Abstract
We herein review available computational and experimental data pointing to the abundance of structural disorder within the nucleoprotein (N) and phosphoprotein (P) from three paramyxoviruses, namely the measles (MeV), Nipah (NiV) and Hendra (HeV) viruses. We provide a detailed molecular description of the mechanisms governing the disorder-to-order transition that the intrinsically disordered C-terminal domain (NTAIL) of their N proteins undergoes upon binding to the C-terminal X domain (PXD) of the homologous P proteins. We also show that NTAIL–PXD complexes are “fuzzy”, i.e., they possess a significant residual disorder, and discuss the possible functional significance of this fuzziness. Finally, we emphasize the relevance of N–P interactions involving intrinsically disordered proteins as promising targets for new antiviral approaches, and end up summarizing the general functional advantages of disorder for viruses.
1. Overview of the Replicative Complex of Paramyxoviruses
Negative-stranded RNA viruses (NSRVs) are causative agents of a large number of human and animal diseases with some of them being identified as potential agents of bioterrorism, and several being included in the NIAID (National Institute of Allergy and Infectious Diseases) and CDC (Center for Disease Control and Prevention) priority pathogen lists. Research on these viruses, as well as efforts aimed at the development of vaccines and antiviral drugs, have been paid an increased attention for many years. NSRVs can be divided into viruses with segmented RNA genomes and those with non-segmented RNA genomes. The latter are grouped within the Mononegavirales order. Among the viral families belonging to this order is the Paramyxoviridae family. The Nipah (NiV), Hendra (HeV) and measles (MeV) viruses belong to the Paramyxovirinae sub-family within the Paramyxoviridae family, where the latter also embraces the Pneumovirinae subfamily. Based on distinguishing nucleotide sequence features, the MeV and the NiV and HeV and have been classified into two distinct genera, the Morbillivirus genus and the Henipavirus genus, respectively [1,2,3].
Paramyxoviruses display a pleomorphic structure. The viral particule contains the non-segmented, negative-stranded RNA genome, which encodes for at least six proteins (Figure 1A). The genome of paramyxoviruses indeed encodes the fusion (F) and the attachment (H) glycoproteins, which are responsible for virus entry, the matrix (M), which is required for virus assembly and budding, and the proteins of the replicative complex (Figure 1B). In paramyxoviruses, RNA transcription and replication require an intricate interplay between three components: the RNA-dependent RNA polymerase (L), the phosphoprotein (P), and the nucleoprotein (N). As in all Mononegavirales members, the genome is encapsidated by N within a helical nucleocapsid. The N:RNA complex, rather than naked RNA, is the template for both transcription and replication. During RNA synthesis, P tethers L onto the N–RNA template through the N-P interaction. The complex formed by the N, P and L proteins constitutes the viral replicative unit, and these proteins are necessary and sufficient to sustain replication of viral RNA in Paramyxovirinae (Figure 1C) [4,5].
The N protein is the most abundant viral protein (Figure 1A). Within infected cells, the N protein from Paramyxoviridae members is found in a soluble, monomeric form (referred to as N°) and in a nucleocapsid assembled form (referred to as NNUC) [6,7]. Following synthesis of the N protein, a chaperone is required to maintain this latter protein in the unassembled form in the cytoplasm. This role is played by the P protein, whose association prevents illegitimate self-assembly of N and retains N in the cytoplasm [8,9]. This soluble N°-P complex is used as the substrate for the encapsidation of the nascent genomic RNA chain during replication. The assembled form of N also forms complexes with P, either isolated (NNUC-P) or bound to L (NNUC-P-L), which are essential to RNA synthesis by the viral polymerase. Hence, the components of the viral replication machinery, namely P, N and L, engage in a complex macromolecular ballet (Figure 1C) (see [2,5,10,11,12,13,14] for reviews on transcription and replication).
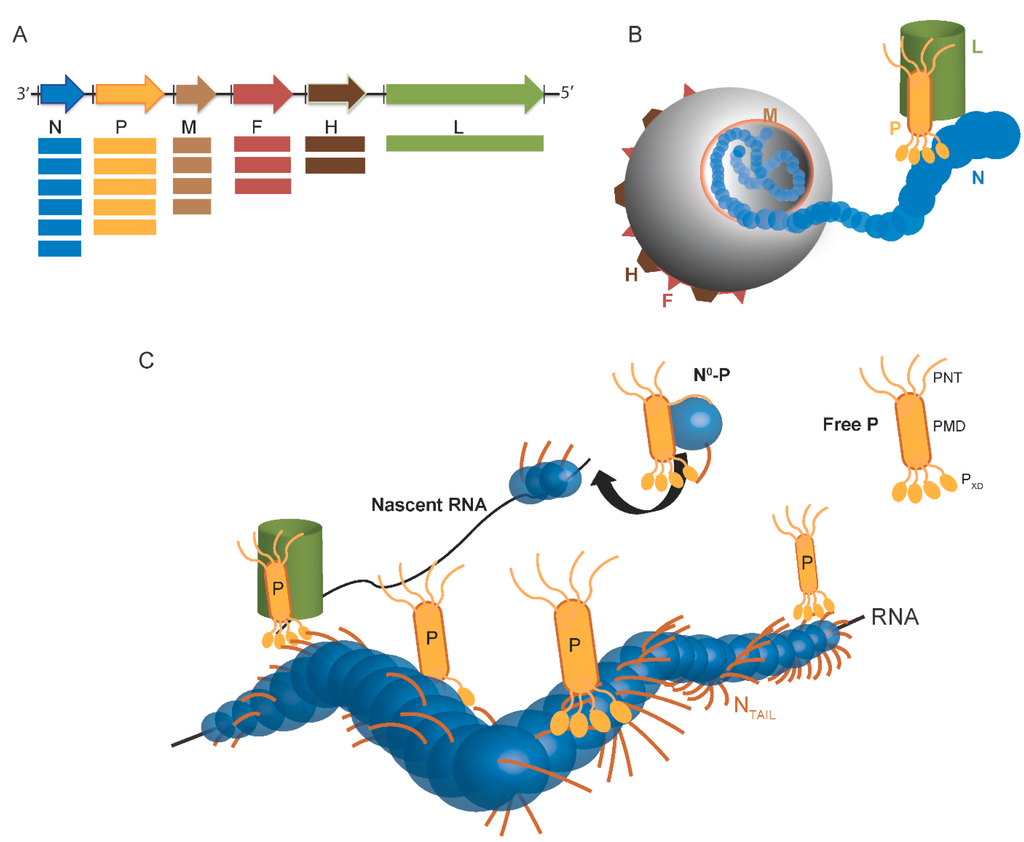
Figure 1.
Scheme of Paramyxovirinae genome and viral particule. (A) Schematic representation of the genome of Paramyxovirinae. The negative-sense genomic RNA is presented in the 3′ to 5′ orientation. The open reading frames are represented by colored arrows and encode the nucleocapsid protein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), attachment protein (H) and large polymerase protein (L). Vertical lines represent gene start and stop signals. Below the genome, shown is a schematic representation of the expression gradient of the encoded proteins as a result of the stop and re-initiation mechanism of the polymerase during transcription [2]; (B) Schematic illustration of the virion. The viral membrane is decorated by the F and H glycoproteins and M is located beneath the membrane. N is bound to genomic RNA and together with P and L forms the viral replication unit; (C) Schematic illustration of the Paramyxoviridae replicative complex. The RNA is represented as a solid black line. The neo-synthetized RNA is shown already partially encapsidated by N. The N and P intrinsically disordered regions are symbolized by lines. The extended conformation of the disordered regions is thought to allow the formation of a tripartite complex between N°, P and L required for nucleocapsid assembly. The P/L complex forms the RNA-dependent RNA polymerase (RdRp) complex that cartwheels onto the nucleocapsid complex via the X domain of P (PXD). P is shown as a tetramer to reflect the prevalence of this oligomeric state in paramyxoviral P proteins.
Figure 1.
