
Pacemaker Activity of the Human Sinoatrial Node: An Update on the Effects of Mutations in HCN4 on the Hyperpolarization-Activated Current



Abstract
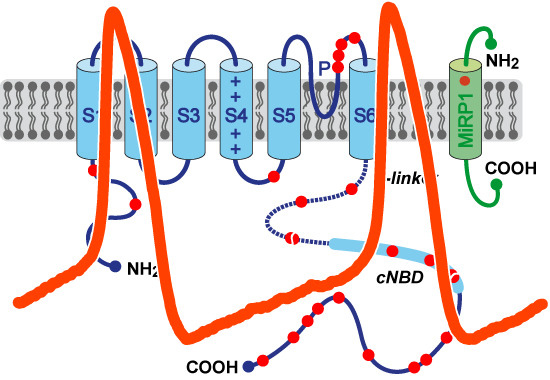
:
1. HCN4 and Familial Sick Sinus Syndrome
2. Mutations in HCN4 and KCNE2 Associated with Sinus Node Dysfunction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Mutation Carriers | Clinical Presentation | Study | |
|---|---|---|---|---|
| Mutations in HCN4 | ||||
| P257S | single index patient (65-year-old male) | diagnosed with paroxysmal AF at age 29; AF became permanent at age 43 years; sinus node dysfunction during ajmaline test in proband and proband’s father; 73 pauses >2.0 s on 24-h Holter monitoring | Macri et al. [26] | |
| A414G | 3 members of a single family | AF and LVNC in 74-year-old male index patient; combined sinus bradycardia and LVNC in his two mutation-carrying sons; severe sinus bradycardia involving 12 episodes of standstill on Holter monitoring in one of the sons | Milano et al. [27] | |
| G480R | 8 members of a single family | asymptomatic sinus bradycardia from a young age, with normal chronotropic and exercise capacity; minimum, average and maximum heart rates of 31 ± 8, 48 ± 12 and 101 ± 21 beats/min, respectively, in the 8 mutation carriers vs. 55 ± 9, 73 ± 11 and 126 ± 16 beats/min, respectively, in the 8 non-carriers | Nof et al. [21] | |
| Y481H | 4 members of two families with a common ancestral haplotype | combined sinus bradycardia and LVNC; frequent episodes of severe bradycardia (heart rate < 30 beats/min) and pacemaker implantation in the index patient of the first family; severe sinus bradycardia (40 beats/min) in the index patient of the second family and pacemaker implantation in his mutation-carrying mother because of bradyarrhythmias | Milano et al. [27] | |
| G482R | 6 members of a single family | combined sinus bradycardia and LVNC; pacemaker implanted in three mutation carriers because of bradycardia-related symptoms (average heart rate of 46 beats/min); MVP in two individuals | Milano et al. [27] | |
| G482R | 3 members of a single German family | combined sinus bradycardia, LVNC, and MVP; minimum and average heart rates of 21 and 34 beats/min, respectively, and pacemaker implantation in the index patient | Schweizer et al. [28] | |
| A485V | 14 members of three Moroccan Jewish decent families | symptomatic familial sinus bradycardia with normal chronotropic and exercise capacity; minimum, average and maximum heart rates of 37 ± 3, 58 ± 6 and 117 ± 27 beats/min in the 14 mutation carriers, respectively, vs. 49 ± 11, 77 ± 12 and 140 ± 32 beats/min, respectively, in the 6 non-carriers | Laish-Farkash et al. [23] | |
| K530N | 6 members of a single family | mild, asymptomatic sinus bradycardia (50–60 beats/min) in the index patient; familial age-dependent tachycardia-bradycardia syndrome and persistent AF; no AF or any other relevant cardiac arrhythmia in non-carriers | Duhme et al. [24] | |
