
Perturbation of Brain Oscillations after Ischemic Stroke: A Potential Biomarker for Post-Stroke Function and Therapy

Abstract
:1. Introduction
2. EEG Signals and the Spectrum of Oscillations
3. EEG in Normal Conditions
3.1. Generators of Oscillations
3.2. Oscillations and Behavior
3.2.1. In Humans
3.2.2. In Animals
3.2.3. Synchronized vs. Desynchronized Cortical State and Behavior
4. EEG and the Cellular Origins of Oscillations
4.1. Under Physiological Conditions
Cellular Mechanisms

4.2. Under Pathological Conditions of Energy Failure
4.2.1. Cellular Events after Ischemia
4.2.2. Cerebral Blood Flow (CBF) and EEG

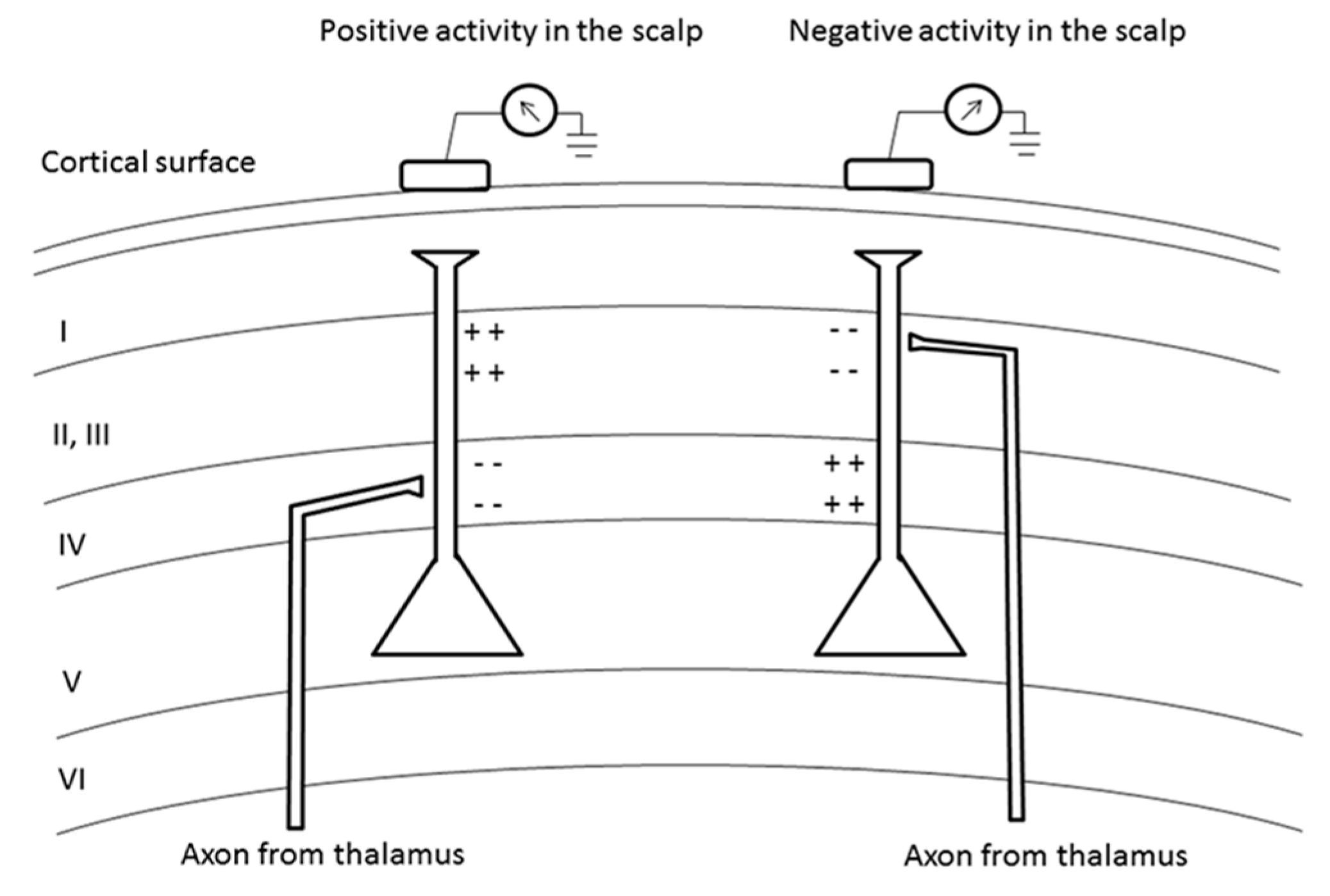{kind=link}
{kind=link}
| CBF Level (mL/100 g/min) | EEG Abnormality | Cellular Response | Degree of Neuronal Injury |
|---|---|---|---|
| 35–70 | Normal | Decreased protein synthesis | No injury |
| 25–35 | Loss of fast β frequencies and decreased amplitude of somatosensory evoked potentials |
| Reversible |
| 18–25 | Slowing of θ rhythm and loss of fast frequencies |
| Reversible |
| 12–18 | Slowing of δ rhythm, increases in slow frequencies and loss of post synaptic evoked responses |
| Reversible |
| <8–10 | Suppression of all frequencies, loss of presynaptic evoked responses |
| Neuronal death |
4.2.3. Penumbra and Core
5. EEG in Stroke Conditions
5.1. Modifications of the Brain Oscillations in Experimental Stroke
5.2. Clinical Applications of Continuous EEG Monitoring during Acute Ischemic Stroke
| Stroke Subtypes | Summary | Time Frame of EEG Detection Relative to Stroke Onset | EEG/qEEG Characteristics |
|---|---|---|---|
| Large (Cortical, including ACA, MCA, PCA territories) | EEG abnormalities following cortical infarction depended on infarct location | <2 weeks (<24 h (34%), <1 week (50%)) | Lateralized EEG abnormalities 80% in MCA territory, 86% in cortical watershed zone, but 50% in PCA territory [177] |
| Strong association between EEG mapping of δ power and lesion locations by CT | <24 h | Close correlation between EEG abnormalities (increased δ power) except striatocapsular in 85% patients [182] | |
| EEG monitoring is useful in all ischemic strokes regardless of locations. Also, pdBSI predicted radiologically (CT, MRI) confirmed stroke with an accuracy higher than the National Institute of Health stroke score (NIHSS) score at admission | <7 days (<72 h (81%)) | Increased pdBSI, DTABR, even in PCS and LACS [184] | |
| Small (subcortical, lacunar) | EEG has relatively low sensitivity in patients with subcortical infarcts | <2 weeks (<24 h (34%), <1 week (50%)) | 82% normal or non-lateralized EEG changes in subcortical lesions [177] |
| EEG has relatively low sensitivity in patients with first lacunar infarcts | <7 days | Abnormal EEG in 43% patients with first lacunar stroke [183] | |
| EEG abnormalities depend on affected lesions in subcortical regions | <24 h | Normal EEG in striatocapsular regions 70% abnormal EEG in other subcortical regions [182] | |
| TIA | EEG has low sensitivity in patients with TIA | <24 h | Non-significant difference between TIA and control by using pdBSI and DTABR [185] |
| DCI in SAH | ADRs may allow earlier detection of DCI in patients with severe SAH | Post-operative day two to post-SAH day 14 | ADR decrease in patients with DCI [186] |
| EEG changes preceded detection of vasospasm/DCI in standard procedures by 2.3 days | 2–12 days (median 5.2 days) | Decrease in α or θ power few days before vasospasm/DCI [187] | |
| Malignant MCA infarction | Emergence of high-voltage contralateral hemisphere δ activity might represent midline shift due to substantial edema in ipsilateral hemisphere and increased intracranial pressure | <25 h | Increasing δ power in contralateral hemisphere in malignant course [188] |
| EEG and brain stem auditory evoked potentials have prognostic value for patients who develop malignant edema | <24 h | Diffuse generalized slowing and slow δ activity in the ischemic hemisphere pointed to a malignant course [190] |
5.3. Continuous EEG Monitoring during Thrombolysis
5.4. Biomarkers of Prediction after Stroke
6. EEG, Oscillations Coupling and Perspectives

7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sanei, S.; Chambers, J.A. EEG Signal Processing; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 35–125. [Google Scholar]
- Acar, E.; Aykut-Bingol, C.; Bingol, H.; Bro, R.; Yener, B. Multiway analysis of epilepsy tensors. Bioinformatics 2007, 23, i10–i18. [Google Scholar] [CrossRef] [PubMed]
- Normann, R.A.; Maynard, E.M.; Rousche, P.J.; Warren, D.J. A neural interface for a cortical vision prosthesis. Vis. Res. 1999, 39, 2577–2587. [Google Scholar] [CrossRef]
- Lebedev, M.A.; Nicolelis, M.A. Brain-machine interfaces: Past, present and future. Trends Neurosci. 2006, 29, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Beck, A. Die Bestimmung der Localisation der Gehirn- und Rückenmarksfunctionen vermittelst der elektrischen Erscheinungen. [The determination of the localisation of the brain and spinal cord functions by way of electrical appearances]. Centralblatt für Physiologie 1890, 4, 473–476. (In Germany) [Google Scholar]
- Berger, H. Über das elektrenkephalogramm des menschen. Eur. Arch. Psychiatry Clin. Neurosci. 1929, 87, 527–570. [Google Scholar] [CrossRef]
- Buzsaki, G.; Draguhn, A. Neuronal oscillations in cortical networks. Science 2004, 304, 1926–1929. [Google Scholar] [CrossRef] [PubMed]
- Steriade, M.; Nunez, A.; Amzica, F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: Depolarizing and hyperpolarizing components. J. Neurosci. 1993, 13, 3252–3265. [Google Scholar] [PubMed]
- Bragin, A.; Engel, J., Jr.; Wilson, C.L.; Fried, I.; Buzsaki, G. High-frequency oscillations in human brain. Hippocampus 1999, 9, 137–142. [Google Scholar] [CrossRef]
- Ferri, R.; Cosentino, F.I.; Elia, M.; Musumeci, S.A.; Marinig, R.; Bergonzi, P. Relationship between δ, ς, β, and γ EEG bands at REM sleep onset and REM sleep end. Clin. Neurophysiol. 2001, 112, 2046–2052. [Google Scholar] [CrossRef]
- Engel, J., Jr.; Bragin, A.; Staba, R.; Mody, I. High-frequency oscillations: What is normal and what is not? Epilepsia 2009, 50, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, M.; Beyeler, A.; Leinekugel, X. Neural circuits underlying the generation of θ oscillations. J. Physiol. Paris 2012, 106, 81–92. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.A.; Pape, H.C. Noradrenergic and serotonergic modulation of a hyperpolarization-activated cation current in thalamic relay neurones. J. Physiol. 1990, 431, 319–342. [Google Scholar] [CrossRef] [PubMed]