Scheme of Paramyxovirinae genome and viral particule. (A) Schematic representation of the genome of Paramyxovirinae. The negative-sense genomic RNA is presented in the 3′ to 5′ orientation. The open reading frames are represented by colored arrows and encode the nucleocapsid protein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), attachment protein (H) and large polymerase protein (L). Vertical lines represent gene start and stop signals. Below the genome, shown is a schematic representation of the expression gradient of the encoded proteins as a result of the stop and re-initiation mechanism of the polymerase during transcription [2]; (B) Schematic illustration of the virion. The viral membrane is decorated by the F and H glycoproteins and M is located beneath the membrane. N is bound to genomic RNA and together with P and L forms the viral replication unit; (C) Schematic illustration of the Paramyxoviridae replicative complex. The RNA is represented as a solid black line. The neo-synthetized RNA is shown already partially encapsidated by N. The N and P intrinsically disordered regions are symbolized by lines. The extended conformation of the disordered regions is thought to allow the formation of a tripartite complex between N°, P and L required for nucleocapsid assembly. The P/L complex forms the RNA-dependent RNA polymerase (RdRp) complex that cartwheels onto the nucleocapsid complex via the X domain of P (PXD). P is shown as a tetramer to reflect the prevalence of this oligomeric state in paramyxoviral P proteins.
Although in vitro L can synthesize short RNA transcripts using naked RNA as substrate in the absence of P [15], in infected cells P is required to stabilize L and to allow recognition of the N:RNA template. L is thought to carry out most (if not all) enzymatic activities required for transcription and replication, including nucleotide polymerization, mRNA capping and polyadenylation. It is found in low amounts in infected cells and is unstable unless bound to the P protein, thus making its full characterization challenging [16]. Since no functional paramyxoviral polymerase has been biochemically characterized so far, most of our present knowledge arises from bioinformatics studies. Among Paramyxovirinae members, the only exceptions are represented by the L/P complex from two Paramyxovirinae members, namely Rinderpest virus (RDV) whose polymerase has been partially purified [17], and Sendai virus (SeV) whose polymerase was shown to possess a methyltransefrase activity in its C-terminal region [18]. Among Pneumovirinae, the only exception is the L protein from respiratory syncytial virus (RSV) that could be partially purified and whose RNA polymerase activity was documented in vitro [19]. In addition, minireplicon studies allowed the identification within RSV L of a flexible hinge region tolerating insertion and demonstrated the crucial role of the GDNQ motif [20].
Accordingly, most of our present knowledge of the replicative complex of paramyxoviruses concerns the N and P proteins. In the last decades, many efforts have been devoted to the molecular characterization of paramyxoviral N and P proteins. The N–P interaction has attracted much interest not only from a fundamental point of view, but also from a more applied perspective: in fact, since abolishing the N–P interaction prevents the recruitment of L onto the nucleocapsid template, this interaction is regarded as a potential target for antiviral approaches.
In the course of a thorough structural and functional characterization of paramyxoviral N and P proteins that made use of a wide range of bioinformatics and experimental approaches, we showed that these proteins are enriched in intrinsically disordered regions (IDRs) and that these IDRs play key roles in the formation of the tripartite N–P–L complex and in the establishment of a broad molecular partnership (for reviews see [11,21,22,23,24,25,26,27]). Our seminal observations on MeV P and N proteins fostered subsequent studies that brought awareness of the prevalence and conservation of structural disorder within paramyxoviruses N and P proteins thereby pointing to its functional significance.
Intrinsically disordered proteins (IDPs) and IDRs are widespread functional proteins/regions that lack stable structures under physiological conditions. Behind their inability to fold resides an important raison d’être, which is tightly coupled to specific features of their amino acid sequence. Indeed, a specific imbalance in the content of hydrophobic vs. polar residues in IDP/Rs, confers them the ability to populate a wide conformational space with conformations ranging from completely extended (i.e., random coils, RC) to more compact (i.e., pre-molten globules, PMGs, and molten globules, MGs). The nature of the conformational ensemble that IDP/Rs could sample is coupled to their function and, hence, to the interactions they establish with their partners. Indeed, in many cases, IDP/Rs fold upon binding leading to either stable complexes amenable to crystallization, or, more often, to fuzzy complexes [28], i.e., complexes with significant residual disorder. These peculiar characteristics award IDP/Rs a number of advantages over folded proteins that promote their frequent involvement in particular functions, such as for instance, hubs in protein interaction networks and cell signaling (for a recent review on IDP/Rs see [29]).
In the current review, we provide a detailed description of the molecular information that exist to date on the N and P proteins from paramyxoviruses while highlighting the unique role of structural disorder in ensuring an efficient replication and transcription of the paramyxoviral genome. We accordingly discuss through the manuscript the functional role of induced folding and residual flexibility in terms of transcription, replication and molecular partnership. We also underscore how targeting the N–P interaction holds promises for new antiviral approaches. We finally conclude by highlighting the functional implications and general advantages of structural disorder within viruses.
2. Abundance of Structural Disorder in Paramyxoviral N and P Proteins
2.1. Paramyxoviral P Proteins Are Highly Disordered
The P gene of Paramyxovirinae members illustrates how a virus encodes as much information as possible in a single gene. Indeed, the P gene can give rise to a number of different polypeptide products by means of either overlapping reading frames, or of a peculiar transcription process whereby one or more non-templated nucleotides are inserted, resulting in a shift of the reading frame during translation. Accordingly, the P genes encode at least two non-structural proteins (C and V) in addition to the P protein.
Although paramyxoviruses share a similar P modular organization, Henipavirus P proteins (707 amino acids in HeV and 709 amino acids in NiV), are much larger than those of other paramyxoviruses (507 amino acids in MeV) (Figure 2A) [3]. In all cases, computational analyses showed that the P proteins consist of an N-terminal domain (PNT) that is predicted to be intrinsically disordered (Figure 2B) and is also found in the V protein, and a C-terminal domain (PCT) that can be further subdivided in various regions [30,31]. The disordered state of MeV, NiV and HeV PNT was confirmed using a wide range of biochemical and biophysical approaches. Indeed, PNT domains were found (i) to be highly sensitive to proteolysis; (ii) To possess NOESY and circular dichroism (CD) spectra typical of IDPs (Figure 2C,D); (iii) To possess Stokes radii (RS) much larger than those expected for globular proteins with the same size and (iv) to gain structure upon addition of the secondary structure stabilizer 2,2,2-trifluoroethanol (TFE) (Figure 2C) [30,31,32]. Beyond PNT, other IDRs occur within the P protein. In fact, PCT is composed of alternating disordered and ordered regions (Figure 2A) [30,32]. As a result, as much as 70%–80% of the residues in MeV, NiV and HeV P proteins are disordered. Amongst the ordered domains in PCT are the P multimerisation domain (PMD) and the C-terminal X domain (PXD), which are responsible respectively for the oligomerization of P and for binding to the C-terminal domain of N (i.e., NTAIL) [33,34,35,36,37]. PMD and PXD are separated by a flexible linker region predicted to be poorly ordered [31]. Indeed, in the case of SeV, NMR studies carried out on the C-terminal region of P showed that the region upstream PXD is disordered [38,39,40]. Besides, an additional flexible region (referred to as “spacer”) occurs upstream PMD in MeV, NiV and HeV [30,31,41].
Paramyxoviridae P proteins are phosphorylated at multiple sites, with these phosphorylation sites being interestingly located within the disordered PNT domains [31,42,43,44]. This is in good agreement with the findings by Iakoucheva et al. [45], pointing out the importance of structural disorder for post-translational modifications (PTMs) such as phosphorylation. Indeed, PTM of proteins has three structural requirements: an appropriate local sequence, structural exposure, and flexibility of the site so that it can be productively accommodated within the active site of the modifying enzyme. These requirements are in an intimate relationship with structural disorder.