| D553N | single index patient (43-year-old female) and two family members | wide spectrum of cardiac arrhythmias, including severe bradycardia (24-h average of 39 beats/min), QT prolongation and Torsade de Pointes in the index patient; QT prolongation in family members | Ueda et al. [19] | |
| 573X | single index patient (66-year-old female) | idiopathic sinus bradycardia of 41 beats/min; chronotropic incompetence; intermittent episodes of AF | Schulze-Bahr et al. [2] | |
| S672R | 15 members of a single Italian family | asymptomatic sinus bradycardia; average resting heart rate, corrected for age and gender, of 52.2 ± 1.4 beats/min (range 43–60 beats/min), in the 15 mutation carriers vs. 73.2 ± 1.6 beats/min (range 64–81 beats/min) in the 12 non-affected family members | Milanesi et al. [20] | |
| 695X | 8 members of a single German family | marked sinus bradycardia with no signs of chronotropic incompetence; basal heart rate of 45.9 ± 4.6 beats/min (range 38–51 beats/min) in the 8 mutation carriers vs. 66.5 ± 9.1 beats/min in the 6 non-carriers; minimum heart rate of 35.9 ± 5.6 vs. 47.2 ± 5.9 beats/min; maximum heart rates of 160.3 ± 12.6 vs. 171.8 ± 18.7 beats/min; LVNC in 5 of the mutation carriers | Schweizer et al. [22,28] | |
| P883R | single male patient | sinus bradycardia (35 to 40 beats/min), paroxysmal AF (tachycardia-bradycardia syndrome) and LVNC; pacemaker implantation | Schweizer et al. [28] | |
| G1097W | single index patient (69-year-old male) | complete AV block with wide QRS, but no sinus nodal dysfunction; atrial rate of 132 beats/min; ventricular rate of 33 beats/min; pacemaker implantation at the age of 51 years | Zhou et al. [25] | |
| Mutations in KCNE2 | ||||
| M54T | single index patient (55-year-old Caucasian male) | history of marked sinus bradycardia; average heart rate of 43 beats/min (range 30–125 beats/min), along with pauses; daughter died suddenly at the age of 13, and post-mortem genetic testing revealed the M54T mutation | Nawathe et al. [29] | |
2.1. HCN4-K189R
| Mutation | Type of Expression | Expression System | Shift in V1/2 or Activation Threshold (mV) | Slope Factor (mV) | Time Constant of Activation | Time Constant of Deactivation | Reversal Potential | Full-Activated Current Density | Channel Expression | Sensitivity to cAMP | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutations and Variants in HCN4 | |||||||||||
| K189R | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| P257S | heteromeric | CHO | = | = | ? | ? | ? | = | ≈50% | ? | Macri et al. [26] |
| homomeric | CHO | 0% | ↓ | Macri et al. [26] | |||||||
| A414G | heteromeric | CHO | −23.9 | = | ? | ? | ? | = | ? | ? | Milano et al. [27] |
| G480R | heteromeric | oocyte, HEK | ≈−15 | ? | ↑ a | ? | = | ≈50% | ? | = b | Nof et al. [21] |
| homomeric | oocyte, HEK | ≈−30 | ? | ↑ a | ? | = | ≈12% | ↓ | = b | Nof et al. [21] | |
| oocyte | ? | ? | ? | ? | ? | ≈20% | ? | ? | Laish-Farkash et al. [23] | ||
| Y481H | heteromeric | CHO | −43.9 | = | ? | ? | ? | = | ? | ? | Milano et al. [27] |