- Ball, G.J.; Gloor, P.; Schaul, N. The cortical electromicrophysiology of pathological δ waves in the electroencephalogram of cats. Electroencephalogr. Clin. Neurophysiol. 1977, 43, 346–361. [Google Scholar] [CrossRef]
- Plouin, P.; Kaminska, A.; Moutard, M.L.; Soufflet, C. Developmental aspects of normal EEG. Handb. Clin. Neurol. 2013, 111, 79–85. [Google Scholar] [PubMed]
- Lu, X.C.; Williams, A.J.; Tortella, F.C. Quantitative electroencephalography spectral analysis and topographic mapping in a rat model of middle cerebral artery occlusion. Neuropathol. Appl. Neurobiol. 2001, 27, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Ke, Z.; Li, L.; Yip, S.P.; Tong, K.Y. EEG patterns from acute to chronic stroke phases in focal cerebral ischemic rats: Correlations with functional recovery. Physiol. Meas. 2013, 34, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Steriade, M. Impact of network activities on neuronal properties in corticothalamic systems. J. Neurophysiol. 2001, 86, 1–39. [Google Scholar] [PubMed]
- Csicsvari, J.; Jamieson, B.; Wise, K.D.; Buzsaki, G. Mechanisms of γ oscillations in the hippocampus of the behaving rat. Neuron 2003, 37, 311–322. [Google Scholar] [PubMed]
- Olejniczak, P. Neurophysiologic basis of EEG. J. Clin. Neurophysiol. 2006, 23, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Llinas, R.R. The intrinsic electrophysiological properties of mammalian neurons: Insights into central nervous system function. Science 1988, 242, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Crunelli, V.; Hughes, S.W. The slow (<1 Hz) rhythm of non-REM sleep: A dialogue between three cardinal oscillators. Nat. Neurosci. 2010, 13, 9–17. [Google Scholar] [PubMed]
- Green, J.D.; Arduini, A.A. Hippocampal electrical activity in arousal. J. Neurophysiol. 1954, 17, 533–557. [Google Scholar] [PubMed]
- Sirota, A.; Montgomery, S.; Fujisawa, S.; Isomura, Y.; Zugaro, M.; Buzsaki, G. Entrainment of neocortical neurons and γ oscillations by the hippocampal θ rhythm. Neuron 2008, 60, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Monmaur, P.; Allix, M.; Schoevaert-Brossault, D.; Houcine, O.; Plotkine, M.; Willig, F. Effects of transient cerebral ischemia on the hippocampal dentate θ profile in the acute rat: A study 4–5 months following recirculation. Brain Res. 1990, 508, 124–134. [Google Scholar] [CrossRef]
- Adey, W.R. EEG patterns in sleep and wakefulness in high spinal cord injuries. Proc. Annu. Clin. Spinal. Cord. Inj. Conf. 1967, 16, 2–9. [Google Scholar] [PubMed]
- Mitchell, S.J.; Ranck, J.B., Jr. Generation of θ rhythm in medial entorhinal cortex of freely moving rats. Brain Res. 1980, 189, 49–66. [Google Scholar] [CrossRef]
- Alonso, A.; Garcia-Austt, E. Neuronal sources of θ rhythm in the entorhinal cortex of the rat. II. Phase relations between unit discharges and θ field potentials. Exp. Brain Res. 1987, 67, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.W.; Borst, J.G. Electrical activity of the cingulate cortex. I. Generating mechanisms and relations to behavior. Brain Res. 1987, 407, 68–80. [Google Scholar] [CrossRef]
- Pare, D.; Collins, D.R. Neuronal correlates of fear in the lateral amygdala: Multiple extracellular recordings in conscious cats. J. Neurosci. 2000, 20, 2701–2710. [Google Scholar] [PubMed]
- Hari, R.; Salmelin, R.; Makela, J.P.; Salenius, S.; Helle, M. Magnetoencephalographic cortical rhythms. Int. J. Psychophysiol. 1997, 26, 51–62. [Google Scholar] [CrossRef]
- Buffalo, E.A.; Fries, P.; Landman, R.; Buschman, T.J.; Desimone, R. Laminar differences in γ and α coherence in the ventral stream. Proc. Natl. Acad. Sci. USA 2011, 108, 11262–11267. [Google Scholar] [CrossRef] [PubMed]
- Roopun, A.K.; Middleton, S.J.; Cunningham, M.O.; LeBeau, F.E.; Bibbig, A.; Whittington, M.A.; Traub, R.D. A β2-frequency (20–30 Hz) oscillation in nonsynaptic networks of somatosensory cortex. Proc. Natl. Acad. Sci. USA 2006, 103, 15646–15650. [Google Scholar] [CrossRef] [PubMed]
- Basar, E.; Schurmann, M.; Basar-Eroglu, C.; Karakas, S. Α oscillations in brain functioning: An integrative theory. Int. J. Psychophysiol. 1997, 26, 5–29. [Google Scholar] [CrossRef]
- Steriade, M.; McCormick, D.A.; Sejnowski, T.J. Thalamocortical oscillations in the sleeping and aroused brain. Science 1993, 262, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Sauseng, P.; Klimesch, W.; Gruber, W.R.; Hanslmayr, S.; Freunberger, R.; Doppelmayr, M. Are event-related potential components generated by phase resetting of brain oscillations? A critical discussion. Neuroscience 2007, 146, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, F.H.; van Lierop, T.H.; Schrijer, C.F.; van Leeuwen, W.S. Organization of thalamic and cortical α rhythms: Spectra and coherences. Electroencephalogr. Clin. Neurophysiol. 1973, 35, 627–639. [Google Scholar] [CrossRef]
- Ohmoto, T.; Mimura, Y.; Baba, Y.; Miyamoto, T.; Matsumoto, Y.; Nishimoto, A.; Matsumoto, K. Thalamic control of spontaneous α-rhythm and evoked responses. Appl. Neurophysiol. 1978, 41, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Terao, Y.; Sakurai, Y.; Sakuta, M.; Ishii, K.; Sugishita, M. FDG-PET in an amnestic and hypersomnic patient with bilateral paramedian thalamic infarction. Rinsho Shinkeigaku 1993, 33, 951–956. [Google Scholar] [PubMed]
- Goldman, R.I.; Stern, J.M.; Engel, J., Jr.; Cohen, M.S. Simultaneous EEG and fMRI of the α rhythm. Neuroreport 2002, 13, 2487–2492. [Google Scholar] [CrossRef] [PubMed]
- Feige, B.; Scheffler, K.; Esposito, F.; di Salle, F.; Hennig, J.; Seifritz, E. Cortical and subcortical correlates of electroencephalographic α rhythm modulation. J. Neurophysiol. 2005, 93, 2864–2872. [Google Scholar] [CrossRef] [PubMed]
- Sadato, N.; Nakamura, S.; Oohashi, T.; Nishina, E.; Fuwamoto, Y.; Waki, A.; Yonekura, Y. Neural networks for generation and suppression of α rhythm: A PET study. Neuroreport 1998, 9, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Crone, N.E.; Miglioretti, D.L.; Gordon, B.; Lesser, R.P. Functional mapping of human sensorimotor cortex with electrocorticographic spectral analysis. II. Event-related synchronization in the γ band. Brain 1998, 121, 2301–2315. [Google Scholar] [CrossRef] [PubMed]
