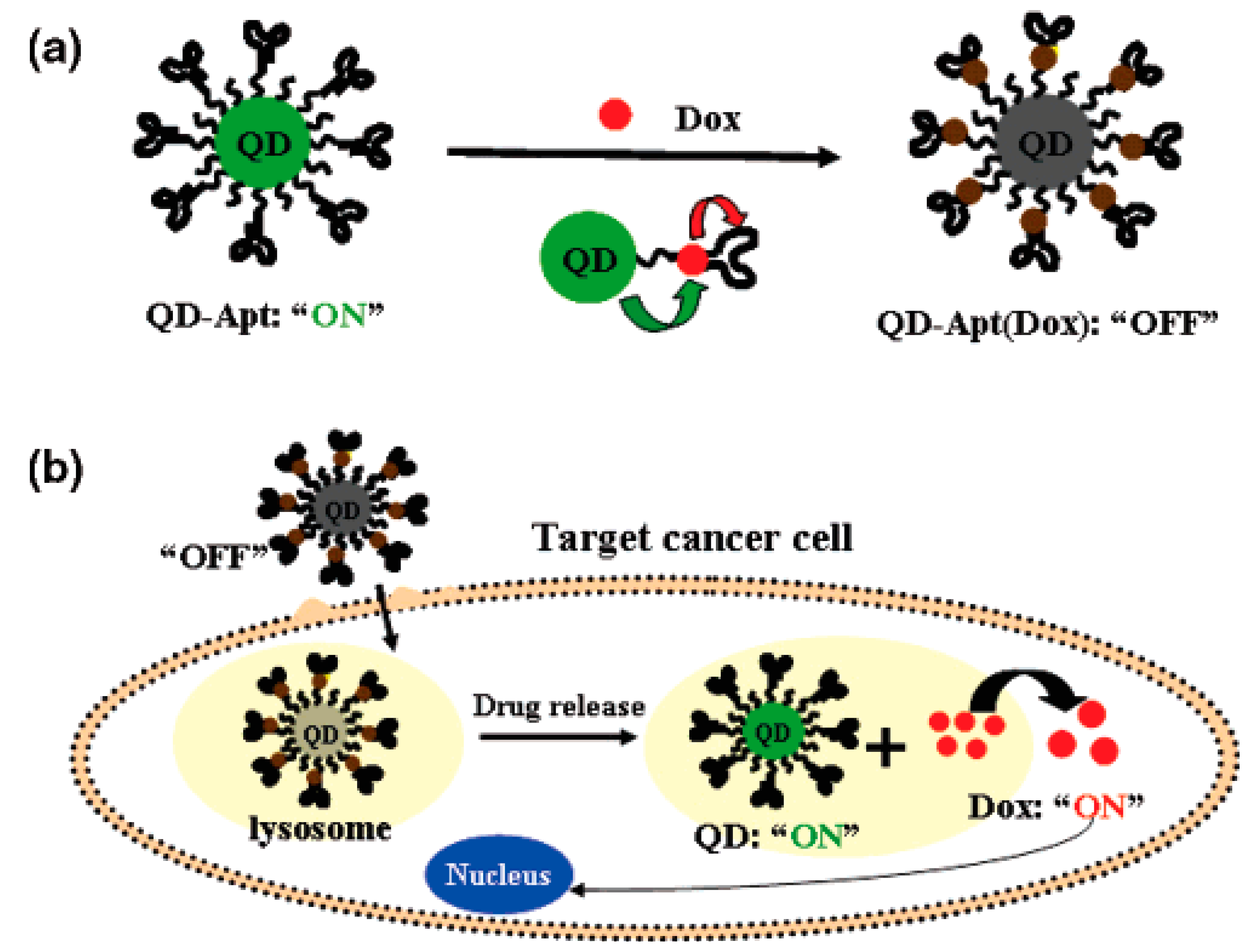
4.1.5. Quantum Dots

4.2. Organic Nanomaterials
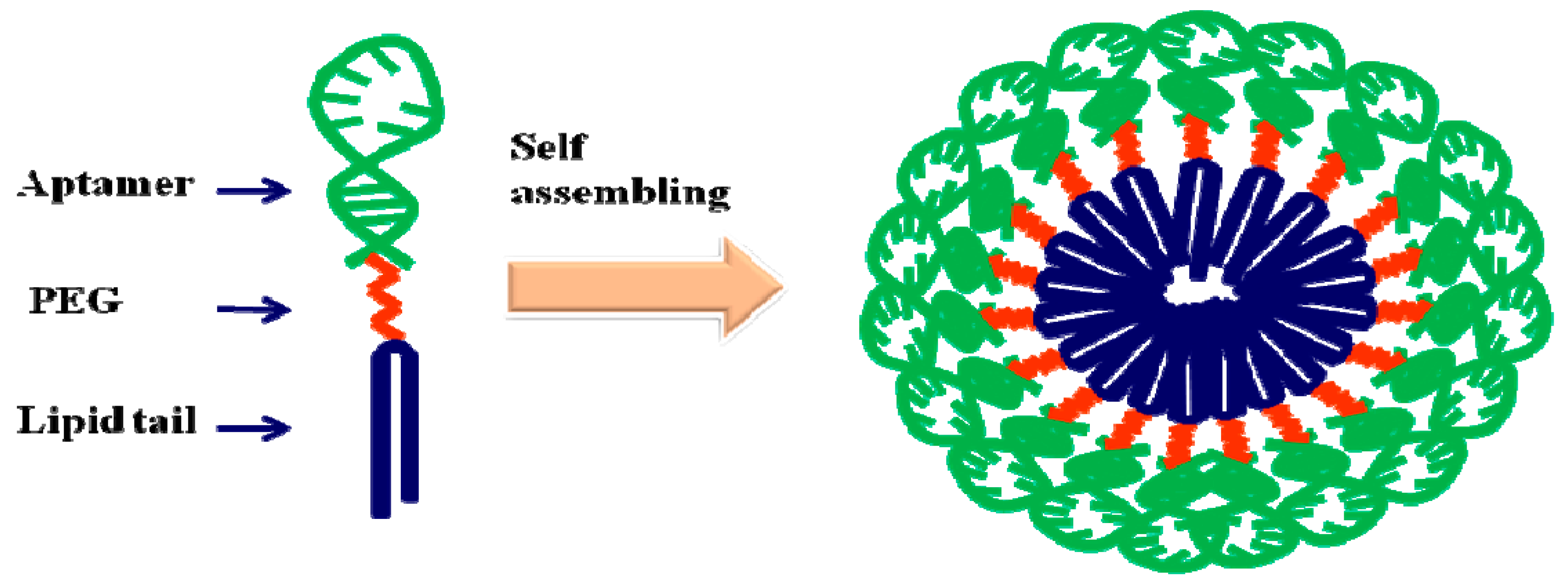
4.2.1. Liposomes
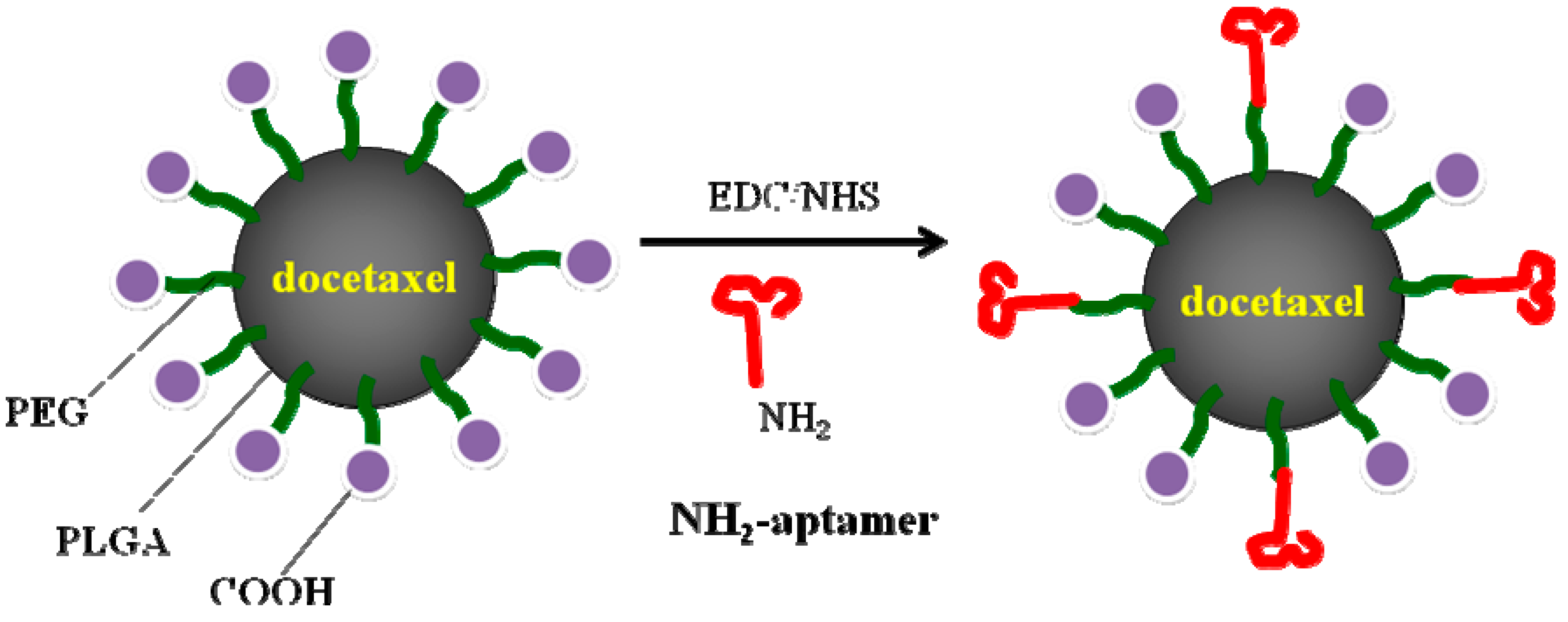
4.2.2. Poly(lactide-co-glycolic acid) Nanoparticles

4.2.3. Polymeric Micelles

4.2.4. Dendrimers
4.2.5. Serum Albumin Nanoparticles
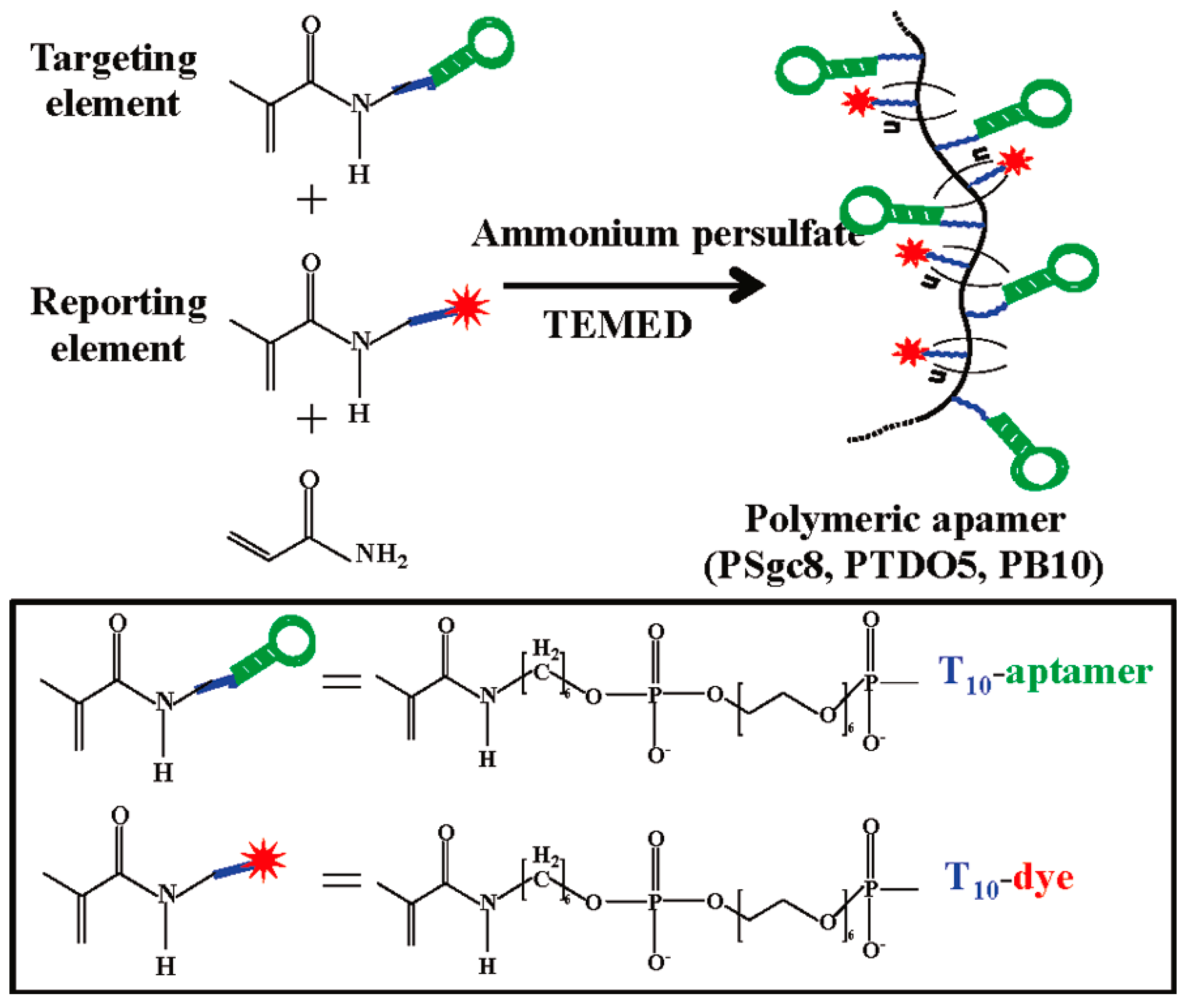
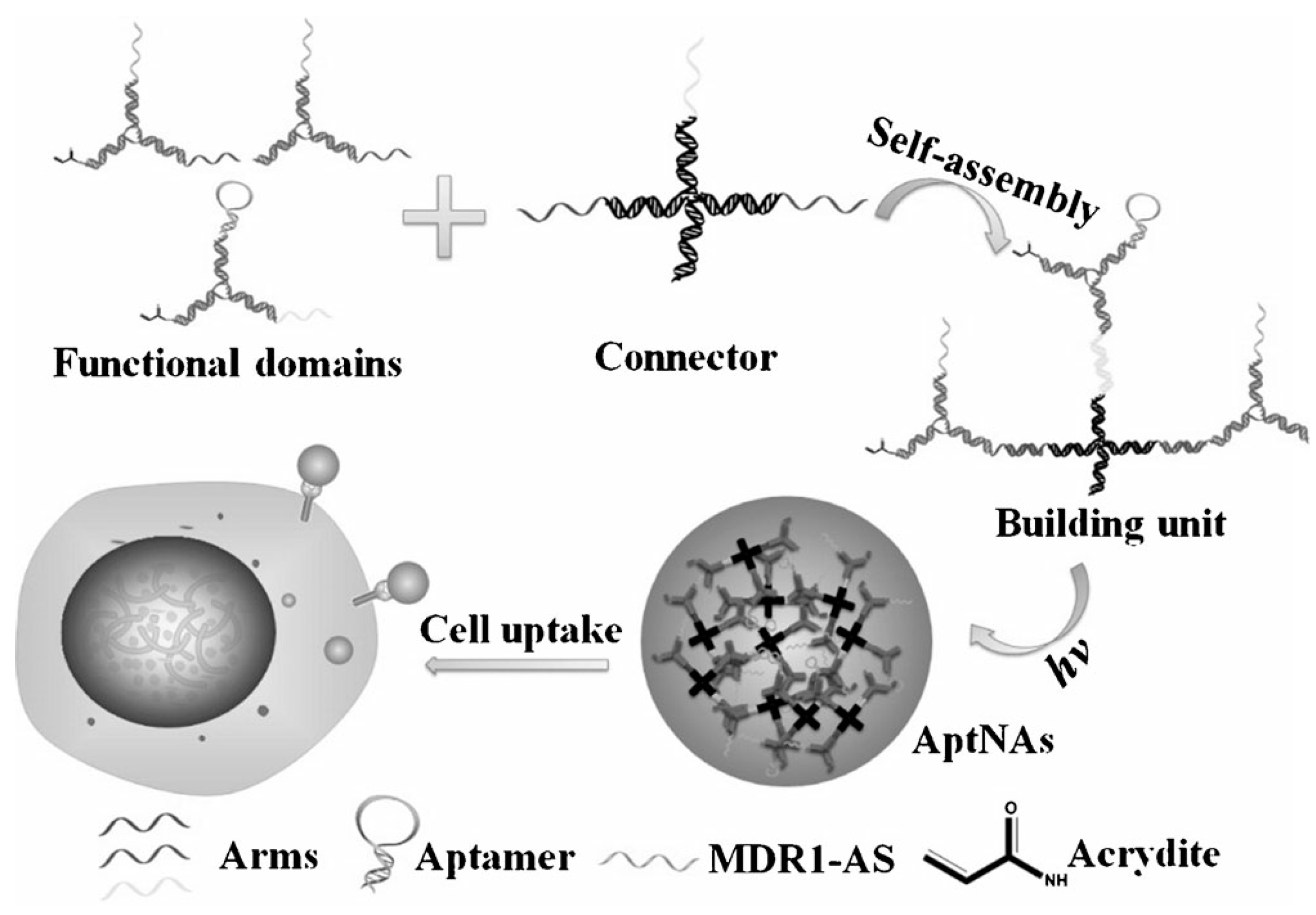
4.2.6. DNA Nanomaterials


5. Conclusions
| Aptamer-Functionalized Targeted Drug Delivery Systems | Advantage | Disadvantage |
|---|---|---|
| Aptamer-small molecule conjugated systems | Good chemical stability, isotropic properties | Low drug loading, complex and costly procedures |
| Aptamer-nanomaterial conjugated systems | High loading capacity | Unpredictable risks, complex and costly procedures |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muro, S. Challenges in design and characterization of ligand-targeted drug delivery systems. J. Control. Release 2012, 164, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef] [PubMed]
- Jaracz, S.; Chen, J.; Kuznetsova, L.V.; Ojima, I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.L.; Zhang, B.; Sun, X.R.; Zhou, Z.X.; Ma, X.P.; Sun, Q.H.; Tang, J.B.; Shen, Y.Q.; Kirk, E.V.; Murdoch, W.J.; et al. Acid active cell-penetrating peptides for in vivo tumor-targeted drug delivery. J. Am. Chem. Soc. 2013, 135, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Jarosch, F.; Buchner, K.; Klussmann, S. In vitro selection using a dual RNA library that allows primerless selection. Nucleic Acids Res. 2006, 34, e86. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.Y.; Byun, J. Nucleic acid aptamers: New methods for selection, stabilization, and application in biomedical science. Biomol. Ther. 2013, 21, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, X.B.; Lv, Y.; Gong, L.; Wang, R.; Zhu, X.; Yang, R.; Tan, W. Functional DNA-containing nanomaterials: Cellular applications in biosensing, imaging, and targeted therapy. Acc. Chem. Res. 2014, 47, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Q.; Qiu, L. Smart ligand: Aptamer-mediated targeted delivery of chemotherapeutic drugs and siRNA for cancer therapy. J. Control. Release 2013, 171, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Mosing, R.K.; Bowser, M.T. Isolating aptamers using capillary electrophoresis-SELEX (CE-SELEX). Methods Mol. Biol. 2009, 535, 33–43. [Google Scholar] [PubMed]
- Glökler, J.; Schütze, T.; Konthur, Z. Automation in the high-throughput selection of random combinatorial libraries—different approaches for select applications. Molecules 2010, 15, 2478–2490. [Google Scholar] [CrossRef] [PubMed]
- Blind, M.; Blank, M. Aptamer selection technology and recent advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef]
- Elle, I.C.; Karlsen, K.K.; Terp, M.G.; Larsen, N.; Nielsen, R.; Derbyshire, N.; Mandrup, S.; Ditzel, H.J.; Wengel, J. Selection of LNA-containing DNA aptamers against recombinant human CD73. Mol. Biosyst. 2015, 11, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murgha, Y.E.; Rouillard, J.M.; Gulari, E. Methods for the preparation of large quantities of complex single-stranded oligonucleotide libraries. PLoS ONE 2014, 9, e94752. [Google Scholar] [PubMed]
- Yang, J.; Bowser, M.T. Capillary electrophoresis-SELEX selection of catalytic DNA aptamers for a small-molecule porphyrin target. Anal. Chem. 2013, 85, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, D.; Schluesener, H.J.; Zhang, Z. Advances in SELEX and application of aptamers in the central nervous system. Biomol. Eng. 2007, 24, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Tok, J.; Lai, J.; Leung, T.; Li, S.F. Selection of aptamers for signal transduction proteins by capillary electrophoresis. Electrophoresis 2010, 31, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Bruno, J.G.; Phillips, T.; Montez, T.; Garcia, A.; Sivils, J.C.; Mayo, M.W.; Greis, A. Development of a fluorescent enzyme-linked DNA aptamer-magnetic bead sandwich assay and portable fluorometer for sensitive and rapid listeria detection. J. Fluoresc. 