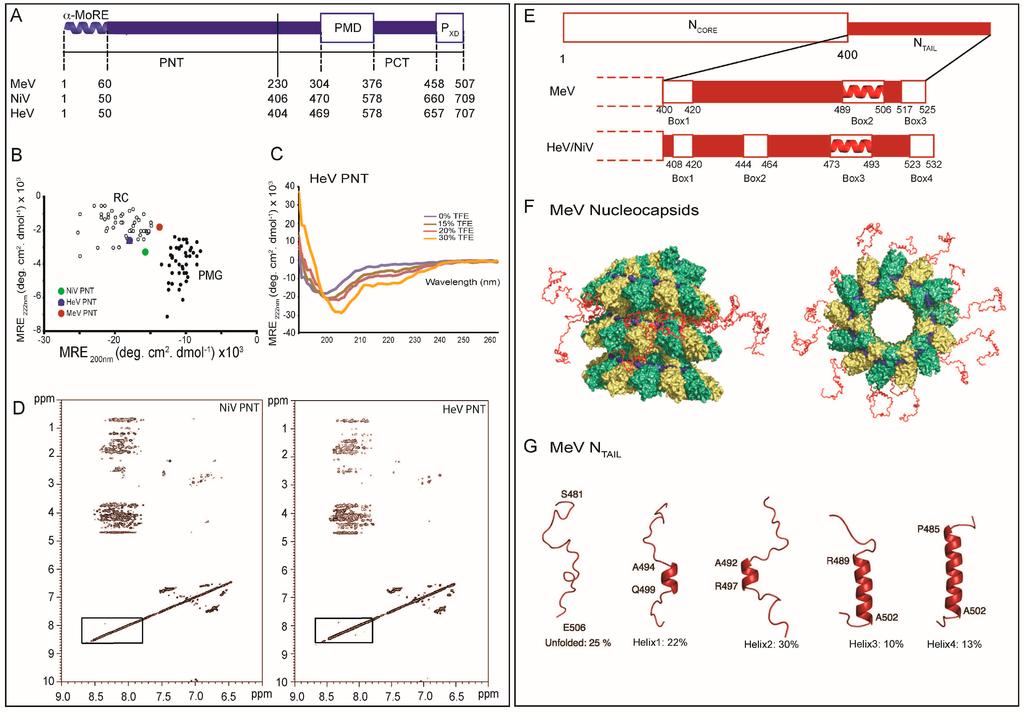
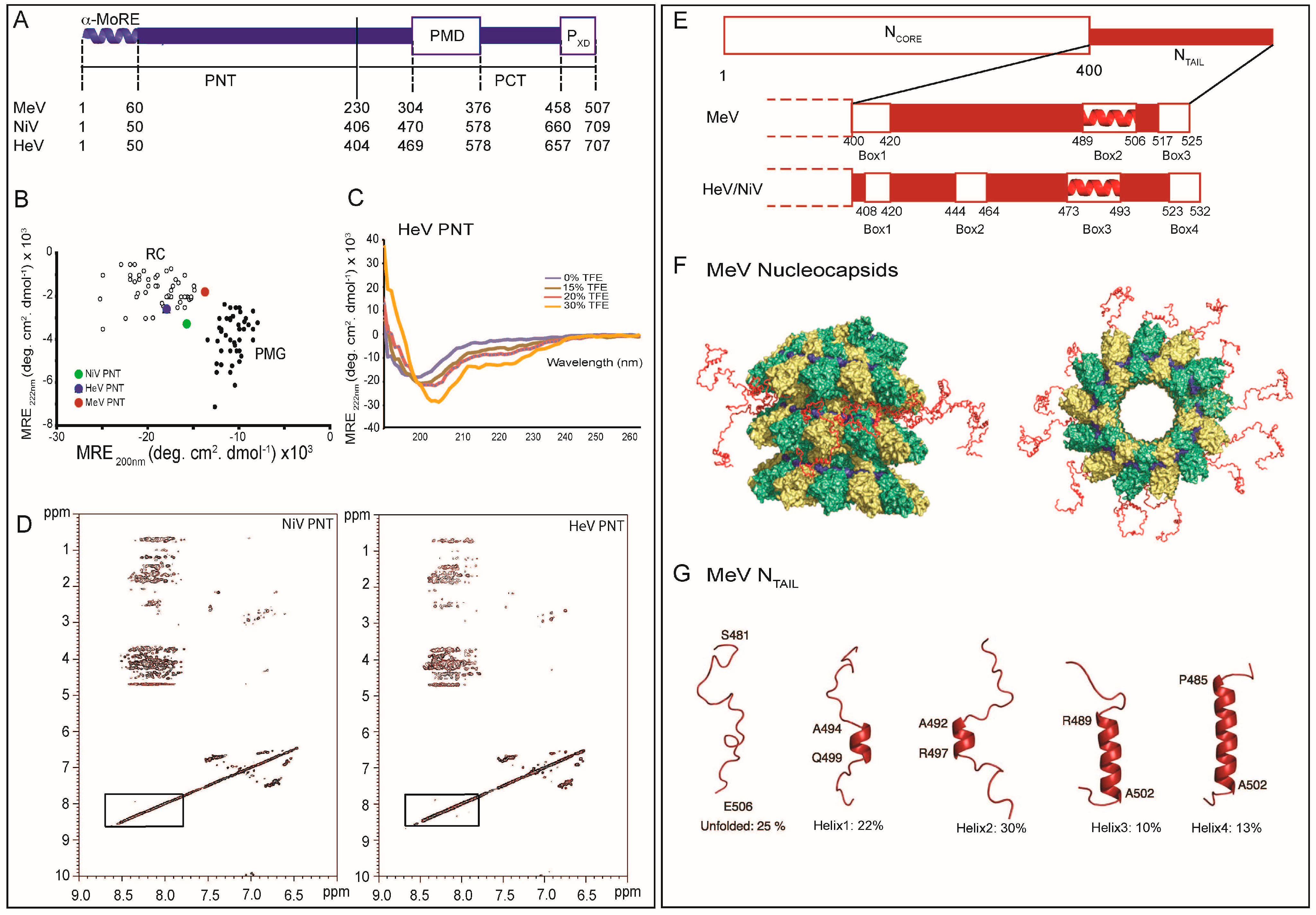
Figure 2.
Structural disorder within paramyxoviral N and P proteins. (A) Modular organization of the MeV, NiV and HeV P proteins. Domain organization of P showing that it is composed in the three viruses of two moieties, PNT and PCT, that are separated by a vertical solid line. Dotted lines indicate the borders of the different domains in the three viruses. Structured and disordered regions are represented as large or narrow boxes, respectively. PNT: N-terminal region of P; PCT: C-terminal region of P; PMD: P multimerization domain; PXD: X domain of P. The α-MoRE at the N-terminus of PNT, which is partly preconfigured in solution as an α-helix, is shown; (B) 222–200 nm ellipticity plot (modified from [27]). The mean residue ellipticity values at 222 nm of a set of well-characterized unfolded or pre-molten proteins have been plotted against the mean residue ellipticity values at 200 nm. The position in the plot of MeV and Henipavirus PNT is highlighted. RC: random coil; PMG: premolten globule-like. Note that HeV PNT is the most extended domain; (C) Far-UV circular dichroism spectra of HeV PNT in the absence and the presence of increasing concentrations of TFE (15%, 20% and 30%) showing that HeV PNT is able to fold into an α-helical conformation; Data were taken from [27] (D); Two-dimensional 1H NMR NOESY spectra of Henipavirus PNT proteins (modified from [30]); (E) Modular organization of the MeV, NiV and HeV N proteins. Domain organization of N showing that it is composed in the three viruses of two moieties, NCORE that is ordered and NTAIL that is disordered. Ordered and disordered regions are represented as large or narrow boxes, respectively. The NTAIL region from the three viruses is zoomed out to show the various boxes that correspond to putative or experimentally proven MoREs. The α-MoRE that is involved in the interaction with the PXD is shown as a red helix; (F) NMR-based model to locate MeV NTAIL within the viral nucleocapsid. NCORE monomers are colored in green and yellow, while the RNA is shown in blue. Front (left) and top (right) views of 13 NTAIL conformers (shown in red) sampling an ensemble of conformations that point out from the surface of the viral nucleocapsid. (Adapted with permission from [46]). Copyright 2011 National Academy of Sciences; (G) Quantitative ensemble description of MeV NTAIL in isolation. Four conformational states of the intrinsically disordered NTAIL, as derived by NMR spectroscopy, are represented by cartoon structures. Adapted with permission from [46]). Copyright 2011 National Academy of Sciences.
Figure 2.