- Tallon-Baudry, C.; Bertrand, O.; Delpuech, C.; Pernier, J. Stimulus specificity of phase-locked and non-phase-locked 40 Hz visual responses in human. J. Neurosci. 1996, 16, 4240–4249. [Google Scholar] [PubMed]
- Basar, E.; Basar-Eroglu, C.; Karakas, S.; Schurmann, M. Brain oscillations in perception and memory. Int. J. Psychophysiol. 2000, 35, 95–124. [Google Scholar] [CrossRef]
- Basar, E.; Rahn, E.; Demiralp, T.; Schurmann, M. Spontaneous EEG θ activity controls frontal visual evoked potential amplitudes. Electroencephalogr. Clin. Neurophysiol. 1998, 108, 101–109. [Google Scholar] [CrossRef]
- Buhl, E.H.; Tamas, G.; Fisahn, A. Cholinergic activation and tonic excitation induce persistent γ oscillations in mouse somatosensory cortex in vitro. J. Physiol. 1998, 513, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.M.; McCormick, D.A. Chattering cells: Superficial pyramidal neurons contributing to the generation of synchronous oscillations in the visual cortex. Science 1996, 274, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Whittington, M.A.; Traub, R.D.; Jefferys, J.G. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature 1995, 373, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, K.D.; Fifkova, E.; Jones, M.S.; Barth, D.S. Focal stimulation of the thalamic reticular nucleus induces focal γ waves in cortex. J. Neurophysiol. 1998, 79, 474–477. [Google Scholar] [PubMed]
- Bringuier, V.; Fregnac, Y.; Baranyi, A.; Debanne, D.; Shulz, D.E. Synaptic origin and stimulus dependency of neuronal oscillatory activity in the primary visual cortex of the cat. J. Physiol. 1997, 500, 751–774. [Google Scholar] [CrossRef] [PubMed]
- Cardin, J.A.; Palmer, L.A.; Contreras, D. Stimulus-dependent γ (30–50 Hz) oscillations in simple and complex fast rhythmic bursting cells in primary visual cortex. J. Neurosci. 2005, 25, 5339–5350. [Google Scholar] [CrossRef] [PubMed]
- Whittington, M.A.; Traub, R.D.; Faulkner, H.J.; Stanford, I.M.; Jefferys, J.G. Recurrent excitatory postsynaptic potentials induced by synchronized fast cortical oscillations. Proc. Natl. Acad. Sci. USA 1997, 94, 12198–12203. [Google Scholar] [CrossRef] [PubMed]
- Traub, R.D.; Whittington, M.A.; Buhl, E.H.; Jefferys, J.G.; Faulkner, H.J. On the mechanism of the γ→β frequency shift in neuronal oscillations induced in rat hippocampal slices by tetanic stimulation. J. Neurosci. 1999, 19, 1088–1105. [Google Scholar] [PubMed]
- Colling, S.B.; Stanford, I.M.; Traub, R.D.; Jefferys, J.G. Limbic γ rhythms. I. Phase-locked oscillations in hippocampal CA1 and subiculum. J. Neurophysiol. 1998, 80, 155–161. [Google Scholar] [PubMed]
- Steriade, M.; Timofeev, I. Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron 2003, 37, 563–576. [Google Scholar] [CrossRef]
- Basar, E.; Basar-Eroglu, C.; Karakas, S.; Schurmann, M. γ, α, δ, and θ oscillations govern cognitive processes. Int. J. Psychophysiol. 2001, 39, 241–248. [Google Scholar] [CrossRef]
- Tononi, G.; Cirelli, C. Time to be SHY? Some comments on sleep and synaptic homeostasis. Neural Plast. 2012, 2012, 415250. [Google Scholar] [CrossRef] [PubMed]
- Assenza, G.; Pellegrino, G.; Tombini, M.; di Pino, G.; di Lazzaro, V. Wakefulness δ waves increase after cortical plasticity induction. Clin. Neurophysiol. 2015, 126, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Tononi, G.; Cirelli, C. Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 2014, 81, 12–34. [Google Scholar] [CrossRef] [PubMed]
- Gorgoni, M.; D’Atri, A.; Lauri, G.; Rossini, P.M.; Ferlazzo, F.; de Gennaro, L. Is sleep essential for neural plasticity in humans, and how does it affect motor and cognitive recovery? Neural Plast. 2013, 2013, 103949. [Google Scholar] [CrossRef] [PubMed]
- Llinas, R.; Ribary, U. Coherent 40-Hz oscillation characterizes dream state in humans. Proc. Natl. Acad. Sci. USA 1993, 90, 2078–2081. [Google Scholar] [CrossRef] [PubMed]
- Destexhe, A.; Sejnowski, T.J. Interactions between membrane conductances underlying thalamocortical slow-wave oscillations. Physiol. Rev. 2003, 83, 1401–1453. [Google Scholar] [CrossRef] [PubMed]
- Singer, W. Neuronal synchrony: A versatile code for the definition of relations? Neuron 1999, 24, 111–125. [Google Scholar] [CrossRef]
- Varela, F.; Lachaux, J.P.; Rodriguez, E.; Martinerie, J. The brainweb: Phase synchronization and large-scale integration. Nat. Rev. Neurosci. 2001, 2, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Bragin, A.; Engel, J., Jr.; Staba, R.J. High-frequency oscillations in epileptic brain. Curr. Opin. Neurol. 2010, 23, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.K.; Fries, P. Β-band oscillations—Signalling the status quo? Curr. Opin. Neurobiol. 2010, 20, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Kisley, M.A.; Cornwell, Z.M. γ and β neural activity evoked during a sensory gating paradigm: Effects of auditory, somatosensory and cross-modal stimulation. Clin. Neurophysiol. 2006, 117, 2549–2563. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Haenschel, C.; Nikolic, D.; Singer, W. The role of oscillations and synchrony in cortical networks and their putative relevance for the pathophysiology of schizophrenia. Schizophr. Bull. 2008, 34, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Haenschel, C.; Baldeweg, T.; Croft, R.J.; Whittington, M.; Gruzelier, J. γ and β frequency oscillations in response to novel auditory stimuli: A comparison of human electroencephalogram (EEG) data with in vitro models. Proc. Natl. Acad. Sci. USA 2000, 97, 7645–7650. [Google Scholar] [CrossRef] [PubMed]
- Marco-Pallares, J.; Cucurell, D.; Cunillera, T.; Garcia, R.; Andres-Pueyo, A.; Munte, T.F.; Rodriguez-Fornells, A. Human oscillatory activity associated to reward processing in a gambling task. Neuropsychologia 2008, 46, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Hasselmo, M.E. What is the function of hippocampal θ rhythm?—Linking behavioral data to phasic properties of field potential and unit recording data. Hippocampus 2005, 15, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Kahana, M.J.; Seelig, D.; Madsen, J.R. θ returns. Curr. Opin. Neurobiol. 2001, 11, 739–744. [Google Scholar] [CrossRef]