| G482R | heteromeric | HEK | = | = | = | = | = | 35% | = | ? | Schweizer et al. [28] |
| CHO | −38.7 | = | ? | ? | ? | = | ? | ? | Milano et al. [27] | ||
| homomeric | HEK | ? | ? | ? | ? | ? | 6% | = | ? | Schweizer et al. [28] | |
| A485V | heteromeric | oocyte, HEK | ≈−30 | ? | ↑ a | ↑ a | = | ≈33% | = | ? | Laish-Farkash et al. [23] |
| homomeric | oocyte, HEK | ≈−60 | ? | ↑ a | ↑ a | = | ≈5% | = | ↓ | Laish-Farkash et al. [23] | |
| K530N | heteromeric | HEK | ≈−14 | = | 237% | = | = | = | ? | ↑ | Duhme et al. [24] |
| homomeric | HEK | = | = | = | = | = | = | ? | = | Duhme et al. [24] | |
| D553N | heteromeric | COS | = | = | ≈90% | ≈110% | ? | ≈37% | ↓ | ? | Ueda et al. [19] |
| oocyte, COS | ? | ? | ? | ? | ? | ≈54% | = | ? | Netter et al. [34] | ||
| homomeric | COS | = | = | ≈90% | ≈110% | ? | ≈8% | ↓ | ? | Ueda et al. [19] | |
| oocyte, COS | = | = | = | = | ? | ≈12% | = | ↓ | Netter et al. [34] | ||
| 573X | heteromeric | COS | = c | −1.9 c | = | ? | ? | ? | = | ↓ | Schulze-Bahr et al. [2] |
| homomeric | COS | −4.6 c | = | = | ? a | ? | ? | = | ↓ | Schulze-Bahr et al. [2] | |
| S672R | heteromeric | HEK | −4.9 | = | = | ≈74% | ? | ? | ? | ? | Milanesi et al. [20] |
| homomeric | HEK | −8.4 | = | = | ≈63% | ? | ? | ? | = | Milanesi et al. [20] | |
| oocyte | −6.1 | ? | ≈180% | ≈90% | ? | ? | ? | ↓ | Xu et al. [32] | ||
| N688S | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| 695X | heteromeric | HEK | = | = | = | ? | ? | = | ? | ↓ | Schweizer et al. [22] |
| homomeric | HEK | = | −3.5 | 72% | = | = | ? | ? | ↓ | Schweizer et al. [22] | |
| T822M | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| G885R | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| P945S | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| A1045V | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| R1068H | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| G1077S | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| G1097W | heteromeric | CHO | −7.6 | = | ? | 81% | ? | 55% | ? | ? | Zhou et al. [25] |
| homomeric | CHO | −12 | = | ? | 79% | ? | 47% | ? | = | Zhou et al. [25] | |
| E1193Q | homomeric | CHO | = | = | ? | ? | ? | = | ? | ? | Macri et al. [26] |
| Mutations in KCNE2 | |||||||||||
| M54T | homomeric | NRVM | = | = | 192% | = | ? | 18% | ? | ? | Nawathe et al. [29] |
2.2. HCN4-P257S
2.3. HCN4-A414G
2.4. HCN4-G480R
2.5. HCN4-Y481H
2.6. HCN4-G482R
2.7. HCN4-A485V
2.8. HCN4-K530N
2.9. HCN4-D553N
2.10. HCN4-573X
2.11. HCN4-S672R
2.12. HCN4-N688S
2.13. HCN4-695X
2.14. HCN4-T822M
2.15. HCN4-P883R
2.16. HCN4-G885R, -P945S, -A1045V, -R1068H and -G1077S
2.17. HCN4-G1097W
2.18. HCN4-E1193Q
2.19. KCNE2-M54T
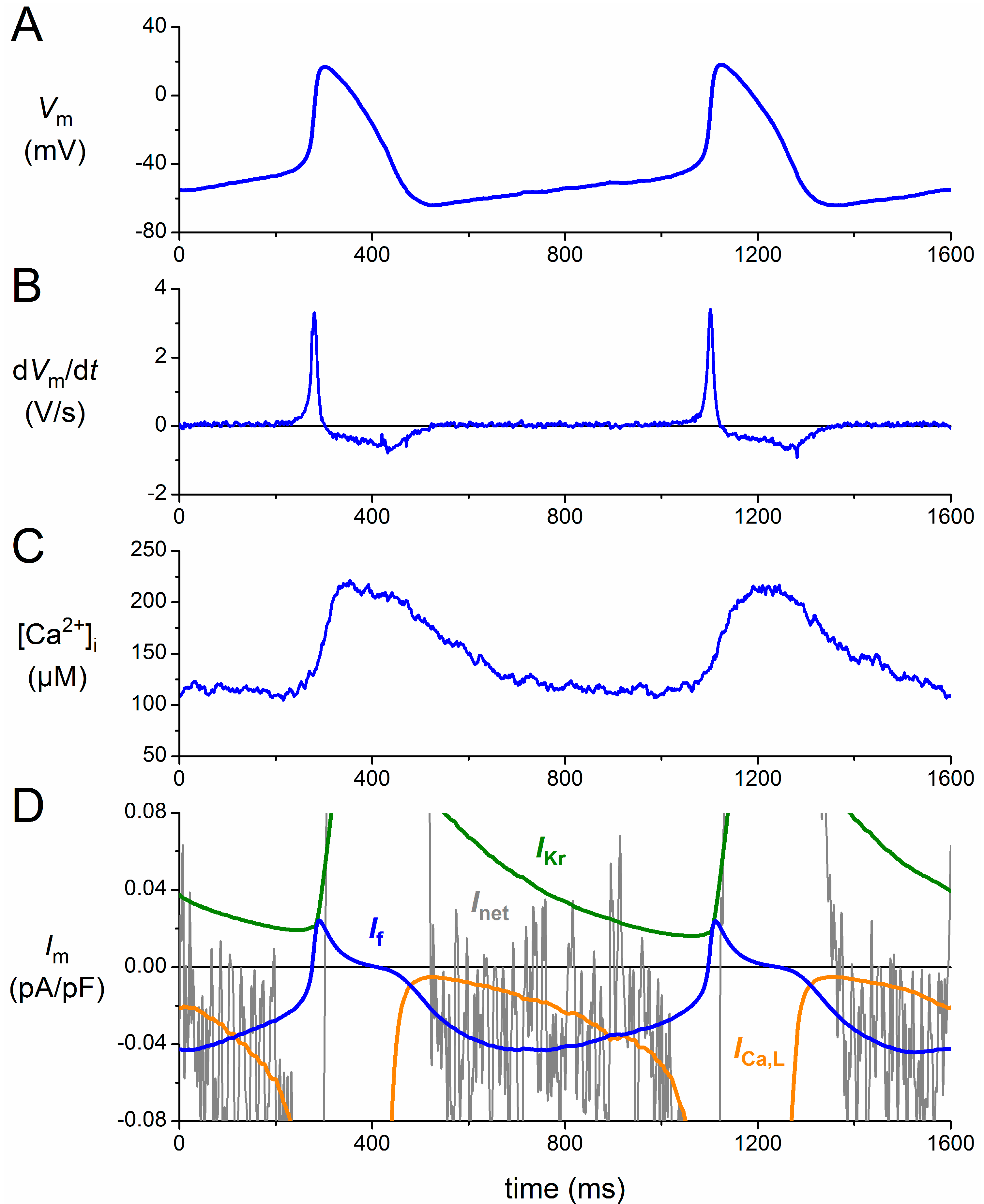
3. Functional Effects of Novel HCN4 Mutations on Human If
3.1. Numerical Reconstruction of If

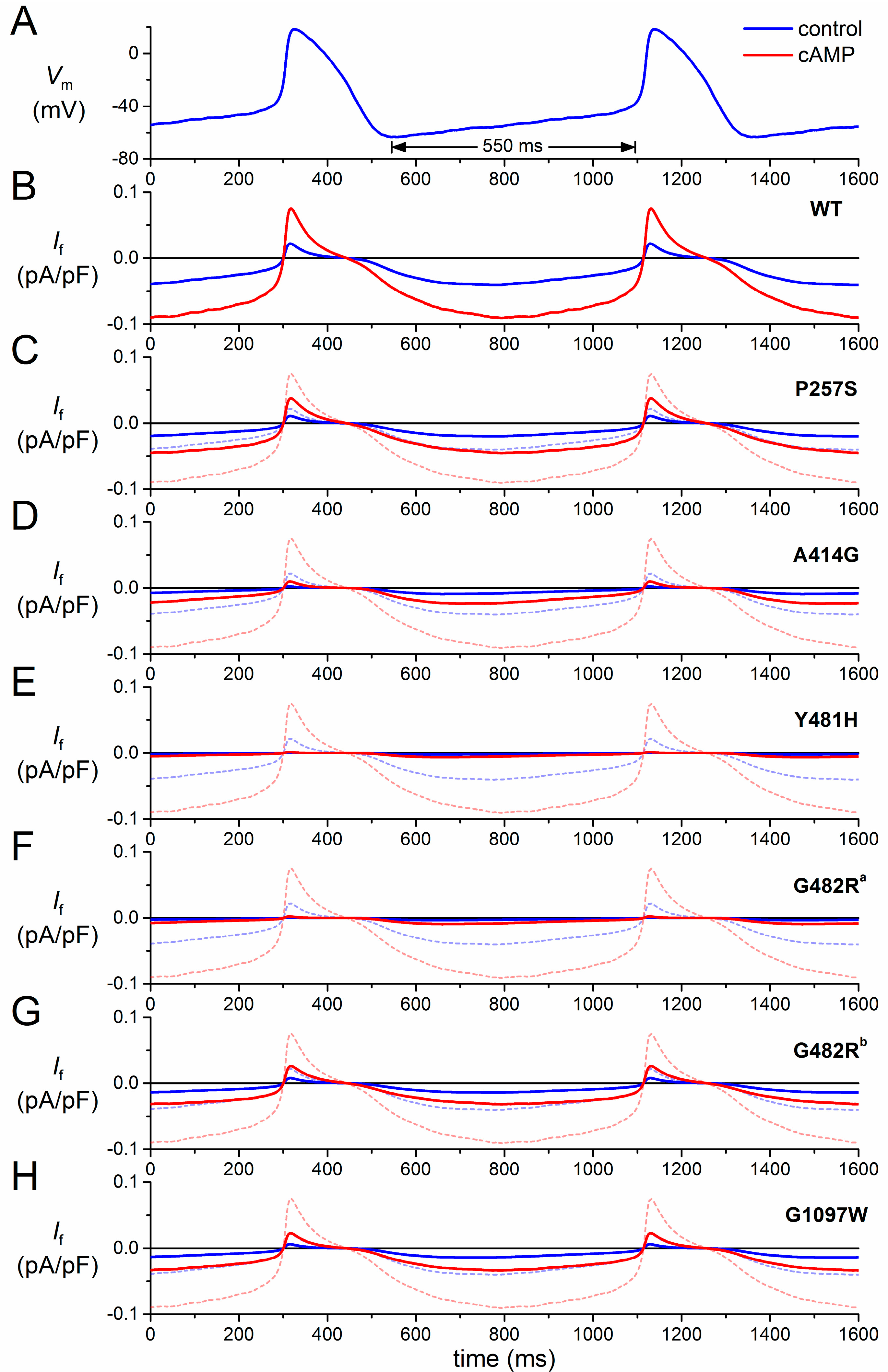
3.2. HCN4-P257S
3.3. HCN4-A414G
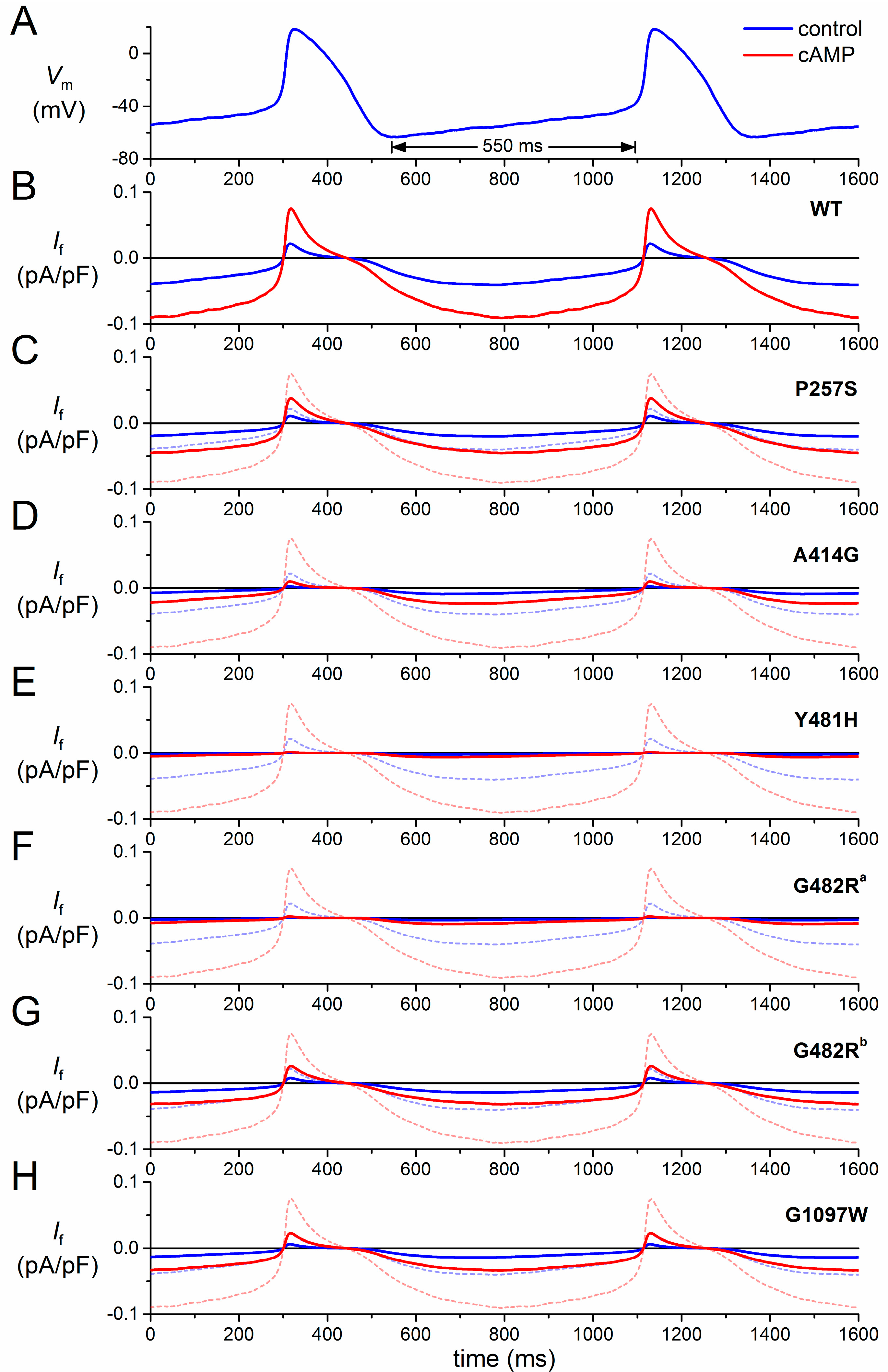

| Mutation | Scaling Factor for If Conductance | Shift (mV) | Shift with cAMP (mV) |
|---|---|---|---|
| HCN4-P257S | 0.50 | 0 | +15 |