Structural disorder within paramyxoviral N and P proteins. (A) Modular organization of the MeV, NiV and HeV P proteins. Domain organization of P showing that it is composed in the three viruses of two moieties, PNT and PCT, that are separated by a vertical solid line. Dotted lines indicate the borders of the different domains in the three viruses. Structured and disordered regions are represented as large or narrow boxes, respectively. PNT: N-terminal region of P; PCT: C-terminal region of P; PMD: P multimerization domain; PXD: X domain of P. The α-MoRE at the N-terminus of PNT, which is partly preconfigured in solution as an α-helix, is shown; (B) 222–200 nm ellipticity plot (modified from [27]). The mean residue ellipticity values at 222 nm of a set of well-characterized unfolded or pre-molten proteins have been plotted against the mean residue ellipticity values at 200 nm. The position in the plot of MeV and Henipavirus PNT is highlighted. RC: random coil; PMG: premolten globule-like. Note that HeV PNT is the most extended domain; (C) Far-UV circular dichroism spectra of HeV PNT in the absence and the presence of increasing concentrations of TFE (15%, 20% and 30%) showing that HeV PNT is able to fold into an α-helical conformation; Data were taken from [27] (D); Two-dimensional 1H NMR NOESY spectra of Henipavirus PNT proteins (modified from [30]); (E) Modular organization of the MeV, NiV and HeV N proteins. Domain organization of N showing that it is composed in the three viruses of two moieties, NCORE that is ordered and NTAIL that is disordered. Ordered and disordered regions are represented as large or narrow boxes, respectively. The NTAIL region from the three viruses is zoomed out to show the various boxes that correspond to putative or experimentally proven MoREs. The α-MoRE that is involved in the interaction with the PXD is shown as a red helix; (F) NMR-based model to locate MeV NTAIL within the viral nucleocapsid. NCORE monomers are colored in green and yellow, while the RNA is shown in blue. Front (left) and top (right) views of 13 NTAIL conformers (shown in red) sampling an ensemble of conformations that point out from the surface of the viral nucleocapsid. (Adapted with permission from [46]). Copyright 2011 National Academy of Sciences; (G) Quantitative ensemble description of MeV NTAIL in isolation. Four conformational states of the intrinsically disordered NTAIL, as derived by NMR spectroscopy, are represented by cartoon structures. Adapted with permission from [46]). Copyright 2011 National Academy of Sciences.
2.3. Folding Propensities of the Disordered NTAIL and PNT Domains
Although NTAIL and PNT are mostly unfolded in solution, they have been shown to retain some degree of compaction based upon their Stokes radii (RS) and their ellipticity values at 200 and 222 nm, which are consistent with a PMG state [26,32,57]. PMGs are characterized by a conformational state between the RC and the MG state, and possess a certain degree of compaction due to the presence of fluctuating secondary and/or tertiary structures. Indeed, the addition of urea increased the RS of both NTAIL and PNT supporting a conformational state that is not completely unfolded in solution [30].
In general, PMGs possess more hydrophobic residues than RCs. This is readily appreciable in the HCA plots of NTAIL and PNT that are characterized by the presence of short regions locally enriched in hydrophobic clusters (see Figure 2 and Figure 3 in [26]). These regions correspond to putative Molecular Recognition Elements (MoREs). MoREs are short, order-prone regions within IDPs that have a certain propensity to bind to a partner and thereby to undergo induced folding (i.e., a disorder-to-order transition) [67,68,69,70]. In fact, it is currently accepted that the crux of molecular recognition by IDPs is ensured by MoREs. This phenomenon could be explained by the fact that MoREs in the PMG state present a significant interest from an energetic point of view as they facilitate the folding upon binding process [70]. In other words, the residual structure restrains the conformational space sampled by IDPs, thereby reducing the number of interconverting conformers in solution and rendering the structural transition of the IDP to the (partially) folded conformation energetically less demanding.
In the case of PNT, predictions pointed out the presence of a short (40–50 amino acids) order-prone segment at its N-terminus (Figure 2A). This N-terminal module with α-helical propensity corresponds to a conserved region amongst Avulavirus and Rubulavirus members [31]. The involvement of the N-terminal PNT region in N°-binding has been experimentally confirmed in the case of rubulaviruses and of NiV [66,71]. Using computational approaches, it has been shown that all Paramyxovirinae P proteins share a short (11–16 residues) sequence motif within their first 40 residues [72]. It has been proposed that this region would be conserved in all Mononegavirales P proteins as a result of divergent evolution, and would be involved in binding to N° [72]. In agreement, a similar N-terminal module, globally disordered yet containing transient α-helices (aa 1–60) has also been identified and characterized in the P protein from the vesicular stomatitis virus (a rhabdovirus) [73], and subsequently shown to fold upon binding to N° [74]. The N-terminal region of Paramyxovirinae P likely corresponds to an α-helical MoRE (α-MoRE). The folding potential of MeV, NiV and HeV PNT domains was also confirmed by far-UV CD studies, where increasing concentrations of TFE were shown to induce a pronounced gain of α-helicity [30,32]. In addition, in the case of MeV PNT, limited proteolysis experiments in the presence of TFE led to the identification of a thermolysin-resistant fragment. This fragment, spanning residues 27–99, contains a protein region (aa 27–38) with a strong propensity to fold as an α-helix. The extent of residual compaction within the three PNT domains follows the order NiV PNT > HeV PNT > MeV PNT (Figure 2B). Although it is more than plausible that the N-terminal region of MeV and HeV PNT folds upon binding to N°, assessment of the effective folding upon binding abilities of this putative α-MoRE awaits however the isolation and purification of a binding partner. Beyond N°, one such a possible binding partner can be L and/or SNAP29, by analogy with the closely related RDV and human parainfluenza type 3 virus, respectively [75,76].
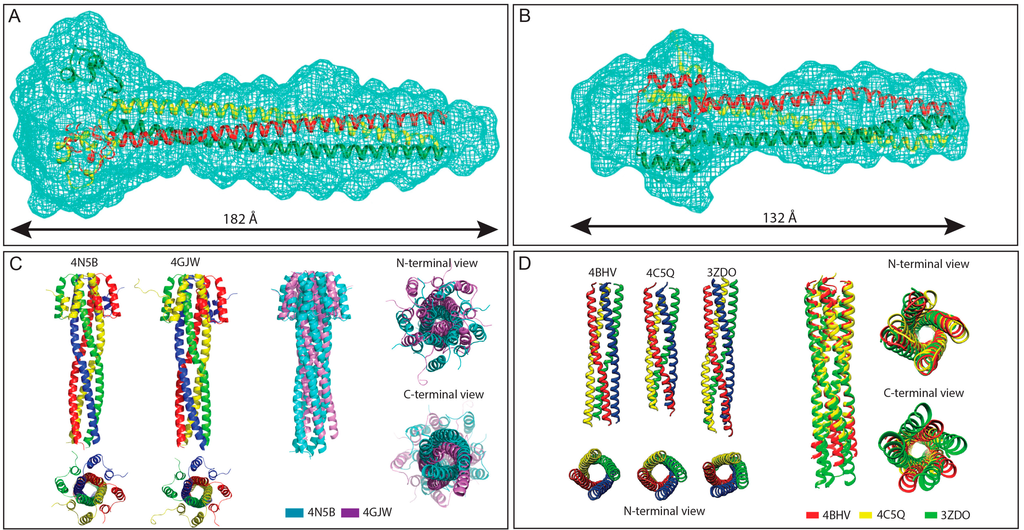
Figure 3.