2015, 25, 173–183. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. FluMag-SELEX as an advantageous method for DNA aptamer selection. Anal. Bioanal. Chem. 2005, 383, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Li, D.; Zhang, G.; Li, H.; Shao, N.; Liang, Z.; Zhang, L.; Lu, A.; Zhang, G. Comparison of the methods for generating single-stranded DNA in SELEX. Analyst 2015, 140, 3439–3444. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Cell-type-specific, aptamer-functionalized agents for targeted disease. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.X.; Zhang, K.H.; Zou, X.S.; Chen, Y.Q.; Li, J.G. Screening and identification of the nucleic acid aptamers in nasopharyngeal carcinoma. Genet. Mol. Res. 2013, 12, 6850–6857. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, H.; Lukasser, M.; Mayer, G.; Hüttenhofer, A. Cell-SELEX: In vitro selection of synthetic small specific ligands. Methods Mol. Biol. 2015, 1296, 213–224. [Google Scholar] [PubMed]
- Zhu, H.; Li, J.; Zhang, X.B.; Ye, M.; Tan, W. Nucleic acid aptamer-mediated drug delivery for targeted cancer therapy. ChemMedChem 2015, 10, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, E.Y.; Kim, S.Y.; Byun, S.K.; Lee, D.; Oh, K.J.; Kim, W.K.; Han, B.S.; Chi, S.W.; Lee, S.C.; et al. Identification of DNA aptamers toward epithelial cell adhesion molecule via cell-SELEX. Mol. Cells 2014, 37, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Guo, B.; Wu, H.; Shao, N.; Li, D.; Liu, J.; Dang, L.; Wang, C.; Li, H.; Li, S.; et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. Nat. Med. 2015, 21, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Conidi, A.; van den Berghe, V.; Huylebroeck, D. Aptamers and their potential to selectively target aspects of EGF, Wnt/β-catenin and TGFβ-smad family signaling. Int. J. Mol. Sci. 2013, 14, 6690–6719. [Google Scholar] [CrossRef] [PubMed]
- Hoinka, J.; Zotenko, E.; Friedman, A.; Sauna, Z.E.; Przytycka, T.M. Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics 2012, 28, i215–i223. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.C.; Ellington, A.D. Automated selection of anti-protein aptamers. Bioorg. Med. Chem. 2001, 9, 2525–2531. [Google Scholar] [CrossRef]
- Eulberg, D.; Buchner, K.; Maasch, C.; Klussmann, S. Development of an automated in vitro selection protocol to obtain RNA-based aptamers: Identification of a biostable substance P antagonist. Nucleic Acids Res. 2005, 33, e45. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Tan, W. Aptamers generated from cell-SELEX for molecular medicine: A chemical biology approach. Acc. Chem. Res. 2010, 43, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Chen, S.; Nilsen-Hamilton, M.; Levine, H.A. A mathematical analysis of multiple-target SELEX. Bull. Math. Biol. 2010, 72, 1623–1665. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.N.; Jensen, K.B.; Julin, C.M.; Weil, M.; Larry, G. High affinity ligands from in vitro selection: Complex targets. Proc. Natl. Acad. Sci. USA 1998, 95, 2902–2907. [Google Scholar] [CrossRef] [PubMed]
- Shamah, S.M.; Healy, J.M.; Cload, S.T. Complex target SELEX. Acc. Chem. Res. 2008, 41, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T.; Feinsod, M.; Guyer, D.R. Pegaptanib for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and Development of the G-rich Oligonucleotide AS1411 as a Novel Treatment for Cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, D.; Cao, Z.; Meng, L.; Mallikaratchy, P.; Sefah, K.; Wang, H.; Li, Y.; Tan, W. Cell-Specific Aptamer Probes for Membrane Protein Elucidation in Cancer Cells. J. Prot. Res. 2008, 7, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Mallikaratchy, P.; Tang, Z.; Kwame, S.; Meng, L.; Shangguan, D.; Tan, W. Aptamer Directly Evolved from Live Cells Recognizes Membrane Bound Immunoglobin Heavy Mu Chain in Burkitt’s Lymphoma Cells. Mol. Cell. Proteom. 