| HCN4-A414G | 1 | −23.9 | −8.9 |
| HCN4-G480R | 0.50 | −15 | 0 |
| HCN4-Y481H | 1 | −43.9 | −28.9 |
| HCN4-G482R a | 1 | −38.7 | −23.7 |
| HCN4-G482R b | 0.35 | 0 | +15 |
| HCN4-A485V | 0.33 | −30 | −15 |
| HCN4-K530N | 1 | −14 | +7.8 |
| HCN4-D553N | 0.37 | 0 | +15 |
| HCN4-573X | 1 | 0 | 0 |
| HCN4-S672R | 1 | −4.9 | +10.1 |
| HCN4-695X | 1 | 0 | 0 |
| HCN4-G1097W | 0.55 | −7.6 | +7.4 |
| KCNE2-M54T | 0.18 | 0 | +15 |
3.4. HCN4-Y481H
3.5. HCN4-G482R
3.6. HCN4-G1097W
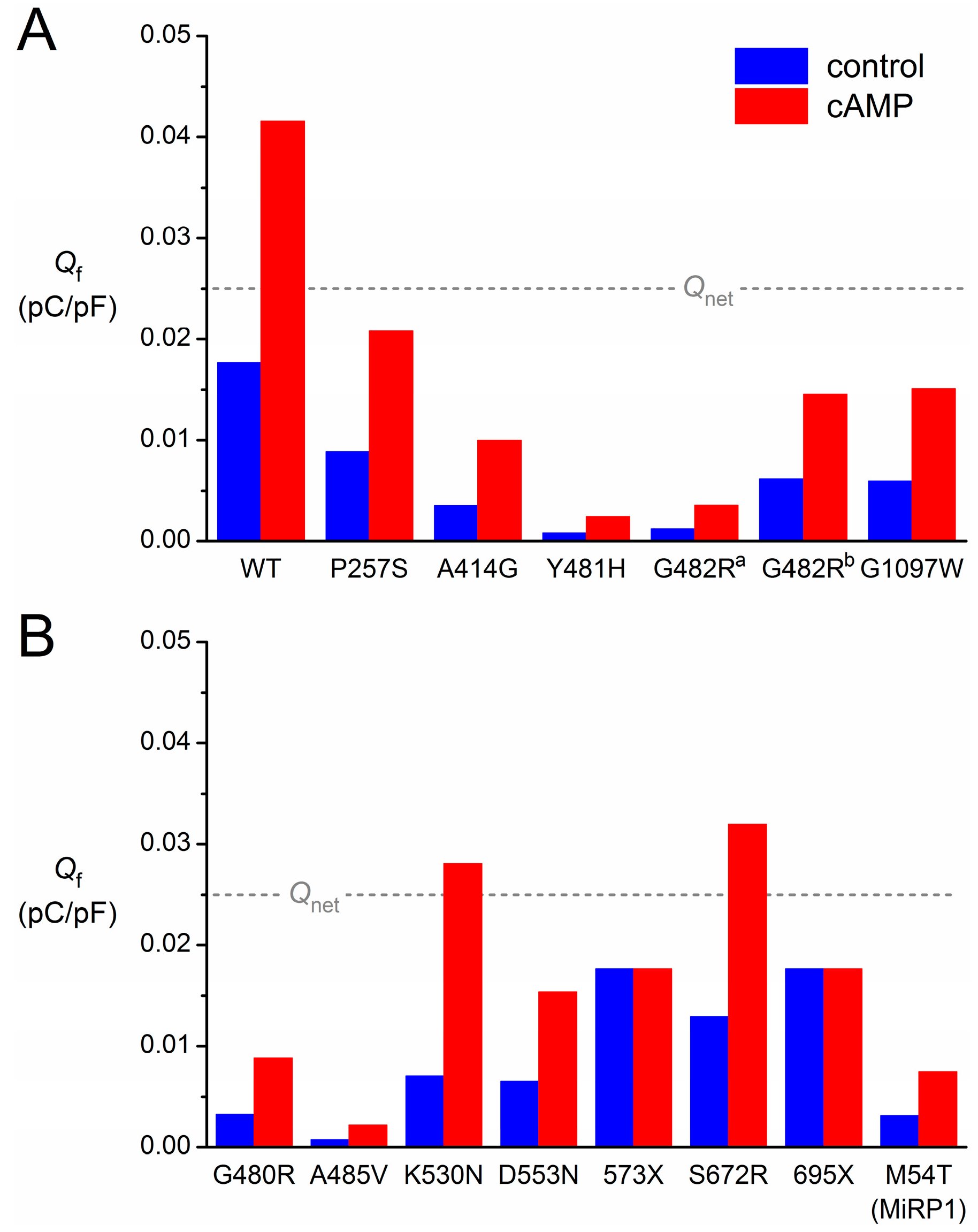
3.7. Limitations in the Reconstruction of If
4. Some Concluding Remarks
Author Contributions
Conflicts of Interest
References
- Bigger, J.T., Jr.; Reiffel, J.A. Sick sinus syndrome. Annu. Rev. Med. 1979, 30, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Bahr, E.; Neu, A.; Friederich, P.; Kaupp, U.B.; Breithardt, G.; Pongs, O.; Isbrandt, D. Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Investig. 2003, 111, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Stieber, J.; Hofmann, F.; Ludwig, A. Pacemaker channels and sinus node arrhythmia. Trends Cardiovasc. Med. 2004, 14, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; van Ginneken, A.C.G.; Wilders, R. Pacemaker activity of the human sinoatrial node: Role of the hyperpolarization-activated current, If. Int. J. Cardiol. 2009, 132, 318–336. [Google Scholar] [CrossRef] [PubMed]
- Nof, E.; Antzelevitch, C.; Glikson, M. The contribution of HCN4 to normal sinus node function in humans and animal models. Pacing Clin. Electrophysiol. 2010, 33, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bottelli, G.; Milanesi, R.; DiFrancesco, J.C.; DiFrancesco, D. HCN-related channelopathies. Pflüg. Arch. 2010, 460, 405–415. [Google Scholar] [CrossRef]
- DiFrancesco, D. Funny channel gene mutations associated with arrhythmias. J. Physiol. 2013, 591, 4117–4124. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Chen, F.; Li, B.; Hu, Z. Neurophysiology of HCN channels: From cellular functions to multiple regulations. Prog. Neurobiol. 2014, 112, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M. Cortical HCN channels: Function, trafficking and plasticity. J. Physiol. 2014, 592, 2711–2719. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Schnorr, S.; Ludwig, A. HCN channels—Modulators of cardiac and neuronal excitability. Int. J. Mol. Sci. 2015, 16, 1429–1447. [Google Scholar] [CrossRef] [PubMed]
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; DiFrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation 2009, 119, 1562–1575. [Google Scholar] [CrossRef] [PubMed]
- Den Hoed, M.; Eijgelsheim, M.; Esko, T.; Brundel, B.J.J.M.; Peal, D.S.; Evans, D.M.; Nolte, I.M.; Segrè, A.V.; Holm, H.; Handsaker, R.E.; et al. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat. Genet. 2013, 45, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, A.; Bucchi, A.; Johnsen, A.B.; Logantha, S.J.R.J.; Monfredi, O.; Yanni, J.; Prehar, S.; Hart, G.; Cartwright, E.; Wisloff, U.; et al. Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat. Commun. 