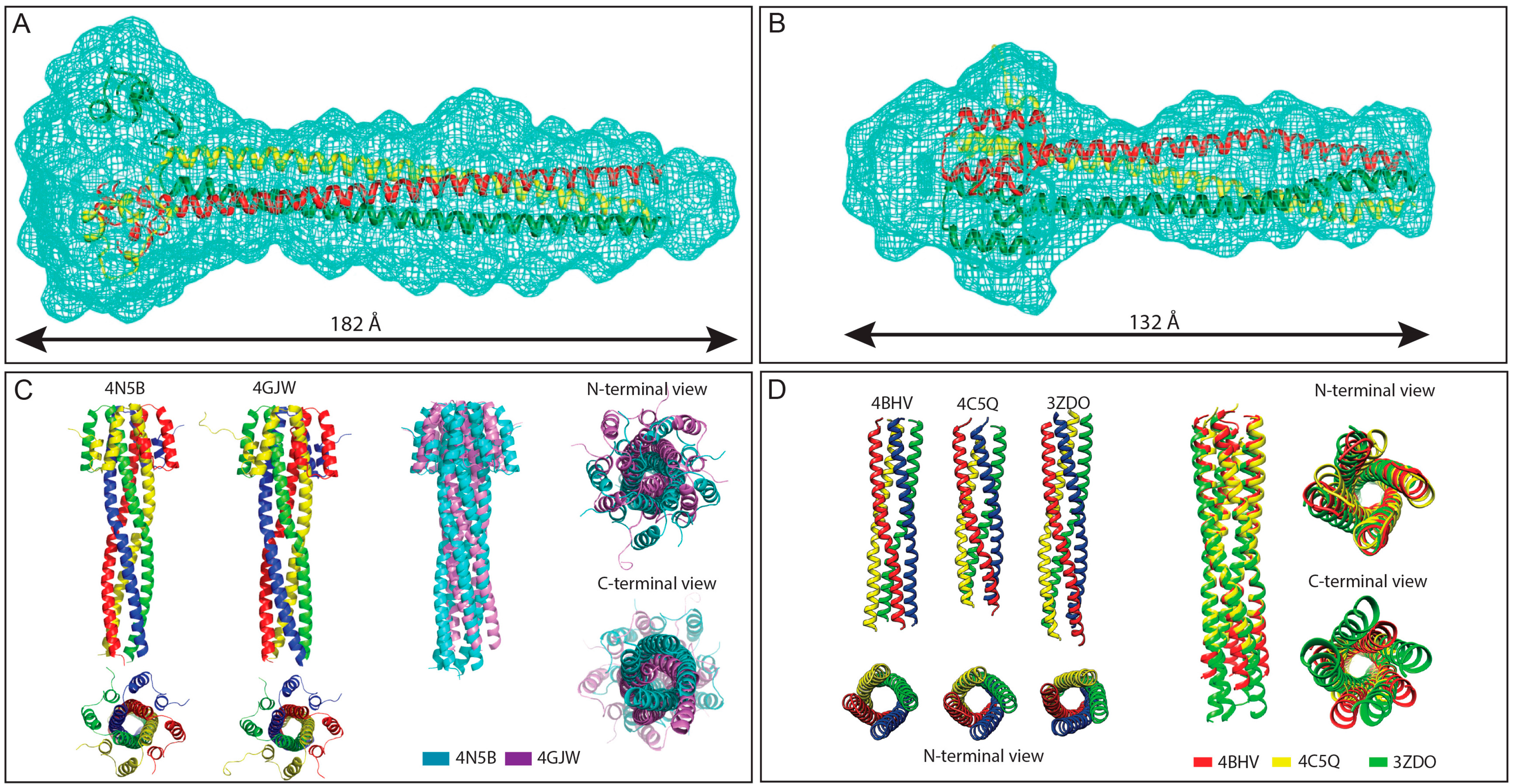
Structures of Henipavirus and MeV P multimerization domains. (A,B) SAXS-derived ab initio envelopes of NiV (A) and HeV (B) PMD; The structures of a trimeric coiled-coil model of NiV (A) and HeV (B) PMD are shown embedded in their respective envelope, with the three chains being displayed in three different colors. Data from panel A and B were taken from [77,78], respectively; (C) Structural comparison among NiV PMD crystal structures. Left: Ribbon model of the two crystal structures of NiV PMD solved so far (pdb codes 4N5B and 4GJW), with the four chains being displayed in four different colors; Right: superimposition of the two NiV PMD tetramers, with PDB entries 4N5B and 4GJW shown in cyan and violet, respectively; and (D) Structural comparison among MeV PMD structures. Left: ribbon representations of the crystal structures of the MeV PMD tetramers as observed in the three different MeV PMD forms solved to date; Right: superimposition of the three MeV PMD tetramers, with PDB entries 4BHV, 4C5Q and 3ZDO shown in red, yellow and green, respectively. Data from panel (C) were taken from [79] and were reproduced with permission of the International Union of Crystallography.
Figure 3.
Structures of Henipavirus and MeV P multimerization domains. (A,B) SAXS-derived ab initio envelopes of NiV (A) and HeV (B) PMD; The structures of a trimeric coiled-coil model of NiV (A) and HeV (B) PMD are shown embedded in their respective envelope, with the three chains being displayed in three different colors. Data from panel A and B were taken from [77,78], respectively; (C) Structural comparison among NiV PMD crystal structures. Left: Ribbon model of the two crystal structures of NiV PMD solved so far (pdb codes 4N5B and 4GJW), with the four chains being displayed in four different colors; Right: superimposition of the two NiV PMD tetramers, with PDB entries 4N5B and 4GJW shown in cyan and violet, respectively; and (D) Structural comparison among MeV PMD structures. Left: ribbon representations of the crystal structures of the MeV PMD tetramers as observed in the three different MeV PMD forms solved to date; Right: superimposition of the three MeV PMD tetramers, with PDB entries 4BHV, 4C5Q and 3ZDO shown in red, yellow and green, respectively. Data from panel (C) were taken from [79] and were reproduced with permission of the International Union of Crystallography.
In the case of NTAIL domains, the structural properties of the MoREs have been unraveled using conformational and spectroscopic analyses. For both NiV and HeV, the latter analyses have been applied on full and truncated forms of the NTAIL regions bearing various combinations of the four predicted MoREs (Figure 2E) [80]. The firth and fourth MoREs (amino acids 408–422 and 523–532, respectively) were found to possess irregular forms (i.e., I-MoRE), while the MoREs encompassing residues 444–464 (i.e., Box2) and 473-493 (i.e., Box3) were found to have clear α-helical propensities (i.e., α-MoREs) [80]. These results contrast the sequence-based secondary structure predictions that pointed out the occurrence of an α-helix within Box1 and of a β-strand within Box2 [30]. However, it has been frequently reported that some MoREs possess ambiguous amino acid sequences thus misleading secondary structure predictions [81]. In addition, some IDPs possess the capacity to adapt to the structure of distinct partners [82]. Whether these erroneous predictions of the secondary structure content of Box1 and Box2 reflect an intrinsic limitation of the predictors or rather intrinsic plasticity of these Boxes allowing them to adopt different conformations in a template-dependent manner remains to be unraveled and awaits the identification of their corresponding partners. Those studies also showed that Box1 is a major determinant of protein compaction and Box4 is likely involved in the stabilization of the α-helices located in Box2 and Box3 [80]. Furthermore, site-directed spin-labeling (SDSL) EPR spectroscopy studies unveiled a considerable conformational heterogeneity within Box3, consistent with the occurrence of multiple helical conformers of different length [83]. In agreement, analysis of the Cα chemical shifts of the free form of HeV and NiV NTAIL domains showed that Box3 is at least transiently populated as an α-helix, and in the case of NiV, a second, short α-helical region could be detected within Box2 [63,64].
In the case of MeV NTAIL, an α-MoRE (aa 488–499 of N) has been identified within one (namely Box2) out of three regions conserved in Morbillivirus members (Figure 2E). The role of this α-MoRE in binding to PXD and in the α-helical induced folding of NTAIL was confirmed by spectroscopic and biochemical experiments carried out on a truncated NTAIL form devoid of the 489–525 region [33] and subsequently by structural studies [36]. Interestingly, using MD simulations, the isolated α-MoRE was found to behave like a MG [84]. Indeed, the distribution of the radius of gyration (Rg) of the α-MoRE was found to be smaller in the unbound state than in the bound state, indicating that the majority of the conformations of the free form of the α-MoRE are more compact than the bound conformation (the folded state). Analysis of the Cα chemical shifts of NTAIL and of the mobility of spin labels grafted within Box2 showed that the α-MoRE of MeV NTAIL is partly preconfigured as an α-helix in the absence of PXD [85,86,87]. More recently, an atomic-resolution ensemble description of the α-MoRE of MeV NTAIL could be obtained using recently developed tools designed to provide quantitative descriptions of conformational equilibria in IDPs on the basis of experimental NMR data (Figure 2G) [38,88]. By combining residual dipolar couplings (RDCs) measurements and ensemble optimization methods [38,88], the α-MoRE was shown to exist in a rapidly interconverting conformational equilibrium between an unfolded form and conformers containing four discrete α-helical elements situated around the interaction site [46]. All of these α-helices are stabilized by N-capping interactions mediated by side chains of four different aspartic acids or serines that precede the observed helices [89]. N-capping stabilization of helices or turns represents an important mechanism by which the primary sequence encodes pre-recognition states, and has already been observed in other IDPs including SeV NTAIL [88]. In this latter case, the α-MoRE was shown to possess a similar conformational behavior, although in this case the α-MoRE samples an extended conformation and only three helical conformers [51,88].
Comparison between NiV, HeV and MeV NTAIL protein sequences pointed out the occurrence of an additional MoRE in the case of Henipavirus NTAIL (i.e., Box2), where the Box3 region corresponds functionally to Box2 of MeV NTAIL based on binding abilities towards PXD (Figure 2E) [35,36,80,90]. This finding further emphasizes the plasticity of IDP/Rs, which tolerates insertions/deletions in functionally relevant regions.