2007, 6, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Tope, S.; Maske, S.; Nagulwar, V.; Sufi, J.; Welankiwar, A. Aptamers as therapeutics. Indo Am. J. Pharm. Res. 2013, 3, 2718–2743. [Google Scholar]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Wang, H.; Chen, Y.; Zhang, X.; Zhu, H.; Yang, C.; Yang, R.; Liu, C. Molecular aptamers for drug delivery. Trends Biotechnol. 2011, 29, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Famulok, M.; Hartig, J.S.; Mayer, G. Functional Aptamers and Aptazymes in Biotechnology, Diagnostics, and Therapy. Chem. Rev. 2007, 107, 3715–3743. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Polisky, B.; Uhlenbeck, O.; Yarus, M. Diversity of Oligonucleotide Functions. Annu. Rev. Biochem. 1995, 64, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, P.E.; Lewis, S.D.; Silva, R.F.; Preiss, J.R.; Horwitz, L.R.; Pendergrast, P.S.; McCauley, T.G.; Kurz, J.C.; Epstein, D.M.; Wilson, C.; et al. Direct in Vitro Selection of a 2′-O-Methyl Aptamer to VEGF. Chem. Biol. 2005, 12, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Kahlon, J.B.; Thomas, S.D.; Trent, J.O.; Miller, D.M. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem. 1999, 274, 26369–26377. [Google Scholar] [CrossRef] [PubMed]
- Dapić, V.; Abdomerović, V.; Marrington, R.; Peberdy, J.; Rodger, A.; Trent, J.O.; Bates, P.J. Biophysical and biological properties of quadruplex oligodeoxyribonucle otides. Nucleic Acids Res. 2003, 31, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Dapić, V.; Bates, P.J.; Trent, J.O.; Rodger, A.; Thomas, S.D.; Miller, D.M. Antiproliferative activity of G-quartetforming oligonucleotides with backbone and sugar modifications. Biochemistry 2002, 41, 3676–3685. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reyes, E.M.; Teng, Y.; Bates, P.J. A new paradigm for aptamer therapeutic AS1411 action: Uptake by macropinocytosis and its stimulation by a nucleolin-dependent mechanism. Cancer Res. 2010, 70, 8617–8629. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Shangguan, D.; Liu, H.; Phillips, J.A.; Zhang, X.; Chen, Y.; Tan, W. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. ChemBioChem 2009, 10, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.W.; Zhu, G.; Mei, L.; Xie, Y.; Ma, H.; Ye, M.; Qing, F.L.; Tan, W. Automated modular synthesis of aptamer-drug conjugates for targeted drug delivery. J. Am. Chem. Soc. 2014, 136, 2731–2734. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V.J. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D.; Dunne, T.S.; Siegel, M.M.; Chang, C.C.; Morton, G.O.; Borders, D.B. Calichemicins, a novel family of antitumor antibiotics. 1. Chemistry and partial structure of calichemicin γ1. J. Am. Chem. Soc. 1987, 109, 3464–3466. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Bai, R.; Petit, G.R.; Hame, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal: Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Hartley, J.A.; Hamaguchi, A.; Suggitt, M.; Gregson, S.J.; Thurston, D.E.; Howard, P.W. DNA inter strand cross-linking and in vivo antitumor activity of the extended pyrrolo [2,1-c][1,4]benzodiazepine dimer SG2057. Investig. New Drugs 2012, 30, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, G.; Salnikov, A.V.; Luttgau, S.; Herr, I.; Anderl, J.; Faulstich, H. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesionmolecule monoclonal antibody against pancreatic carcinoma. J. Natl. Cancer Inst. 2012, 104, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Kemshead, J.T.; Hopkins, K. Uses and limitations of monoclonal antibodies (MoAbs) in the treatment of malignant disease: A review. J. R. Soc. Med. 1993, 86, 219–224. [Google Scholar] [PubMed]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellstrom, K.E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef] [PubMed]