- Burgess, A.P.; Gruzelier, J.H. Short duration power changes in the EEG during recognition memory for words and faces. Psychophysiology 2000, 37, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.M.; Sillanmaki, L.; Koivisto, M.; Saarela, C.; Haggqvist, A.; Laine, M.; Hamalainen, H. The effects of memory load on event-related EEG desynchronization and synchronization. Clin. Neurophysiol. 2000, 111, 2071–2078. [Google Scholar] [CrossRef]
- Palva, S.; Palva, J.M. New vistas for α-frequency band oscillations. Trends Neurosci. 2007, 30, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Steriade, M. Arousal: Revisiting the reticular activating system. Science 1996, 272, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.M. Patterns of arousal in comatose patients. J. Neurol. Neurosurg. Psychiatry 1976, 39, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rill, E.; Kezunovic, N.; Hyde, J.; Simon, C.; Beck, P.; Urbano, F.J. Coherence and frequency in the reticular activating system (RAS). Sleep Med. Rev. 2013, 17, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.D.; Thiele, A. Cortical state and attention. Nat. Rev. Neurosci. 2011, 12, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Buzsaki, G. θ oscillations in the hippocampus. Neuron 2002, 33, 325–340. [Google Scholar] [CrossRef]
- Vanderwolf, C.H. Recovery from large medial thalamic lesions as a result of electroconvulsive therapy. J. Neurol. Neurosurg. Psychiatry 1968, 31, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Vertes, R.P. Hippocampal θ rhythm: A tag for short-term memory. Hippocampus 2005, 15, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.; Fox, S.E. Hippocampal θ activity in monkeys. Brain Res. 1991, 538, 59–63. [Google Scholar] [CrossRef]
- Chrobak, J.J.; Buzsaki, G. High-frequency oscillations in the output networks of the hippocampal-entorhinal axis of the freely behaving rat. J. Neurosci. 1996, 16, 3056–3066. [Google Scholar] [PubMed]
- Jadhav, S.P.; Kemere, C.; German, P.W.; Frank, L.M. Awake hippocampal sharp-wave ripples support spatial memory. Science 2012, 336, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Girardeau, G.; Benchenane, K.; Wiener, S.I.; Buzsaki, G.; Zugaro, M.B. Selective suppression of hippocampal ripples impairs spatial memory. Nat. Neurosci. 2009, 12, 1222–1223. [Google Scholar] [CrossRef] [PubMed]
- Schurmann, M.; Basar-Eroglu, C.; Basar, E. γ responses in the EEG: Elementary signals with multiple functional correlates. Neuroreport 1997, 8, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Bragin, A.; Jando, G.; Nadasdy, Z.; Hetke, J.; Wise, K.; Buzsaki, G. γ (40–100 Hz) oscillation in the hippocampus of the behaving rat. J. Neurosci. 1995, 15, 47–60. [Google Scholar] [PubMed]
- Poulet, J.F.; Petersen, C.C. Internal brain state regulates membrane potential synchrony in barrel cortex of behaving mice. Nature 2008, 454, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Crochet, S.; Petersen, C.C. Correlating whisker behavior with membrane potential in barrel cortex of awake mice. Nat. Neurosci. 2006, 9, 608–610. [Google Scholar] [CrossRef] [PubMed]
- Niell, C.M.; Stryker, M.P. Modulation of visual responses by behavioral state in mouse visual cortex. Neuron 2010, 65, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Clement, E.A.; Richard, A.; Thwaites, M.; Ailon, J.; Peters, S.; Dickson, C.T. Cyclic and sleep-like spontaneous alternations of brain state under urethane anaesthesia. PLoS ONE 2008, 3, e2004. [Google Scholar] [CrossRef] [PubMed]
- Renart, A.; de la Rocha, J.; Bartho, P.; Hollender, L.; Parga, N.; Reyes, A.; Harris, K.D. The asynchronous state in cortical circuits. Science 2010, 327, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, T.L.; Copelli, M.; Caixeta, F.; Belchior, H.; Chialvo, D.R.; Nicolelis, M.A.; Ribeiro, S. Spike avalanches exhibit universal dynamics across the sleep-wake cycle. PLoS ONE 2010, 5, e14129. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.; Naim, A.; Lampl, I. The subthreshold relation between cortical local field potential and neuronal firing unveiled by intracellular recordings in awake rats. J. Neurosci. 2010, 30, 4440–4448. [Google Scholar] [CrossRef] [PubMed]
- Poulet, J.F.; Fernandez, L.M.; Crochet, S.; Petersen, C.C. Thalamic control of cortical states. Nat. Neurosci. 2012, 15, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Munk, M.H.; Roelfsema, P.R.; Konig, P.; Engel, A.K.; Singer, W. Role of reticular activation in the modulation of intracortical synchronization. Science 1996, 272, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Fries, P.; Reynolds, J.H.; Rorie, A.E.; Desimone, R. Modulation of oscillatory neuronal synchronization by selective visual attention. Science 2001, 291, 1560–1563. [Google Scholar] [CrossRef] [PubMed]
- Chalk, M.; Herrero, J.L.; Gieselmann, M.A.; Delicato, L.S.; Gotthardt, S.; Thiele, A. Attention reduces stimulus-driven γ frequency oscillations and spike field coherence in V1. Neuron 2010, 66, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Puig, M.V.; Watakabe, A.; Ushimaru, M.; Yamamori, T.; Kawaguchi, Y. Serotonin modulates fast-spiking interneuron and synchronous activity in the rat prefrontal cortex through 5-HT1A and 5-HT2A receptors. J. Neurosci. 2010, 30, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Gervasoni, D.; Lin, S.C.; Ribeiro, S.; Soares, E.S.; Pantoja, J.; Nicolelis, M.A. Global forebrain dynamics predict rat behavioral states and their transitions. J. Neurosci. 2004, 24, 11137–11147. [Google Scholar] [CrossRef] [PubMed]
- Cantero, J.L.; Atienza, M.; Stickgold, R.; Kahana, M.J.; Madsen, J.R.; Kocsis, B. Sleep-dependent θ oscillations in the human hippocampus and neocortex. J. Neurosci. 2003, 23, 10897–10903. [Google Scholar] [PubMed]
- Watrous, A.J.; Lee, D.J.; Izadi, A.; Gurkoff, G.G.; Shahlaie, K.; Ekstrom, A.D. A comparative study of human and rat hippocampal low-frequency oscillations during spatial navigation. Hippocampus 2013, 23, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Axmacher, N.; Henseler, M.M.; Jensen, O.; Weinreich, I.; Elger, C.E.; Fell, J. Cross-frequency coupling supports multi-item working memory in the human hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Canolty, R.T.; Edwards, E.; Dalal, S.S.; Soltani, M.; Nagarajan, S.S.; Kirsch, H.E.; Berger, M.S.; Barbaro, N.M.; Knight, R.T. High γ power is phase-locked to θ oscillations in human neocortex. Science 2006, 313, 1626–1628. [Google Scholar] [CrossRef] [PubMed]