NTAIL and PNT domains are disordered not only in isolation but also within the full-length N and P proteins. Indeed, N and P proteins have been found to be highly sensitive to proteolysis [90] (Beltrandi, Habchi, Longhi and Cavalli, unpublished data). In addition, as already mentioned, many detectable peaks in the Heteronuclear Single Quantum Correlation (HSQC) spectra of intact nucleocapsids well superimpose onto those of the free NTAIL regions indicating that NTAIL is disordered within nucleocapsids-like particles [46,63,64].
From a functional point of view, beyond imposing structural constraints to the conformational ensemble of IDPs, the main function of MoREs is to allow engagement in a broad molecular partnership. Indeed, in the case of MeV, the flexible NTAIL domain has been shown to bind via its MoRE to numerous partners including the X domain of the P protein [33,35,57,91], the major inducible heat shock protein hsp70 [92,93,94], the interferon regulatory factor 3 [95,96], a yet unidentified protein cell receptor involved in MeV-induced immunosuppression [97,98], a nuclear export protein [99], the matrix protein [100], peroxiredoxin 1 [101] and possibly, components of the cell cytoskeleton [102,103]. In the case of NiV and HeV, although only one NTAIL partner has been identified and characterized (i.e., PXD) so far, an even broader molecular partnership is expected with respect to MeV NTAIL, based on the presence of the additional MoRE (i.e., Box2). Amongst all the interactions established by NTAIL, the interaction with PXD is critical as it allows the P/L complex to be recruited onto the nucleocapsid in order to allow transcription and replication to take place [22,23,25]. In striking contrast, a recent study by the group of Plemper has challenged the well-established model according to which Box2 is strictly required to recruit the MeV polymerase complex: indeed, Box2 was found to be dispensable for MeV transcription and replication in the absence of the upstream NTAIL region that was found to act as a negative modulator (i.e., to prevent binding of the L–P complex to the nucleocapsid) [104]. Similarly to NTAIL, PNT domains have been reported to interact with multiple partners, including N in both assembled and unassembled forms [105], cellular proteins [106] and the L protein [107,108].
4. Molecular Mechanisms of Paramyxoviral NTAIL–PXD Interactions: A Continuum of Disorder from the Free to the Bound Form
4.1. Molecular Polymorphism in NTAIL–PXD Interactions
As already discussed above, the P protein simultaneously binds to L and to the exposed C-terminal domain of N (NTAIL) via its C-terminal X domain (PXD). The structures of MeV and HeV PXD have been solved and were shown to consist of an anti-parallel triple α-helical bundle (Figure 4A,B). The surface of PXD formed between helices α2 and α3 displays a large hydrophobic cleft [35,37,63,87]. High-resolution structural data are also available for the X domains of the closely related SeV and MuV viruses, the structures of which have been solved by NMR and X-ray crystallography, respectively [39,120]. Interestingly, although the structure of SeV PXD resembles that of MeV PXD, in that it is also formed by three α-helices folded around a hydrophobic core [39], negatively charged residues dominate the surface created by α2 and α3 helices [34]. Another interesting feature is that whilst the fold of MuV PXD is conserved (i.e., three α-helices), it actually exists as a MG in solution as evidenced by CD, NMR and DLS experiments [120]. The observed stable 3D structure in crystals of MuV PXD apparently results from a stabilizing effect brought by the addition of methylamine cosolute during crystallogenesis experiments [120]. Lack of a unique stable 3D structure is not a feature unique to MuV PXD being also shared by the corresponding domains from other rubulaviruses that were found to span a structural continuum ranging from stable α-helical bundles to largely disordered forms in solution [121]. In the same vein, recent electron spray ionization mass spectrometry (ESI–MS) studies unveiled that MeV PXD has a bimodal charge state distribution reflecting the presence of two forms differing in the extent of compaction [122].
Although the structure of NiV PXD has not been solved yet, it is expected to adopt a structure similar to that of its HeV counterpart (Figure 4B) based not only on the high sequence similarity between the two domains (94%), but also on their common spectroscopic features. Indeed, HeV and NiV PXD possess similar CD and NOESY spectra typical of α-helical folded domains [90]. Thus the structural fold of the P X domains is conserved within paramyxoviruses, suggesting that their function is conserved as well (Figure 4C).
Both NiV and HeV PXD form with NTAIL a 1:1 stoichiometric complex that is stable up to 1 M NaCl, and whose KD is in the μM range [90]. The α-MoRE spanning residues 473–493 (i.e., Box3) has been shown to be the PXD binding site [80]. CD, NMR and SDSL EPR studies showed that Henipavirus NTAIL domains undergo PXD-induced α-helical folding within Box3 with the remainder of the chains remaining in the disordered state [63,64,80,90]. Interestingly, the conformational ensemble that is sampled by the free form of NTAIL in solution presages the α-helical conformation of the PXD-bound form, as judged from chemical shift analysis [63,64].