- De Groot, F.M.; Damen, E.W.; Scheeren, H.W. Anticancer prodrugs for application in monotherapy: Targeting hypoxia, tumor-associated enzymes, and receptors. Curr. Med. Chem. 2001, 8, 1093–1122. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Palombo, M.; Sinko, P.J. Recent trends in targeted anticancer prodrug and conjugate design. Curr. Med. Chem. 2008, 15, 1802–1826. [Google Scholar] [CrossRef] [PubMed]
- Ranson, M.; Sliwkowski, M.X. Perspectives on anti-HER monoclonal antibodies. Oncology 2002, 63, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug. Chem. 2010, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blättler, W.A.; Goldmacher, V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Audette, C.; Hoffee, M.; Lambert, J.M.; Blättler, W.A. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J. Pharmacol. Exp. Ther. 2004, 308, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.J.; Hering, M.A.; James, S.F.; Sun, M.M. C.; Doronina, S.O.; Siadak, A.W.; Senter, P.D.; Wahl, A.F. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin. Cancer Res. 2005, 11, 843–852. [Google Scholar] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of β-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, M.; Boven, E.; Scheeren, H.W.; Haisma, H.J.; Pinedo, H.M. Beta-glucuronidase-mediated drug release. Curr. Pharm. Des. 2002, 8, 1391–1403. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Nguyen, M.T.; Moser, R.F.; Meyer, D.L.; Miyamoto, J.B.; Senter, P.D. Minor groove binder antibody conjugates employing a water soluble β-glucuronide linker. Bioorg. Med. Chem. Lett. 2007, 17, 2278–2280. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Senter, P.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.; Toki, B.E.; Manikumar, G.; Wani, M.C.; Kroll, D.J.; Jeffrey, S.C. Design, synthesis, and biological evaluation of antibody-drug conjugates comprised of potent camptothecin analogues. Bioconjug. Chem. 2009, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.L.L.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; Wong, W.L.T.; Jacobson, F.S.; Koeppen, H.; Schwall, R.H.; Kenkare-Mitra, S.R.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Vugts, D.J.; Visser, G.W.; Stigter-van, W.M.; Bolijn, M.; Spiga, M.; Lazzari, P.; Shankar, S.; Sani, M.; Zanda, M.; et al. Development of novel ADCs: Conjugation of tubulysin analogues to trastuzumab monitored by dual radiolabeling. Cancer Res. 2014, 74, 5700–5710. [Google Scholar] [CrossRef] [PubMed]
- Pillow, T.H.; Tien, J.; Parsons-Reponte, K.L.; Bhakta, S.; Li, H.; Staben, L.R.; Li, G.; Chuh, J.; Fourie-O Donohue, A.; Darwish, M.; et al. Site-specific trastuzumab maytansinoid antibody−drug conjugates with improved therapeutic activity through linker and antibody engineering. J. Med. Chem. 2014, 57, 7890–7899. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Kolodych, S.; Koniev, O.; Baatarkhuu, Z.; Bonnefoy, J.Y.; Debaene, F.; Cianférani, S.; Dorsselaer, A.V.; Wagner, A. CBTF: New amine-to-thiol coupling reagent for preparation of antibody conjugates with increased plasma stability. Bioconjug. Chem. 2015, 26, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Pei, S.-N.; Qi, J.; Zeng, Z.; Iyer, S.P.; Lin, P.; Tung, C.-H.; Zu, Y. Oligonucleotide aptamer-drug conjugates for targeted therapy of acute myeloid leukemia. Biomaterials 2015, 67, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.; Frenkel, E. Nanoparticles for drug delivery in cancer treatment. Urol. Oncol. 2008, 26, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Vigderman, L.; Zubarev, E.R. Therapeutic platforms based on gold nanoparticles and their covalent conjugates with drug molecules. Adv. Drug Deliv. Rev. 2013, 65, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Subinoy, R.; Avinash, B.; Rubul, M.; Vincent, M.R. Monolayer coated gold nanoparticles for delivery applications. Adv. Drug Deliv. Rev. 2012, 64, 200–216. [Google Scholar]