2014, 5, 3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zicha, S.; Fernández-Velasco, M.; Lonardo, G.; L’Heureux, N.; Nattel, S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc. Res. 2005, 66, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, P.; Du, Y.; Zhang, J.; Ma, A. Age-related down-regulation of HCN channels in rat sinoatrial node. Basic Res. Cardiol. 2007, 102, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Ellinor, P.T.; Lunetta, K.L.; Albert, C.M.; Glazer, N.L.; Ritchie, M.D.; Smith, A.V.; Arking, D.E.; Müller-Nurasyid, M.; Krijthe, B.P.; Lubitz, S.A.; et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat. Genet. 2012, 44, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Fenske, S.; Krause, S.C.; Hassan, S.I.; Becirovic, E.; Auer, F.; Bernard, R.; Kupatt, C.; Lange, P.; Ziegler, T.; Wotjak, C.T.; et al. Sick sinus syndrome in HCN1-deficient mice. Circulation 2013, 128, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Budde, T.; Stieber, J.; Moosmang, S.; Wahl, C.; Holthoff, K.; Langebartels, A.; Wotjak, C.; Munsch, T.; Zong, X.; et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003, 22, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakamura, K.; Hayashi, T.; Inagaki, N.; Takahashi, M.; Arimura, T.; Morita, H.; Higashiuesato, Y.; Hirano, Y.; Yasunami, M.; et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 2004, 279, 27194–27198. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, R.; Baruscotti, M.; Gnecchi-Ruscone, T.; DiFrancesco, D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 2006, 354, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Nof, E.; Luria, D.; Brass, D.; Marek, D.; Lahat, H.; Reznik-Wolf, H.; Pras, E.; Dascal, N.; Eldar, M.; Glikson, M. Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation 2007, 116, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, P.A.; Duhme, N.; Thomas, D.; Becker, R.; Zehelein, J.; Draguhn, A.; Bruehl, C.; Katus, H.A.; Koenen, M. cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ. Arrhythm. Electrophysiol. 2010, 3, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Laish-Farkash, A.; Glikson, M.; Brass, D.; Marek-Yagel, D.; Pras, E.; Dascal, N.; Antzelevitch, C.; Nof, E.; Reznik, H.; Eldar, M.; et al. A novel mutation in the HCN4 gene causes symptomatic sinus bradycardia in Moroccan Jews. J. Cardiovasc. Electrophysiol. 2010, 21, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Duhme, N.; Schweizer, P.A.; Thomas, D.; Becker, R.; Schröter, J.; Barends, T.R.; Schlichting, I.; Draguhn, A.; Bruehl, C.; Katus, H.A.; et al. Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur. Heart J. 2013, 34, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ding, W.G.; Makiyama, T.; Miyamoto, A.; Matsumoto, Y.; Kimura, H.; Tarutani, Y.; Zhao, J.; Wu, J.; Zang, W.J.; et al. A novel HCN4 mutation, G1097W, is associated with atrioventricular block. Circ. J. 2014, 78, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Macri, V.; Mahida, S.N.; Zhang, M.L.; Sinner, M.F.; Dolmatova, E.V.; Tucker, N.R.; McLellan, M.; Shea, M.A.; Milan, D.J.; Lunetta, K.L.; et