- Kahana, M.J. The cognitive correlates of human brain oscillations. J. Neurosci. 2006, 26, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.J.; Leuthardt, E.C.; Schalk, G.; Rao, R.P.; Anderson, N.R.; Moran, D.W.; Miller, J.W.; Ojemann, J.G. Spectral changes in cortical surface potentials during motor movement. J. Neurosci. 2007, 27, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Ebersole, J.S. Cortical Generators and EEG Voltage Fields; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2003; pp. 557–608. [Google Scholar]
- Freeman, W.J. The physiology of perception. Sci. Am. 1991, 264, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Borgers, C.; Kopell, N. Effects of noisy drive on rhythms in networks of excitatory and inhibitory neurons. Neural Comput. 2005, 17, 557–608. [Google Scholar] [CrossRef] [PubMed]
- Erdemli, G.; Crunelli, V. Response of thalamocortical neurons to hypoxia: A whole-cell patch-clamp study. J. Neurosci. 1998, 18, 5212–5224. [Google Scholar] [PubMed]
- Krnjevic, K. Electrophysiology of cerebral ischemia. Neuropharmacology 2008, 55, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Gloor, P. Neuronal generators and the problem of localization in electroencephalography: Application of volume conductor theory to electroencephalography. J. Clin. Neurophysiol. 1985, 2, 327–354. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Winship, I.R.; Murphy, T.H. In vivo calcium imaging reveals functional rewiring of single somatosensory neurons after stroke. J. Neurosci. 2008, 28, 6592–6606. [Google Scholar] [CrossRef] [PubMed]
- Schiene, K.; Bruehl, C.; Zilles, K.; Qu, M.; Hagemann, G.; Kraemer, M.; Witte, O.W. Neuronal hyperexcitability and reduction of GABAa-receptor expression in the surround of cerebral photothrombosis. J. Cereb. Blood Flow Metab. 1996, 16, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.H.; Taguchi, N.; Ardeshiri, A.; Kuroiwa, M.; Hurn, P.D.; Traystman, R.J.; Herson, P.S. Ischemic insult to cerebellar Purkinje cells causes diminished GABA(A) receptor function and Allopregnanolone neuroprotection is associated with GABA(A) receptor stabilization. J. Neurochem. 2008, 107, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.T.; Chesselet, M.F. Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J. Neurosci. 2002, 22, 6062–6070. [Google Scholar] [PubMed]
- Carmichael, S.T. Cellular and molecular mechanisms of neural repair after stroke: Making waves. Ann. Neurol. 2006, 59, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Li, P.; Boyd, J.D.; Delaney, K.R.; Murphy, T.H. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J. Neurosci. 2007, 27, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Bender, J.E.; Vishwanath, K.; Moore, L.K.; Brown, J.Q.; Chang, V.; Palmer, G.M.; Ramanujam, N. A robust Monte Carlo model for the extraction of biological absorption and scattering in vivo. IEEE Trans. Biomed. Eng. 2009, 56, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Luhmann, H.J.; Heinemann, U. Hypoxia-induced functional alterations in adult rat neocortex. J. Neurophysiol. 1992, 67, 798–811. [Google Scholar] [PubMed]
- Calabresi, P.; Pisani, A.; Mercuri, N.B.; Bernardi, G. Hypoxia-induced electrical changes in striatal neurons. J. Cereb. Blood. Flow Metab. 1995, 15, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Sigworth, F.J.; Haddad, G.G. Oxygen deprivation activates an ATP-inhibitable K+ channel in substantia nigra neurons. J. Neurosci. 1994, 14, 5590–5602. [Google Scholar] [PubMed]
- Spuler, A.; Grafe, P. Adenosine, “pertussis-sensitive” G-proteins, and K+ conductance in central mammalian neurones under energy deprivation. Neurosci. Lett. 1989, 98, 280–284. [Google Scholar] [CrossRef]
- Knopfel, T.; Spuler, A.; Grafe, P.; Gahwiler, B.H. Cytosolic calcium during glucose deprivation in hippocampal pyramidal cells of rats. Neurosci. Lett. 1990, 117, 295–299. [Google Scholar] [CrossRef]
- Harata, N.; Wu, J.; Ishibashi, H.; Ono, K.; Akaike, N. Run-down of the GABAa response under experimental ischaemia in acutely dissociated CA1 pyramidal neurones of the rat. J. Physiol. 1997, 500, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, E.; Yamamoto, S.; Kudo, Y.; Mihara, S.; Higashi, H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J. Neurophysiol. 1997, 78, 891–902. [Google Scholar] [PubMed]
- Rosen, A.S.; Morris, M.E. Depolarizing effects of anoxia on pyramidal cells of rat neocortex. Neurosci. Lett. 1991, 124, 169–173. [Google Scholar] [CrossRef]
- Krnjevic, K.; Xu, Y.Z. Dantrolene suppresses the hyperpolarization or outward current observed during anoxia in hippocampal neurons. Can. J. Physiol. Pharmacol. 1989, 67, 1602–1604. [Google Scholar] [CrossRef] [PubMed]
- Englund, M.; Hyllienmark, L.; Brismar, T. Chemical hypoxia in hippocampal pyramidal cells affects membrane potential differentially depending on resting potential. Neuroscience 2001, 106, 89–94. [Google Scholar] [CrossRef]
- Jordan, K.G. Emergency EEG and continuous EEG monitoring in acute ischemic stroke. J. Clin. Neurophysiol. 2004, 21, 341–352. [Google Scholar] [PubMed]
- Astrup, J.; Siesjo, B.K.; Symon, L. Thresholds in cerebral ischemia—The ischemic penumbra. Stroke 1981, 12, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Foreman, B.; Claassen, J. Quantitative EEG for the detection of brain ischemia. Crit Care 2012, 16, 216. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, R.L.; Poil, S.S.; Brandeis, D.; Klaver, P.; Bollmann, S.; Ghisleni, C.; Luchinger, R.; Martin, E.; Shankaranarayanan, A.; Alsop, D.C.; et al. Coupling between resting cerebral perfusion and EEG. Brain Topogr. 2013, 26, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Lennox, W.G.; Gibbs, F.A.; Gibbs, E.I. The Relationship in Man of Cerebral Activity to Blood Flow and to Blood Constituents. J. Neurol. Psychiatry 1938, 1, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Faught, E. Current role of electroencephalography in cerebral ischemia. Stroke 1993, 24, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Branston, N.M.; Symon, L.; Crockard, H.A.; Pasztor, E. Relationship between the cortical evoked potential and local cortical blood flow following acute middle cerebral artery occlusion in the baboon. Exp. Neurol. 1974, 45, 195–208. [Google Scholar] [CrossRef]