- Luo, Y.L.; Shiao, Y.S.; Huang, Y.F. Release of photoactivatable drugs from plasmonic nanoparticles for targeted cancer therapy. ACS Nano 2011, 5, 7796–7804. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, X.; Liu, Z.; Pu, F.; Ren, J.; Qu, X. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer gated nanovehicles. Adv. Mater. 2012, 24, 2890–2895. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, G.; You, M.; Song, E.; Shukoor, M.I.; Zhang, K.; Altman, M.B.; Chen, Y.; Zhu, Z.; Huang, C.Z.; et al. Assembly of aptamer switch probes and photosensitizer on gold nanorods for targeted photothermal and photodynamic cancer therapy. ACS Nano 2012, 6, 5070–5077. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sefah, K.; Altman, M.B.; Chen, T.; You, M.; Zhao, Z.; Huang, C.Z.; Tan, W. Aptamer-conjugated nanorods for targeted photothermal therapy of prostate cancer stem cells. Chem. Asian J. 2013, 8, 2417–2422. [Google Scholar] [CrossRef] [PubMed]
- Dreaden, E.C.; Austin, L.A.; Mackey, M.A.; El-Sayed, M.A. Size matters: Gold nanoparticles in targeted cancer drug delivery. Ther. Deliv. 2012, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, M.; Anand, S. Synthesis and applications of nano-structured iron oxides/hydroxides—A review. Int. J. Eng. Sci. Technol. 2010, 2, 127–146. [Google Scholar] [CrossRef]
- Chen, T.; Shukoor, M.I.; Wang, R.; Zhao, Z.; Yuan, Q.; Bamrungsap, S.; Xiong, X.; Wei, T. Smart multifunctional nanostructure for targeted cancer chemotherapy and magnetic resonance imaging. ACS Nano 2011, 5, 7866–7873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, W.F.; Sun, H.B. Multifunctional superparamagnetic iron oxide nanoparticles: Design, synthesis and biomedical photonic applications. Nanoscale 2013, 5, 7664–7684. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Sant, S.; Wang, B.; Laurent, S.; Sen, T. Superparamagnetic iron oxide nanoparticles (SPIONs): Development, surface modification and applications in chemotherapy. Adv. Drug Deliv. Rev. 2011, 63, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Hussain, S.M.; Krestin, G.P. Superparamagnetic iron oxide contrast agents: Physicochemical characteristics and applications in MR imaging. Eur. Radiol. 2001, 11, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Weissleder, R.; Elizondo, G.; Wittenberg, J.; Rabito, C.A.; Bengele, H.H.; Josephson, L. Ultrasmall superparamagnetic iron oxide: Characterization of a new class of contrast agents for MR imaging. Radiology 1990, 175, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Leenders, W. Ferumoxtran-10 advanced magnetics. IDrugs 2003, 6, 987–993. [Google Scholar] [PubMed]
- Wang, A.Z.; Bagalkot, V.; Vasilliou, C.C.; Gu, F.; Alexis, F.; Zhang, L.; Shaikh, M.; Yuet, K.; Cima, M.J.; Langer, R.; et al. Superparamagnetic iron oxide nanoparticle-aptamer bioconjugates for combined prostate cancer imaging and therapy. ChemMedChem 2008, 3, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.K.; Kim, D.; Lee, I.H.; So, J.S.; Jeong, Y.Y.; Jon, S. Image-guided prostate cancer therapy using aptamer-functionalized thermally cross-linked superparamagnetic iron oxide nanoparticles. Small 2011, 7, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Saei, A.A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M. Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: Opportunities and challenges. Expert Opin. Drug Deliv. 2014, 11, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Wahajuddin; Arora, S. Superparamagnetic iron oxide nanoparticles: Magnetic nanoplatforms as drug carriers. Int. J. Nanomed. 2012, 7, 3445–3471. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, X.; Lu, Q.; Fei, Z.; Dyson, P.J. Single walled carbon nanotubes as drug delivery vehicles: Targeting DOXorubicin to tumors. Biomaterials 2012, 33, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Tang, Z.; Phillips, J.A.; Yang, R.; Wang, H.; Tan, W. Regulation of singlet oxygen generation using single-walled carbon nanotubes. J. Am. Chem. Soc. 2008, 130, 10856–10857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hou, L.; Jiao, X.; Ji, Y.; Zhu, X.; Li, H.; Chen, X.; Ren, J.; Xia, Y.; Zhang, Z. In vitro and in vivo evaluation of antitumor drug-loaded aptamer targeted single-walled carbon nanotubes system. Curr. Pharm. Biotechnol. 2014, 14, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wu, P.; Yin, Y.; Zhang, H.; Cai, C. Aptamer-functionalized graphene oxide for highly efficient loading and cancer cell-specific delivery of antitumor drug. J. Mater. Chem. B 2014, 2, 3849–3859. [Google Scholar] [CrossRef]