al. A novel trafficking-defective HCN4 mutation is associated with early-onset atrial fibrillation. Heart Rhythm 2014, 11, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Milano, A.; Vermeer, A.M.C.; Lodder, E.M.; Barc, J.; Verkerk, A.O.; Postma, A.V.; van der Bilt, I.A.C.; Baars, M.J.H.; van Haelst, P.L.; Caliskan, K.; et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2014, 64, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, P.A.; Schröter, J.; Greiner, S.; Haas, J.; Yampolsky, P.; Mereles, D.; Buss, S.J.; Seyler, C.; Bruehl, C.; Draguhn, A.; et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J. Am. Coll. Cardiol. 2014, 64, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Nawathe, P.A.; Kryukova, Y.; Oren, R.V.; Milanesi, R.; Clancy, C.E.; Lu, J.T.; Moss, A.J.; DiFrancesco, D.; Robinson, R.B. An LQTS6 MiRP1 mutation suppresses pacemaker current and is associated with sinus bradycardia. J. Cardiovasc. Electrophysiol. 2013, 24, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R. Pacemaker activity of the human sinoatrial node: Effects of HCN4 mutations on the hyperpolarization-activated current. Europace 2014, 16, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.; Wrobel, E.; Strutz-Seebohm, N.; Schulze-Bahr, E.; Seebohm, G. Functional characterization of novel HCN4 mutations associated with cardiac arrhythmia. Acta Physiol. 2014, 210 (Suppl. s695), 224. [Google Scholar] [CrossRef]
- Ueda, K.; Hirano, Y.; Higashiuesato, Y.; Aizawa, Y.; Hayashi, T.; Inagaki, N.; Tana, T.; Ohya, Y.; Takishita, S.; Muratani, H.; et al. Role of HCN4 channel in preventing ventricular arrhythmia. J. Hum. Genet. 2009, 54, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.; Bagnall, R.D.; Duflou, J.; Semsarian, C. Postmortem review and genetic analysis in sudden infant death syndrome: An 11-year review. Hum. Pathol. 2013, 44, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Netter, M.F.; Zuzarte, M.; Schlichthörl, G.; Klöcker, N.; Decher, N. The HCN4 channel mutation D553N associated with bradycardia has a C-linker mediated gating defect. Cell. Physiol. Biochem. 2012, 30, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Singer-Lahat, D.; Lotan, I.; Biel, M.; Flockerzi, V.; Hofmann, F.; Dascal, N. Cardiac calcium channels expressed in Xenopus oocytes are modulated by dephosphorylation but not by cAMP-dependent phosphorylation. Recept. Channels 1994, 2, 215–226. [Google Scholar] [PubMed]
- Xu, X.; Marni, F.; Wu, S.; Su, Z.; Musayev, F.; Shrestha, S.; Xie, C.; Gao, W.; Liu, Q.; Zhou, L. Local and global interpretations of a disease-causing mutation near the ligand entry path in hyperpolarization-activated cAMP-gated channel. Structure 2012, 20, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R.; van Borren, M.M.G.J.; Peters, R.J.G.; Broekhuis, E.; Lam, K.; Coronel, R.; de Bakker, J.M.T.; Tan, H.L. Pacemaker current (If) in the human sinoatrial node. Eur. Heart J. 2007, 28, 2472–2478. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; van Borren, M.M.G.J.; Peters, R.J.G.; Broekhuis, E.; Lam, K.Y.; Coronel, R.; de Bakker, J.M.T.; Tan, H.L.; Wilders, R. Single cells isolated from human sinoatrial node: Action potentials and numerical reconstruction of pacemaker current. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2007, 2007, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R. Relative importance of funny current in human versus rabbit sinoatrial node. J. Mol. Cell. Cardiol. 2010, 48, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; van Borren, M.M.G.J.; Wilders, R. Calcium transient and sodium-calcium exchange current in human versus rabbit sinoatrial node pacemaker cells. Sci. World J. 2013, 2013, 507872. [Google Scholar] [CrossRef]
- Courtemanche, M.; Ramirez, R.J.; Nattel, S. Ionic mechanisms underlying human atrial action potential properties: Insights from a mathematical model. Am. J. Physiol. 1998, 275, H301–H321. [Google Scholar] [PubMed]
- Hoekstra, M.; Verkerk, A.O.; van Ginneken, A.C.G.; Wilders, R. HCN4 current during human sinoatrial node action potentials. Heart Rhythm 2010, 7, S410. [Google Scholar] [CrossRef]
- Wilders, R. Computer modelling of the sinoatrial node. Med. Biol. Eng. Comput. 2007, 45, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.H.; Burstein, B.; Qi, X.Y.; Sakabe, M.; Chartier, D.; Comtois, P.; Wang, Z.; Kuo, C.T.; Nattel, S. Funny current downregulation and sinus node dysfunction associated with atrial tachyarrhythmia: A molecular basis for tachycardia-bradycardia syndrome. Circulation 2009, 119, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Stillitano, F.; Lonardo, G.; Giunti, G.; del Lungo, M.; Coppini, R.; Spinelli, V.; Sartiani, L.; Poggesi, C.; Mugelli, A.; Cerbai, E. Chronic atrial fibrillation alters the functional properties of If in the human atrium. J. Cardiovasc. Electrophysiol. 2013, 24, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Greener, I.D.; Monfredi, O.; Inada, S.; Chandler, N.J.; Tellez, J.O.; Atkinson, A.; Taube, M.A.; Billeter, R.; Anderson, R.H.; Efimov, I.R.; et al. Molecular architecture of the human specialised atrioventricular conduction axis. J. Mol. Cell. Cardiol. 2011, 50, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Viscomi, C.; Mandelli, G.; Consalez, G.; Gnecchi-Rusconi, T.; Montano, N.; Casali, K.R.; Micheloni, S.; Barbuti, A.; et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. USA 2011, 108, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Sizarov, A.; Devalla, H.D.; Anderson, R.H.; Passier, R.; Christoffels, V.M.; Moorman, A.F.M. Molecular analysis of patterning of conduction tissues in the developing human heart. Circ. Arrhythm. Electrophysiol. 2011, 4, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, E.; Jenni, R. Left ventricular non-compaction revisited: A distinct phenotype with genetic heterogeneity? Eur. Heart J. 2011, 32, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Wahl-Schott, C.; Michalakis, S.; Zong, X. Hyperpolarization-activated cation channels: From genes to function. Physiol. Rev. 2009, 89, 847–885. [Google Scholar] [CrossRef] [PubMed]