- Hossmann, K.A. Viability thresholds and the penumbra of focal ischemia. Ann. Neurol. 1994, 36, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Sharbrough, F.W.; Messick, J.M., Jr.; Sundt, T.M., Jr. Correlation of continuous electroencephalograms with cerebral blood flow measurements during carotid endarterectomy. Stroke 1973, 4, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Gallinat, J.; Kunz, D.; Senkowski, D.; Kienast, T.; Seifert, F.; Schubert, F.; Heinz, A. Hippocampal glutamate concentration predicts cerebral θ oscillations during cognitive processing. Psychopharmacology 2006, 187, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Major, S.; Manning, A.; Woitzik, J.; Drenckhahn, C.; Steinbrink, J.; Tolias, C.; Oliveira-Ferreira, A.I.; Fabricius, M.; Hartings, J.A.; et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009, 132, 1866–1881. [Google Scholar] [CrossRef] [PubMed]
- Guyot, L.L.; Diaz, F.G.; O’Regan, M.H.; McLeod, S.; Park, H.; Phillis, J.W. Real-time measurement of glutamate release from the ischemic penumbra of the rat cerebral cortex using a focal middle cerebral artery occlusion model. Neurosci. Lett. 2001, 299, 37–40. [Google Scholar] [CrossRef]
- Nagata, K.; Tagawa, K.; Hiroi, S.; Shishido, F.; Uemura, K. Electroencephalographic correlates of blood flow and oxygen metabolism provided by positron emission tomography in patients with cerebral infarction. Electroencephalogr. Clin. Neurophysiol. 1989, 72, 16–30. [Google Scholar] [CrossRef]
- Powers, W.J. Cerebral hemodynamics in ischemic cerebrovascular disease. Ann. Neurol. 1991, 29, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Von Bornstadt, D.; Houben, T.; Seidel, J.L.; Zheng, Y.; Dilekoz, E.; Qin, T.; Sandow, N.; Kura, S.; Eikermann-Haerter, K.; Endres, M.; et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 2015, 85, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C.; Lauritzen, M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol. Rev. 2015, 95, 953–993. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.; Cuspineda, E.; Valdes, P.; Virues, T.; Llopis, F.; Bosch, J.; Aubert, E.; Hernandez, E.; Pando, A.; Alvarez, M.A.; et al. Assessing acute middle cerebral artery ischemic stroke by quantitative electric tomography. Clin. EEG Neurosci. 2004, 35, 116–124. [Google Scholar] [PubMed]
- Sheorajpanday, R.V.; Nagels, G.; Weeren, A.J.; de Surgeloose, D.; de Deyn, P.P. Additional value of quantitative EEG in acute anterior circulation syndrome of presumed ischemic origin. Clin. Neurophysiol. 2010, 121, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Sheorajpanday, R.V.; Nagels, G.; Weeren, A.J.; van Putten, M.J.; de Deyn, P.P. Quantitative EEG in ischemic stroke: Correlation with functional status after 6 months. Clin. Neurophysiol. 2011, 122, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Huang, J.; Zhu, M.; Guan, Q.; Liu, C. Associations between EEG β power abnormality and diagnosis in cognitive impairment post cerebral infarcts. J. Mol. Neurosci. 2013, 49, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Finnigan, S.; van Putten, M.J. EEG in ischaemic stroke: Quantitative EEG can uniquely inform (sub-)acute prognoses and clinical management. Clin. Neurophysiol. 2013, 124, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Gloor, P.; Ball, G.; Schaul, N. Brain lesions that produce δ waves in the EEG. Neurology 1977, 27, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, D.A.; Pasternak, E.B.; Gurvitch, A.M. Correlation analysis of δ activity generated in cerebral hypoxia. Electroencephalogr. Clin. Neurophysiol. 1977, 42, 445–455. [Google Scholar] [CrossRef]
- Schaul, N.; Gloor, P.; Gotman, J. The EEG in deep midline lesions. Neurology 1981, 31, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Schaul, N.; Gloor, P.; Ball, G.; Gotman, J. The electromicrophysiology of δ waves induced by systemic atropine. Brain Res. 1978, 143, 475–486. [Google Scholar] [CrossRef]
- Moyanova, S.G.; Dijkhuizen, R.M. Present status and future challenges of electroencephalography- and magnetic resonance imaging-based monitoring in preclinical models of focal cerebral ischemia. Brain Res. Bull. 2014, 102, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Lu, X.C.; Hartings, J.A.; Tortella, F.C. Neuroprotection assessment by topographic electroencephalographic analysis: Effects of a sodium channel blocker to reduce polymorphic δ activity following ischaemic brain injury in rats. Fundam. Clin. Pharmacol. 2003, 17, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Moyanova, S.; Kortenska, L.; Kirov, R.; Iliev, I. Quantitative electroencephalographic changes due to middle cerebral artery occlusion by endothelin 1 in conscious rats. Arch. Physiol. Biochem. 1998, 106, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Moyanova, S.G.; Mitreva, R.G.; Kortenska, L.V.; Nicoletti, F.; Ngomba, R.T. Age-dependence of sensorimotor and cerebral electroencephalographic asymmetry in rats subjected to unilateral cerebrovascular stroke. Exp. Transl. Stroke Med. 2013, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Moyanova, S.G.; Kortenska, L.V.; Mitreva, R.G.; Pashova, V.D.; Ngomba, R.T.; Nicoletti, F. Multimodal assessment of neuroprotection applied to the use of MK-801 in the endothelin-1 model of transient focal brain ischemia. Brain Res. 2007, 1153, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Moyanova, S.; Kortenska, L.; Kirov, R.; Itzev, D.; Usunoff, K. Ketanserin reduces the postischemic EEG and behavioural changes following endothelin-1-induced occlusion of the middle cerebral artery in conscious rats. Cent. Eur. J. Med. 2008, 3, 406–416. [Google Scholar] [CrossRef]
- Zhang, S.; Tong, R.; Zhang, H.; Hu, X.; Zheng, X. A pilot studies in dynamic profile of multi parameters of EEG in a rat model of transient middle cerebral artery occlusion. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2006, 1, 1181–1184. [Google Scholar] [PubMed]
- Bhattacharya, P.; Pandey, A.K.; Paul, S.; Patnaik, R. Does Piroxicam really protect ischemic neurons and influence neuronal firing in cerebral ischemia? An exploration towards therapeutics. Med. Hypotheses 2013, 81, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Finger, S.; Koehler, P.J.; Jagella, C. The Monakow concept of diaschisis: Origins and perspectives. Arch. Neurol. 2004, 61, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Von Monakow, C. Die Lokalisation im Grosshirn: und der Abbau der Funktion durch kortikale Herde; Verlag von JF Bergmann: Wiesbaden, German, 1914; pp. 139–149. (In Germany) [Google Scholar]