- Rodriguesand, D.; Elimelech, M. Toxic effects of single-walled carbon nanotubes in the development of E. coli biofilm. Environ. Sci. Technol. 2010, 44, 4583–4589. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Huang, X.-C.; Luo, Y.-L.; Chang, Y.-C.; Hsieh, Y.-Z.; Hsu, H.-Y. Non-metallic nanomaterials in cancer theranostics: A review of silica- and carbon-based drug delivery systems. Sci. Technol. Adv. Mater. 2013, 14, 044407. [Google Scholar] [CrossRef]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.W.; Lin, V.S.Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.L.; Song, X.Y.; Zhou, W.H.; Yang, H.H.; Wen, Y.H.; Wang, X.R. An efficient cell-targeting and intracellular controlled-release drug delivery system based on MSN-PEM-aptamer conjugates. J. Mater. Chem. 2009, 19, 7765–7770. [Google Scholar] [CrossRef]
- Li, L.L.; Yin, Q.; Cheng, J.; Lu, Y. Polyvalent Mesoporous Silica Nanoparticle-Aptamer Bioconjugates Target Breast Cancer Cells. Adv. Healthc. Mater. 2012, 1, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, H.; Shi, J. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv. Mater. 2013, 25, 3144–3176. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.S.; Rao, M.E.B. Quantum dot: Novel carrier for drug delivery. Int. J. Res. Pharm. Biomed. Sci. 2011, 2, 448–458. [Google Scholar]
- Qi, L.; Gao, X. Emerging application of quantum dots for drug delivery and therapy. Expert Opin. Drug Deliv. 2008, 5, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Bagalkot, V.; Zhang, L.; Levy-Nissenbaum, E.; Jon, S.; Kantoff, P.W.; Langer, R.; Farokhzad, O.C. Quantum dot-aptamer conjugates for synchronous cancer imaging, therapy, and sensing of drug delivery based on bi-fluorescence resonance energy transfer. Nano Lett. 2007, 7, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Savla, R.; Taratula, O.; Garbuzenko, O.; Minko, T. Tumor targeted quantum dot-mucin 1 aptamer-doxorubicin conjugate for imaging and treatment of cancer. J. Control. Release 2011, 153, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Winnik, F.M.; Maysinger, D. Quantum dot cytotoxicity and ways to reduce it. Acc. Chem. Res. 2013, 46, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi, S.; Ramesh, B.; Seifalian, A.M. Fluorescence nanoparticles “quantum dots” as drug delivery system and their toxicity: A review. J. Drug Target. 2011, 19, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Hardman, R. A toxicologic review of quantum dots: Toxicity depends on physicochemical and environmental factors. Environ. Health Perspect. 2006, 114, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and Applications. Nanoscale Res. Lett. 2013, 8, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Platzgummer, M.; Kreismayr, G.; Quendler, H.; Stiegler, G.; Ferko, B.; Vecera, G.; Vorauer-Uhl, K.; Katinger, H. GMP production of liposomes—a new industrial approach. J. Liposome Res. 2006, 16, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, Y. Virosomes: Evolution of the liposome as a targeted drug delivery system. Adv. Drug Deliv. Rev. 2000, 43, 197–205. [Google Scholar] [CrossRef]