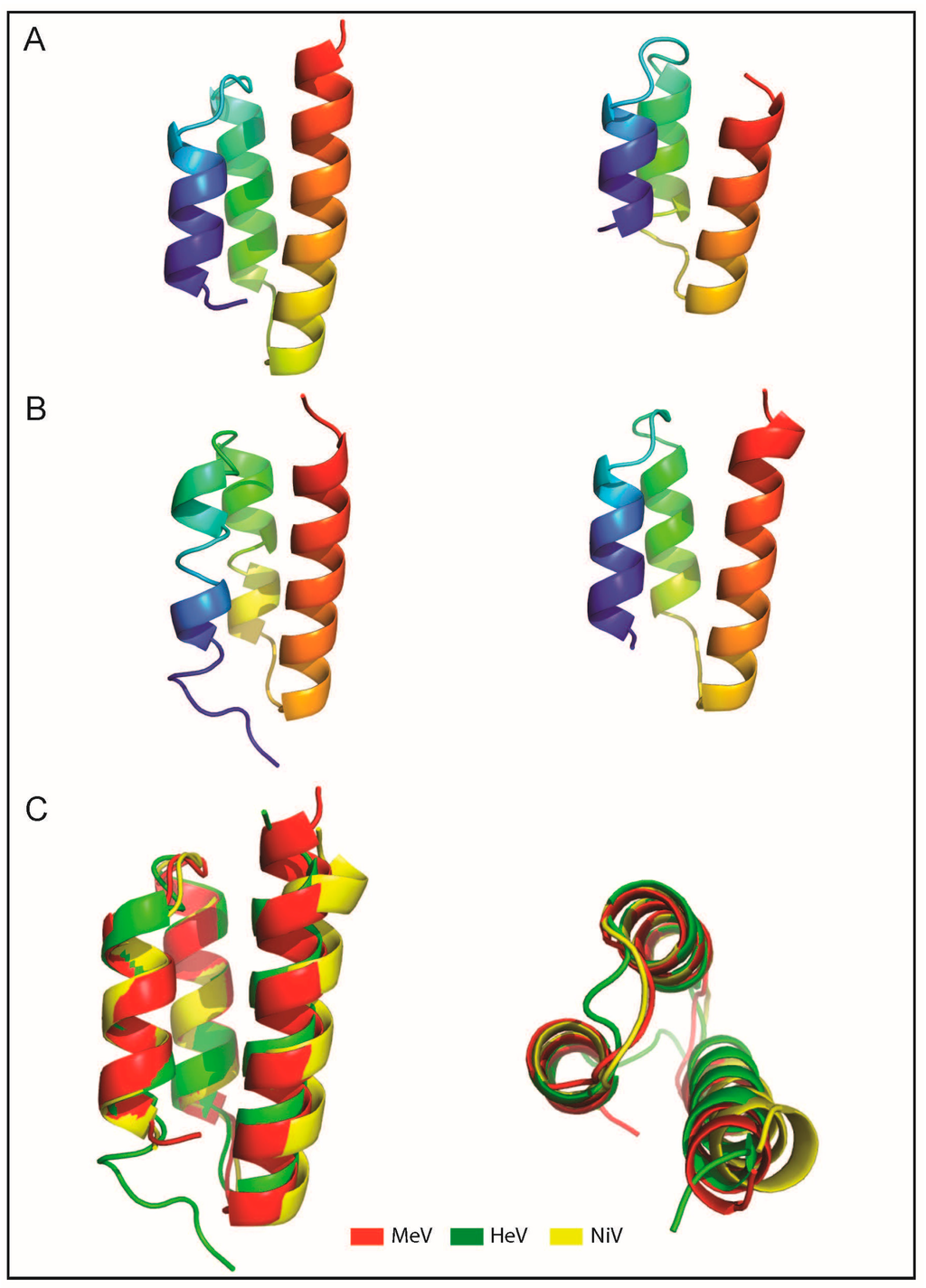
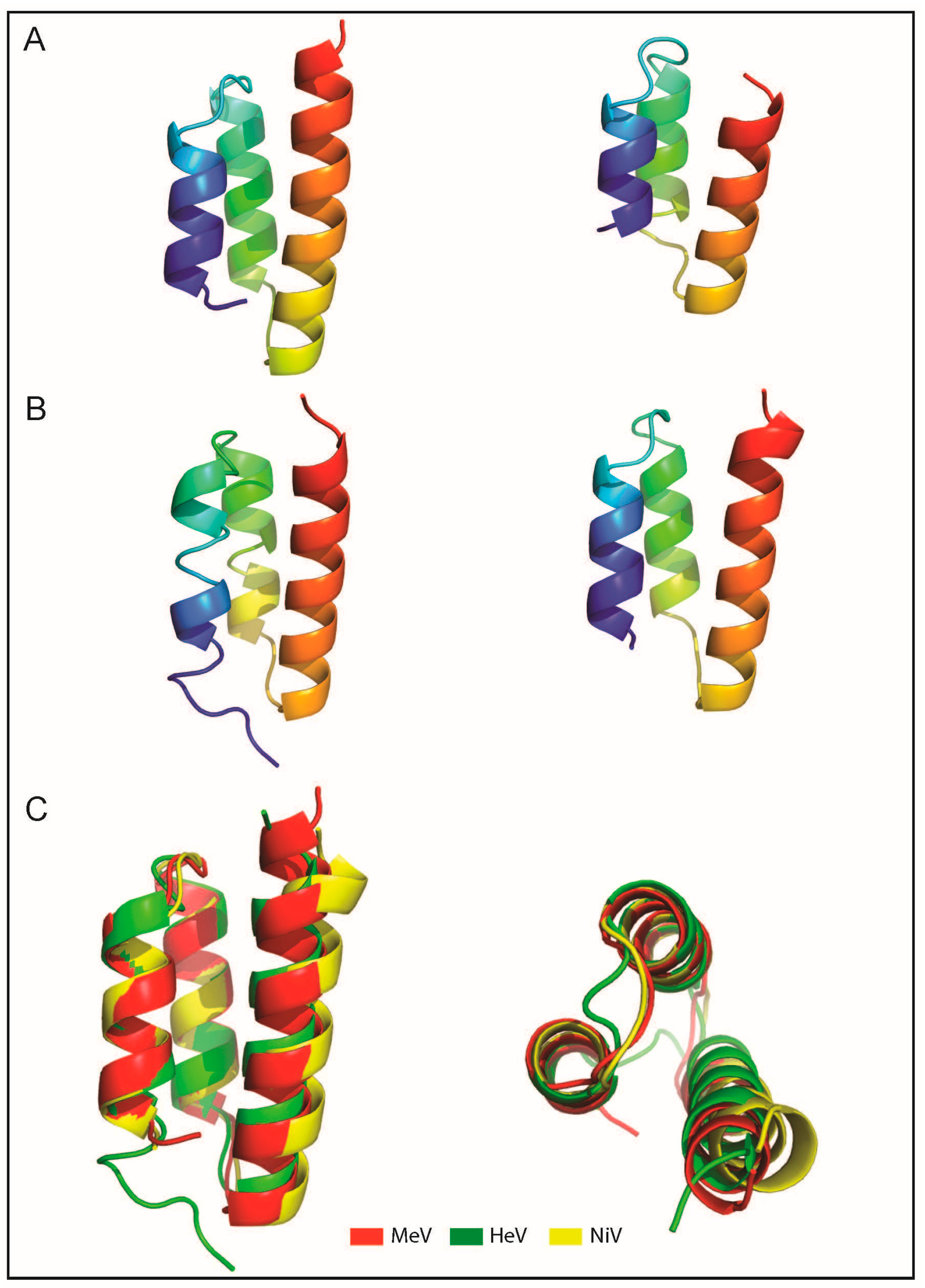
Figure 4.
Structures of MeV and Henipavirus X domains. (A) Ribbon representation of the crystal (left) and solution (right) structure of MeV PXD (pdb codes 1OKS and 2K9D); (B) Ribbon representation of the crystal structure of HeV PXD (left, pdb code 4HEO) and of a model of NiV PXD (right) [90]; (C) Superimposition of the crystal structures of MeV and HeV PXD onto the NiV PXD model. Structures were drawn using Chimera [123] and Pymol [124].
Figure 4.
Structures of MeV and Henipavirus X domains. (A) Ribbon representation of the crystal (left) and solution (right) structure of MeV PXD (pdb codes 1OKS and 2K9D); (B) Ribbon representation of the crystal structure of HeV PXD (left, pdb code 4HEO) and of a model of NiV PXD (right) [90]; (C) Superimposition of the crystal structures of MeV and HeV PXD onto the NiV PXD model. Structures were drawn using Chimera [123] and Pymol [124].
Although direct structural data on Henipavirus NTAIL–PXD complexes are still lacking, recent NMR studies provided first clues on the structure of these complexes. In particular, the vanishing of the resonances of the α-MoRE upon addition of PXD supports the existence of a highly dynamic complex for both NiV and HeV, with the α-MoRE undergoing α-helical fraying at the surface of PXD [63,64,90]. In spite of these similarities, subtle differences distinguish the two Henipavirus NTAIL–PXD complexes. Indeed, the NiV NTAIL–PXD complex is slightly tighter than that of HeV [63,64,80,90], in line with the ability of SEC to document complex formation in the case of NiV but not in the case of HeV [90]. Besides, the α-MoRE region within NiV NTAIL was found to possess a higher degree of pre-configuration with respect to HeV NTAIL, as judged based on the 15N R2 values [64], and to populate longer-lived interconverting α-helical segments that could be detected by EPR measurements [83]. Furthermore, NMR experiments showed in the case of NiV NTAIL an additional involvement of Box2 in the interaction with PXD [64]. That Box2 does participate to some extent to NiV NTAIL binding to PXD was also documented by SDSL EPR measurements showing that in the case of NiV, but not of HeV, the addition of the partner triggers a reduction in the mobility of a spin label grafted within Box2 [83]. The differences that were observed between NiV and HeV NTAIL regions in their free and bound forms, in particular within Box2, might be dictated by a substitution occurring within Box2 at position 457, where an Asp residue in HeV NTAIL is replaced by an Asn in NiV NTAIL.
In the case of HeV, analysis of chemical shift perturbations in reciprocal titration studies and the availability of the crystal structure of PXD allowed identification of the residues involved in the interaction [63]. Those studies revealed that although the binding interface is made of hydrophobic residues, the binding pocket of PXD is surrounded by charged residues that may establish electrostatic interactions with basic residues of Box3. Subsequent isothermal titration calorimetry (ITC) studies carried out at different pH values did in fact confirm the role of electrostatics in complex formation, a conclusion further strengthened by mutational studies that targeted charged residues both within NTAIL and PXD [125]. Collectively, those studies provided direct evidence that charged residues surrounding the hydrophobic binding interface play a crucial role in complex formation, thus arguing for a multiparametric interaction and emphasizing the role of residues located in the neighborhood of the binding interface. Accordingly, it has been proposed that the HeV NTAIL/PXD complex formation may rely on the so-called “electrostatic steering mechanism” [126], where long-range electrostatic forces pull NTAIL toward the relevant acidic patch on the surface of PXD, thus leading to an “electrostatic encounter complex” [127] in which NTAIL is loosely anchored at the periphery of the binding site [125].
The corollary of this model is that HeV NTAIL would fold after binding, a behavior corroborated by quantitative analysis of NMR titration data (see below) [63].
Taking into account the role of electrostatics in the formation of the HeV NTAIL–PXD complex, it is conceivable that the Asp to Asn substitution could be responsible for the observed differences in the role of Box2 in binding to PXD by NiV and HeV NTAIL.
Neither chemical shifts nor electrostatic interactions are able to distinguish rotational symmetry about the axis of the NTAIL helix, although two conformations are most probable, both having the hydrophobic face of the α-MoRE in contact with the hydrophobic interface of PXD. Through a combination of mutational and SAXS studies, experimental evidence was recently gathered supporting a parallel orientation of the MoRE at the surface of HeV PXD [125].