- Andrews, R.J. Transhemispheric diaschisis. A review and comment. Stroke 1991, 22, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Williams, A.J.; Tortella, F.C. Occurrence of nonconvulsive seizures, periodic epileptiform discharges, and intermittent rhythmic δ activity in rat focal ischemia. Exp. Neurol. 2003, 179, 139–149. [Google Scholar] [CrossRef]
- Lammer, A.B.; Beck, A.; Grummich, B.; Forschler, A.; Krugel, T.; Kahn, T.; Schneider, D.; Illes, P.; Franke, H.; Krugel, U. The P2 receptor antagonist PPADS supports recovery from experimental stroke in vivo. PLoS ONE 2011, 6, e19983. [Google Scholar] [CrossRef] [PubMed]
- Huchzermeyer, C.; Albus, K.; Gabriel, H.J.; Otahal, J.; Taubenberger, N.; Heinemann, U.; Kovacs, R.; Kann, O. γ oscillations and spontaneous network activity in the hippocampus are highly sensitive to decreases in pO2 and concomitant changes in mitochondrial redox state. J. Neurosci. 2008, 28, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Huchzermeyer, C.; Kovacs, R.; Wirtz, S.; Schuelke, M. γ oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain 2011, 134, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Tortella, F.C. Neuroprotective effects of the sodium channel blocker RS100642 and attenuation of ischemia-induced brain seizures in the rat. Brain Res. 2002, 932, 45–55. [Google Scholar] [CrossRef]
- Finnigan, S.P.; Rose, S.E.; Walsh, M.; Griffin, M.; Janke, A.L.; McMahon, K.L.; Gillies, R.; Strudwick, M.W.; Pettigrew, C.M.; Semple, J.; et al. Correlation of quantitative EEG in acute ischemic stroke with 30-day NIHSS score: Comparison with diffusion and perfusion MRI. Stroke 2004, 35, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Sheorajpanday, R.V.; Nagels, G.; Weeren, A.J.; van Putten, M.J.; de Deyn, P.P. Reproducibility and clinical relevance of quantitative EEG parameters in cerebral ischemia: A basic approach. Clin. Neurophysiol. 2009, 120, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Sundt, T.M., Jr.; Sharbrough, F.W.; Piepgras, D.G.; Kearns, T.P.; Messick, J.M., Jr.; O’Fallon, W.M. Correlation of cerebral blood flow and electroencephalographic changes during carotid endarterectomy: With results of surgery and hemodynamics of cerebral ischemia. Mayo Clin. Proc. 1981, 56, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Macdonell, R.A.; Donnan, G.A.; Bladin, P.F.; Berkovic, S.F.; Wriedt, C.H. The electroencephalogram and acute ischemic stroke. Distinguishing cortical from lacunar infarction. Arch. Neurol. 1988, 45, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.L.; Jordan, K.G. Regional attenuation without δ (RAWOD): A distinctive EEG pattern that can aid in the diagnosis and management of severe acute ischemic stroke. Am. J. Electroneurodiagn. Technol. 2005, 45, 102–117. [Google Scholar]
- Andraus, M.E.; Alves-Leon, S.V. Non-epileptiform EEG abnormalities: An overview. Arq. Neuropsiquiatr. 2011, 69, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Alberto, P.; Elisabetta, F.; Paola, R.; Uberto, R.; Alfredo, B. The EEG in lacunar strokes. Stroke 1984, 15, 579–580. [Google Scholar] [PubMed]
- Schaul, N. The fundamental neural mechanisms of electroencephalography. Electroencephalogr. Clin. Neurophysiol. 1998, 106, 101–107. [Google Scholar] [CrossRef]
- Murri, L.; Gori, S.; Massetani, R.; Bonanni, E.; Marcella, F.; Milani, S. Evaluation of acute ischemic stroke using quantitative EEG: A comparison with conventional EEG and CT scan. Neurophysiol. Clin. 1998, 28, 249–257. [Google Scholar] [CrossRef]
- Petty, G.W.; Labar, D.R.; Fisch, B.J.; Pedley, T.A.; Mohr, J.P.; Khandji, A. Electroencephalography in lacunar infarction. J. Neurol. Sci. 1995, 134, 47–50. [Google Scholar] [CrossRef]
- Sheorajpanday, R.V.; Nagels, G.; Weeren, A.J.; de Deyn, P.P. Quantitative EEG in ischemic stroke: Correlation with infarct volume and functional status in posterior circulation and lacunar syndromes. Clin. Neurophysiol. 2011, 122, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Sheorajpanday, R.V.; Marien, P.; Weeren, A.J.; Nagels, G.; Saerens, J.; van Putten, M.J.; de Deyn, P.P. EEG in silent small vessel disease: sLORETA mapping reveals cortical sources of vascular cognitive impairment no dementia in the default mode network. J. Clin. Neurophysiol. 2013, 30, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Claassen, J.; Hirsch, L.J.; Kreiter, K.T.; Du, E.Y.; Connolly, E.S.; Emerson, R.G.; Mayer, S.A. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin. Neurophysiol. 2004, 115, 2699–2710. [Google Scholar] [CrossRef] [PubMed]
- Gollwitzer, S.; Groemer, T.; Rampp, S.; Hagge, M.; Olmes, D.; Huttner, H.B.; Schwab, S.; Madzar, D.; Hopfengaertner, R.; Hamer, H.M. Early prediction of delayed cerebral ischemia in subarachnoid hemorrhage based on quantitative EEG: A prospective study in adults. Clin. Neurophysiol. 2015, 126, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Finnigan, S.P.; Rose, S.E.; Chalk, J.B. Contralateral hemisphere δ EEG in acute stroke precedes worsening of symptoms and death. Clin. Neurophysiol. 2008, 119, 1690–1694. [Google Scholar] [CrossRef] [PubMed]
- Burghaus, L.; Hilker, R.; Dohmen, C.; Bosche, B.; Winhuisen, L.; Galldiks, N.; Szelies, B.; Heiss, W.D. Early electroencephalography in acute ischemic stroke: Prediction of a malignant course? Clin. Neurol. Neurosurg. 2007, 109, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Burghaus, L.; Liu, W.C.; Dohmen, C.; Haupt, W.F.; Fink, G.R.; Eggers, C. Prognostic value of electroencephalography and evoked potentials in the early course of malignant middle cerebral artery infarction. Neurol. Sci. 2013, 34, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Finnigan, S.P.; Rose, S.E.; Chalk, J.B. Rapid EEG changes indicate reperfusion after tissue plasminogen activator injection in acute ischaemic stroke. Clin. Neurophysiol. 2006, 117, 2338–2339. [Google Scholar] [CrossRef] [PubMed]