- Cao, Z.; Tong, R.; Mishra, A.; Xu, W.; Wong, G.C.; Cheng, J.; Lu, Y. Reversible cell-specific drug delivery with aptamer-functionalized liposomes. Angew. Chem. Int. Ed. 2009, 48, 6494–6498. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; O’Donoghue, M.B.; Liu, H.; Tan, W. A liposome-based nanostructure for aptamer directed delivery. Chem. Commun. 2010, 46, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Tang, L.; Yang, X.; Hwang, K.; Wang, W.; Yin, Q.; Wong, N.Y.; Dobrucki, L.W.; Yasui, N.; Katzenellenbogen, J.A.; et al. Selective delivery of an anticancer drug with aptamer-functionalized liposomes to breast cancer cells in vitro and in vivo. J. Mater. Chem. B 2013, 1, 5288–5297. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.E.; Lee, K.H.; Park, Y.S.; Oh, D.K.; Oh, S.; Kim, K.S.; Kim, D.E. RNA aptamer-conjugated liposome as an efficient anticancer drug delivery vehicle targeting cancer cells in vivo. J. Control. Release 2014, 196, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Alshaer, W.; Hillaireau, H.; Vergnaud, J.; Ismail, S.; Fattal, E. Functionalizing liposomes with anti-CD44 aptamer for selective targeting of cancer cells. Bioconjug. Chem. 2015, 26, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Langer, R. Drug delivery and targeting. Nature 1998, 392, 5–10. [Google Scholar] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymer nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt (IV) prodrug-PLGA-PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Kolishettib, N.; Lipparda, S.J.; Farokhzad, O.C. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 1850–1855. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Zhang, L.F.; Teply, B.A.; Mann, N.; Wang, A.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc. Natl. Acad. Sci. USA 2008, 105, 2586–2591. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; You, M.; Chen, T.; Zhu, G.; Liang, H.; Tan, W. Self-assembled hybrid nanoparticles for targeted co-delivery of two drugs into cancer cells. Chem. Commun. 2014, 50, 3103–3105. [Google Scholar] [CrossRef] [PubMed]
- Kedar, U.; Phutane, P.; Shidhaye, S.; Kadam, V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine 2010, 6, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Sefah, K.; Liu, H.P.; Wang, R.W.; Tan, W.H. DNA aptamer-micelle as an efficient detection/delivery vehicle toward cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Dave, N.; Hu, J.; Desai, P.; Pauletti, G.; Bai, S.; Hao, J. Solubilization of flurbiprofen into aptamer-modified PEG-PLA micelles for targeted delivery to brain-derived endothelial cells in vitro. J. Microencapsul. 2013, 30, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Siddiqui, I.A.; Nihal, M.; Pilla, S.; Rosenthal, K.; Mukhtar, H.; Gong, S. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted therapy of prostate cancer. Biomaterials 2013, 34, 5244–5253. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, Y.; Ji, Q.; Qiu, L. Targeted delivery of anticancer drugs by aptamer AS1411 mediated Pluronic F127/cyclodextrin-linked polymer composite micelles. Nanomedicine 2015, 11, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, R.; Fang, X.; Chen, F.; Wang, Y.; Chen, M. Nucleolin targeting AS1411 aptamer modified pH-sensitive micelles for enhanced delivery and antitumor efficacy of paclitaxel. Nano Res. 2015, 8, 201–218. [Google Scholar] [CrossRef]
- Deng, C.; Jiang, Y.; Cheng, R.; Meng, F.; Zhong, Z. Biodegradable polymeric micelles for targeted and controlled anticancer drug delivery: Promises, progress and prospects. Nano Today 2012, 7, 467–480. [Google Scholar] [CrossRef]
- Tripathy, S.; Das, M.K. Dendrimers and their applications as novel drug delivery carriers. J. Appl. Pharm. Sci. 2013, 3, 142–149. [Google Scholar]
- Nanjwadea, B.K.; Bechra, H.M.; Derkar, G.K.; Manvi, F.V.; Nanjwade, V.K. Dendrimers: Emerging polymers for drug-delivery systems. Eur. J. Pharm. Sci. 2009, 38, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Pednekar, P.P.; Jadhav, K.R.; Kadam, V.J. Aptamer-dendrimer bioconjugate: A nanotool for therapeutics, diagnosis, and imaging. Expert Opin. Drug Deliv. 2012, 9, 1273–1288. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; An, S.; Yu, M.K.; Kwon, H.K.; Im, S.H.; Jon, S. Targeted chemoimmunotherapy using drug-loaded aptamer-dendrimer bioconjugates. J. Control. Release 2011, 155, 435–441. [Google Scholar] [CrossRef] [PubMed]