- Barbuti, A.; Scavone, A.; Mazzocchi, N.; Terragni, B.; Baruscotti, M.; DiFrancesco, D. A caveolin-binding domain in the HCN4 channels mediates functional interaction with caveolin proteins. J. Mol. Cell. Cardiol. 2012, 53, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Decher, N.; Bundis, F.; Vajna, R.; Steinmeyer, K. KCNE2 modulates current amplitudes and activation kinetics of HCN4: Influence of KCNE family members on HCN4 currents. Pflüg. Arch. 2003, 446, 633–640. [Google Scholar] [CrossRef]
- Pian, P.; Bucchi, A.; Robinson, R.B.; Siegelbaum, S.A. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 2006, 128, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Barbuti, A.; Terragni, B.; Brioschi, C.; DiFrancesco, D. Localization of f-channels to caveolae mediates specific β2-adrenergic receptor modulation of rate in sinoatrial myocytes. J. Mol. Cell. Cardiol. 2007, 42, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Balijepalli, R.C.; Foell, J.D.; Kroboth, S.; Ye, Q.; Luo, Y.H.; Shi, N.Q. Caveolin-3 associates with and affects the function of hyperpolarization-activated cyclic nucleotide-gated channel 4. Biochemistry 2008, 47, 12312–12318. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.J.; Chow, S.S.; Angoli, D.; Nazzari, H.; Cayabyab, F.S.; Morshedian, A.; Accili, E.A. In situ co-distribution and functional interactions of SAP97 with sinoatrial isoforms of HCN channels. J. Mol. Cell. Cardiol. 2009, 46, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Altomare, C.; Terragni, B.; Brioschi, C.; Milanesi, R.; Pagliuca, C.; Viscomi, C.; Moroni, A.; Baruscotti, M.; DiFrancesco, D. Heteromeric HCN1-HCN4 channels: A comparison with native pacemaker channels from the rabbit sinoatrial node. J. Physiol. 2003, 549, 347–359. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkerk, A.O.; Wilders, R. Pacemaker Activity of the Human Sinoatrial Node: An Update on the Effects of Mutations in HCN4 on the Hyperpolarization-Activated Current. Int. J. Mol. Sci. 2015, 16, 3071-3094. https://doi.org/10.3390/ijms16023071
Verkerk AO, Wilders R. Pacemaker Activity of the Human Sinoatrial Node: An Update on the Effects of Mutations in HCN4 on the Hyperpolarization-Activated Current. International Journal of Molecular Sciences. 2015; 16(2):3071-3094. https://doi.org/10.3390/ijms16023071
Chicago/Turabian StyleVerkerk, Arie O., and Ronald Wilders. 2015. "Pacemaker Activity of the Human Sinoatrial Node: An Update on the Effects of Mutations in HCN4 on the Hyperpolarization-Activated Current" International Journal of Molecular Sciences 16, no. 2: 3071-3094. https://doi.org/10.3390/ijms16023071
APA StyleVerkerk, A. O., & Wilders, R. (2015). Pacemaker Activity of the Human Sinoatrial Node: An Update on the Effects of Mutations in HCN4 on the Hyperpolarization-Activated Current. International Journal of Molecular Sciences, 16(2), 3071-3094. https://doi.org/10.3390/ijms16023071