- De Vos, C.C.; van Maarseveen, S.M.; Brouwers, P.J.; van Putten, M.J. Continuous EEG monitoring during thrombolysis in acute hemispheric stroke patients using the brain symmetry index. J. Clin. Neurophysiol. 2008, 25, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Gureyev, T.; Nesterets, Y.; Ma, H.; Thyagarajan, D. Novel application of EEG source localization in the assessment of the penumbra. Cerebrovasc. Dis. 2012, 33, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Bricolo, A.; Turazzi, S.; Faccioli, F. Combined clinical and EEG examinations for assessment of severity of acute head injuries. Acta Neurochir. Suppl. 1979, 28, 35–39. [Google Scholar] [PubMed]
- Bergamasco, B.; Bergamini, L.; Doriguzzi, T.; Sacerdote, I. The sleep cycle in coma: Prognostic value. Electroencephalogr. Clin. Neurophysiol. 1968, 25, 87. [Google Scholar] [PubMed]
- Cillessen, J.P.; van Huffelen, A.C.; Kappelle, L.J.; Algra, A.; van Gijn, J. Electroencephalography improves the prediction of functional outcome in the acute stage of cerebral ischemia. Stroke 1994, 25, 1968–1972. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, M.J.; Tavy, D.L. Continuous quantitative EEG monitoring in hemispheric stroke patients using the brain symmetry index. Stroke 2004, 35, 2489–2492. [Google Scholar] [CrossRef] [PubMed]
- Finnigan, S.P.; Walsh, M.; Rose, S.E.; Chalk, J.B. Quantitative EEG indices of sub-acute ischaemic stroke correlate with clinical outcomes. Clin. Neurophysiol. 2007, 118, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, F.; Pasqualetti, P.; Zappasodi, F.; Tombini, M.; Lupoi, D.; Vernieri, F.; Rossini, P.M. Outcome prediction in acute monohemispheric stroke via magnetoencephalography. J. Neurol. 2007, 254, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Assenza, G.; Zappasodi, F.; Pasqualetti, P.; Vernieri, F.; Tecchio, F. A contralesional EEG power increase mediated by interhemispheric disconnection provides negative prognosis in acute stroke. Restor. Neurol. Neurosci. 2013, 31, 177–188. [Google Scholar] [PubMed]
- Dubovik, S.; Ptak, R.; Aboulafia, T.; Magnin, C.; Gillabert, N.; Allet, L.; Pignat, J.M.; Schnider, A.; Guggisberg, A.G. EEG α band synchrony predicts cognitive and motor performance in patients with ischemic stroke. Behav. Neurol. 2013, 26, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.T.; Wei, L.; Rovainen, C.M.; Woolsey, T.A. New patterns of intracortical projections after focal cortical stroke. Neurobiol. Dis. 2001, 8, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Kilic, E.; Baumann, C.R.; Hermann, D.M.; Bassetti, C.L. γ-hydroxybutyrate accelerates functional recovery after focal cerebral ischemia. Cerebrovasc. Dis. 2008, 26, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Cam, E.; Jaeger, H.; Zunzunegui, C.; Sarnthein, J.; Bassetti, C.L. Sleep disruption aggravates focal cerebral ischemia in the rat. Sleep 2010, 33, 879–887. [Google Scholar] [PubMed]
- Zunzunegui, C.; Gao, B.; Cam, E.; Hodor, A.; Bassetti, C.L. Sleep disturbance impairs stroke recovery in the rat. Sleep 2011, 34, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.S.; Jordan, S.E.; Nuwer, M.R.; Marcus, D.R.; Moore, W.S. Computed electroencephalographic topographic brain mapping. A new and accurate monitor of cerebral circulation and function for patients having carotid endarterectomy. J. Vasc. Surg. 1988, 8, 247–254. [Google Scholar] [CrossRef]
- Vespa, P.M.; Nuwer, M.R.; Juhasz, C.; Alexander, M.; Nenov, V.; Martin, N.; Becker, D.P. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr. Clin. Neurophysiol. 1997, 103, 607–615. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Deschenes, M. Projections to layer VI of the posteromedial barrel field in the rat: A reappraisal of the role of corticothalamic pathways. Cereb. Cortex. 1998, 8, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.K.; Konig, P.; Kreiter, A.K.; Singer, W. Interhemispheric synchronization of oscillatory neuronal responses in cat visual cortex. Science 1991, 252, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, J.J.; Buzsaki, G. γ oscillations in the entorhinal cortex of the freely behaving rat. J. Neurosci. 1998, 18, 388–398. [Google Scholar] [PubMed]
- Tort, A.B.; Komorowski, R.; Eichenbaum, H.; Kopell, N. Measuring phase-amplitude coupling between neuronal oscillations of different frequencies. J. Neurophysiol. 2010, 104, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Azcarate, J.; Nicolas, M.J.; Cordon, I.; Alegre, M.; Valencia, M.; Artieda, J. δ-mediated cross-frequency coupling organizes oscillatory activity across the rat cortico-basal ganglia network. Front. Neural Circuits 2013, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.W.; Crunelli, V. Thalamic mechanisms of EEG α rhythms and their pathological implications. Neuroscientist 2005, 11, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Voytek, B.; Knight, R.T. Dynamic Network Communication as a Unifying Neural Basis for Cognition, Development, Aging, and Disease. Biol. Psychiatry 2015, 77, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Jhung, K.; Lee, J.; An, S.K. θ-γ coupling during a working memory task as compared to a simple vigilance task. Neurosci. Lett. 2013, 532, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Hillis, A.E.; Qureshi, A.; Hillis, A.E. Working Memory Dysfunction in Stroke Patients. The Behavioral and Cognitive Neurology of Stroke; Cambridge University Press: Cambridge, UK, 2013; pp. 297–311. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabiller, G.; He, J.-W.; Nishijima, Y.; Wong, A.; Liu, J. Perturbation of Brain Oscillations after Ischemic Stroke: A Potential Biomarker for Post-Stroke Function and Therapy. Int. J. Mol. Sci. 2015, 16, 25605-25640. https://doi.org/10.3390/ijms161025605
Rabiller G, He J-W, Nishijima Y, Wong A, Liu J. Perturbation of Brain Oscillations after Ischemic Stroke: A Potential Biomarker for Post-Stroke Function and Therapy. International Journal of Molecular Sciences. 2015; 16(10):25605-25640. https://doi.org/10.3390/ijms161025605
Chicago/Turabian StyleRabiller, Gratianne, Ji-Wei He, Yasuo Nishijima, Aaron Wong, and Jialing Liu. 2015. "Perturbation of Brain Oscillations after Ischemic Stroke: A Potential Biomarker for Post-Stroke Function and Therapy" International Journal of Molecular Sciences 16, no. 10: 25605-25640. https://doi.org/10.3390/ijms161025605
APA StyleRabiller, G., He, J.-W., Nishijima, Y., Wong, A., & Liu, J. (2015). Perturbation of Brain Oscillations after Ischemic Stroke: A Potential Biomarker for Post-Stroke Function and Therapy. International Journal of Molecular Sciences, 16(10), 25605-25640. https://doi.org/10.3390/ijms161025605