- MacAdam, A.B.; Shafi, Z.B.; James, S.L.; Marriott, C.; Martin, G.P. Preparation of hydrophobic and hydrophilic albumin microspheres and determination of surface carboxylic acid and amino residues. Int. J. Pharm. 1997, 151, 47–55. [Google Scholar] [CrossRef]
- Wang, G.; Uludag, H. Recent developments in nanoparticle-based drug delivery and targeting systems with emphasis on protein-based nanoparticles. Expert Opin. Drug Deliv. 2008, 5, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Tong, W.; Yu, D.; Xu, J.; Li, J.; Gao, C. Bovine serum albumin nanoparticles modified with multilayers and aptamers for pH-responsive and targeted anti-cancer drug delivery. J. Mater. Chem. 2012, 22, 6053–6060. [Google Scholar] [CrossRef]
- Yang, L.; Meng, L.; Zhang, X.; Chen, Y.; Zhu, G.; Liu, H.; Xiong, X.; Sefah, K.; Tan, W. Engineering polymeric aptamers for selective cytotoxicity. J. Am. Chem. Soc. 2011, 133, 13380–13386. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Han, D.; Chen, T.; Peng, L.; Zhu, G.; You, M.; Qiu, L.; Sefah, K.; Zhang, X.; Tan, W. Building a multifunctional aptamer-based DNA nanoassembly for targeted cancer therapy. J. Am. Chem. Soc. 2013, 135, 18644–18650. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Zhang, X.; Zhao, Z.; Zhu, G.; Chen, T.; Fu, T.; Tan, W. DNA nanoflowers for multiplexed cellular imaging and traceable targeted drug delivery. Angew. Chem. Int. Ed. 2014, 53, 5821–5826. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Hu, R.; Zhao, Z.; Chen, Z.; Zhang, X.; Tan, W. Noncanonical self-assembly of multifunctional DNA nanoflowers for biomedical applications. J. Am. Chem. Soc. 2013, 135, 16438–16445. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zheng, J.; Song, E.; Donovana, M.; Zhang, K.; Liu, C.; Tan, W. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc. Natl. Acad. Sci. USA 2013, 110, 7998–8003. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, H.; Li, S.; Zaia, J.; Rossi, J.J. Novel Dual Inhibitory Function Aptamer–siRNA Delivery System for HIV-1 Therapy. Mol. Ther. 2008, 16, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Jung, Y.; Choi, H.; Yang, J.; Suh, J.-S.; Huh, Y.-M; Kim, K.; Haam, S. Prostate cancer cell death produced by the co-delivery of Bcl-xL shRNA and doxorubicin using an aptamer-conjugated polyplex. Biomaterials 2010, 31, 4592–4599. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Priceman, S.J.; Kujawski, M.; Xin, H.; Cherryholmes, G.A.; Zhang, W.; Zhang, C.; Lahtz, C.; Kowolik, C.; Forman, S.J.; et al. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J. Clin. Investig. 2014, 124, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, F.; Liu, B.; Lu, J.; Li, F.; Li, D.; Liang, C.; Dang, L.; Liu, J.; He, B.; Badshah, S.A.; et al. Progress and Challenges in Developing Aptamer-Functionalized Targeted Drug Delivery Systems. Int. J. Mol. Sci. 2015, 16, 23784-23822. https://doi.org/10.3390/ijms161023784
Jiang F, Liu B, Lu J, Li F, Li D, Liang C, Dang L, Liu J, He B, Badshah SA, et al. Progress and Challenges in Developing Aptamer-Functionalized Targeted Drug Delivery Systems. International Journal of Molecular Sciences. 2015; 16(10):23784-23822. https://doi.org/10.3390/ijms161023784
Chicago/Turabian StyleJiang, Feng, Biao Liu, Jun Lu, Fangfei Li, Defang Li, Chao Liang, Lei Dang, Jin Liu, Bing He, Shaikh Atik Badshah, and et al. 2015. "Progress and Challenges in Developing Aptamer-Functionalized Targeted Drug Delivery Systems" International Journal of Molecular Sciences 16, no. 10: 23784-23822. https://doi.org/10.3390/ijms161023784
APA StyleJiang, F., Liu, B., Lu, J., Li, F., Li, D., Liang, C., Dang, L., Liu, J., He, B., Badshah, S. A., Lu, C., He, X., Guo, B., Zhang, X.-B., Tan, W., Lu, A., & Zhang, G. (2015). Progress and Challenges in Developing Aptamer-Functionalized Targeted Drug Delivery Systems. International Journal of Molecular Sciences, 16(10), 23784-23822. https://doi.org/10.3390/ijms161023784