Similarly to HeV and NiV, MeV NTAIL undergoes α-helical folding upon binding to PXD. The PXD-induced α-helical folding occurs within the predicted α-MoRE located within Box2 (amino acids 486–504) and gives rise to a pseudo-four helix complex of which the crystal structure has been solved at 1.8 Å resolution (Figure 5A) [35,36]. In the structure of the chimeric construct in which PXD and the α-MoRE are covalently linked to each other, the α-MoRE of NTAIL adopts a parallel orientation with respect to PXD and is embedded in a large hydrophobic cleft delimited by helices α2 and α3 [36]. Indeed, the residues that are involved in the interaction are mainly hydrophobic, involving Leu481, Leu484, Ile488, Phe497, Met500, and Ile504 from PXD and Ser491, Ala494, Leu495, Leu498 and Met501 from NTAIL.
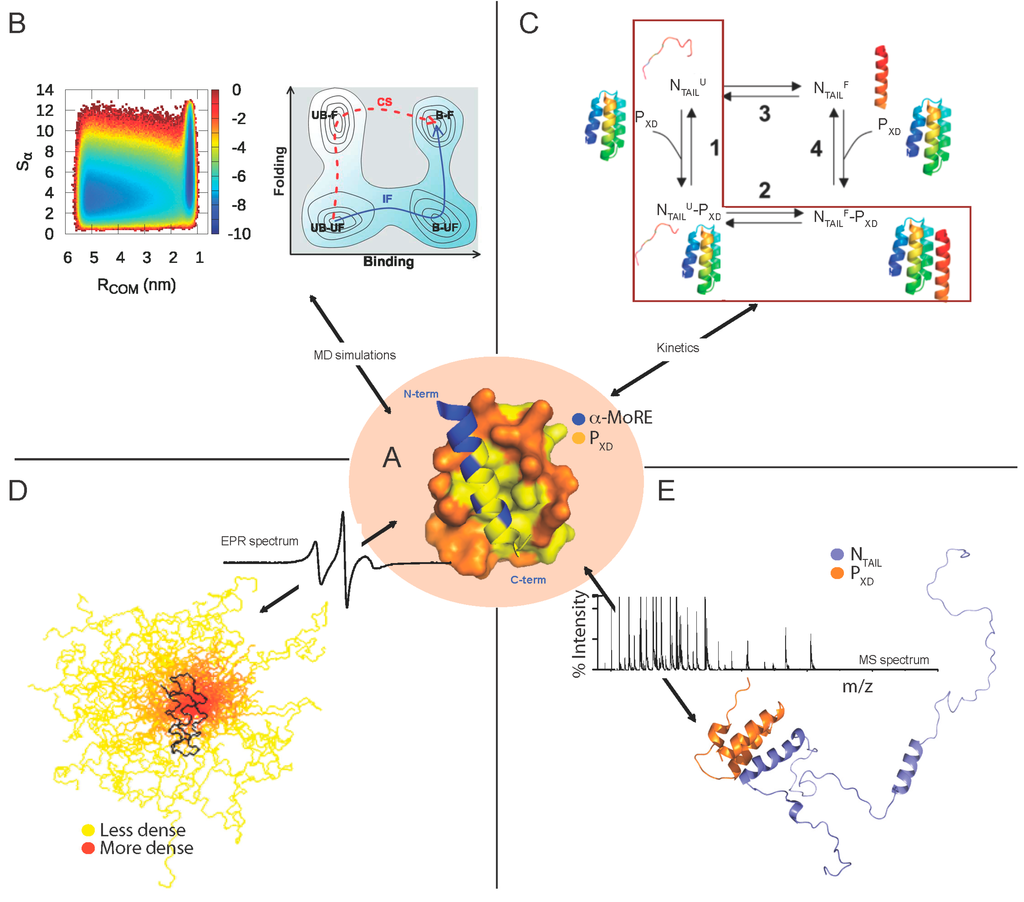
Figure 5.
Molecular mechanisms and fuzziness of the MeV NTAIL–PXD complex as unveiled by independent experimental evidences. (A) Structure of the MeV chimeric NTAIL–PXD construct (PDB code 1T6O) [36]. MeV PXD (amino acids 459–507 of P) is shown in orange with surface presentation while the α-MoRE region (amino acids 486–504 of N) is shown in blue with ribbon representation. Hydrophobic residues are shown in yellow; (B) (Left) Free-energy surface for the interaction between the α-MoRE and MeV PXD as a function of RCOM (binding order parameter) and Sα (folding order parameter); (Right) Schematic free-energy surface showing that folding upon binding takes place according to an induced folding (IF) mechanism and not to a conformational selection (CS) mechanism despite the preexistence of unbound-folded state (UB-F). Note that, in the folding upon binding process, there are four possible states: unbound-unfolded (UB-UF), bound-unfolded (B-UF), unbound-folded (UB-F), and bound-folded (B-F). The schematic picture gives a good illustration that the preexistence of the UB-F state is a necessary but not a sufficient condition for a conformational selection mechanism. (Adapted with permission from [84]). Copyright 2013 National Academy of Sciences; (C) A kinetic-based model showing the folding after binding mechanism of the MeV NTAIL–PXD interaction (highlighted in a red square). Indeed, NTAIL recognizes PXD by first forming a weak encounter complex in a disordered conformation and is then subsequently locked-in by a folding step. (Reprinted with permission from [128]); Copyright 2014 American Chemical Society (D) Model of the partly disordered MeV NTAIL–PXD complex as a conformational ensemble as derived from a combined EPR and modeling approach. 50 best-fit structures of the 488–525 region of NTAIL in complex with PXD. The NTAIL conformers are depicted with a color gradient ranging from yellow to red with increasing structural density, while PXD is shown in black (modified from [129]). A typical EPR spectrum is also shown; and (E) Cartoon representation of the structural model of the NTAIL–PXD complex as derived from a combined ESI-IM-MS and modeling approach. The disordered NTAIL is shown in blue and the ordered X domain of P is shown in orange. A typical MS spectrum is shown. Reprinted with permission from [122]. Copyright 2014 The American Society for Mass Spectrometry. EPR: electron paramagnetic resonance, MS: mass spectrometry, MoRE: Molecular Recognition Element, MD: molecular dynamics. Structures were drawn using Pymol [124].
Figure 5.
Molecular mechanisms and fuzziness of the MeV NTAIL–PXD complex as unveiled by independent experimental evidences. (A) Structure of the MeV chimeric NTAIL–PXD construct (PDB code 1T6O) [36]. MeV PXD (amino acids 459–507 of P) is shown in orange with surface presentation while the α-MoRE region (amino acids 486–504 of N) is shown in blue with ribbon representation. Hydrophobic residues are shown in yellow; (B) (Left) Free-energy surface for the interaction between the α-MoRE and MeV PXD as a function of RCOM (binding order parameter) and Sα (folding order parameter); (Right) Schematic free-energy surface showing that folding upon binding takes place according to an induced folding (IF) mechanism and not to a conformational selection (CS) mechanism despite the preexistence of unbound-folded state (UB-F). Note that, in the folding upon binding process, there are four possible states: unbound-unfolded (UB-UF), bound-unfolded (B-UF), unbound-folded (UB-F), and bound-folded (B-F). The schematic picture gives a good illustration that the preexistence of the UB-F state is a necessary but not a sufficient condition for a conformational selection mechanism. (Adapted with permission from [84]). Copyright 2013 National Academy of Sciences; (C) A kinetic-based model showing the folding after binding mechanism of the MeV NTAIL–PXD interaction (highlighted in a red square). Indeed, NTAIL recognizes PXD by first forming a weak encounter complex in a disordered conformation and is then subsequently locked-in by a folding step. (Reprinted with permission from [128]); Copyright 2014 American Chemical Society (D) Model of the partly disordered MeV NTAIL–PXD complex as a conformational ensemble as derived from a combined EPR and modeling approach. 50 best-fit structures of the 488–525 region of NTAIL in complex with PXD. The NTAIL conformers are depicted with a color gradient ranging from yellow to red with increasing structural density, while PXD is shown in black (modified from [129]). A typical EPR spectrum is also shown; and (E) Cartoon representation of the structural model of the NTAIL–PXD complex as derived from a combined ESI-IM-MS and modeling approach. The disordered NTAIL is shown in blue and the ordered X domain of P is shown in orange. A typical MS spectrum is shown. Reprinted with permission from [122]. Copyright 2014 The American Society for Mass Spectrometry. EPR: electron paramagnetic resonance, MS: mass spectrometry, MoRE: Molecular Recognition Element, MD: molecular dynamics. Structures were drawn using Pymol [124].
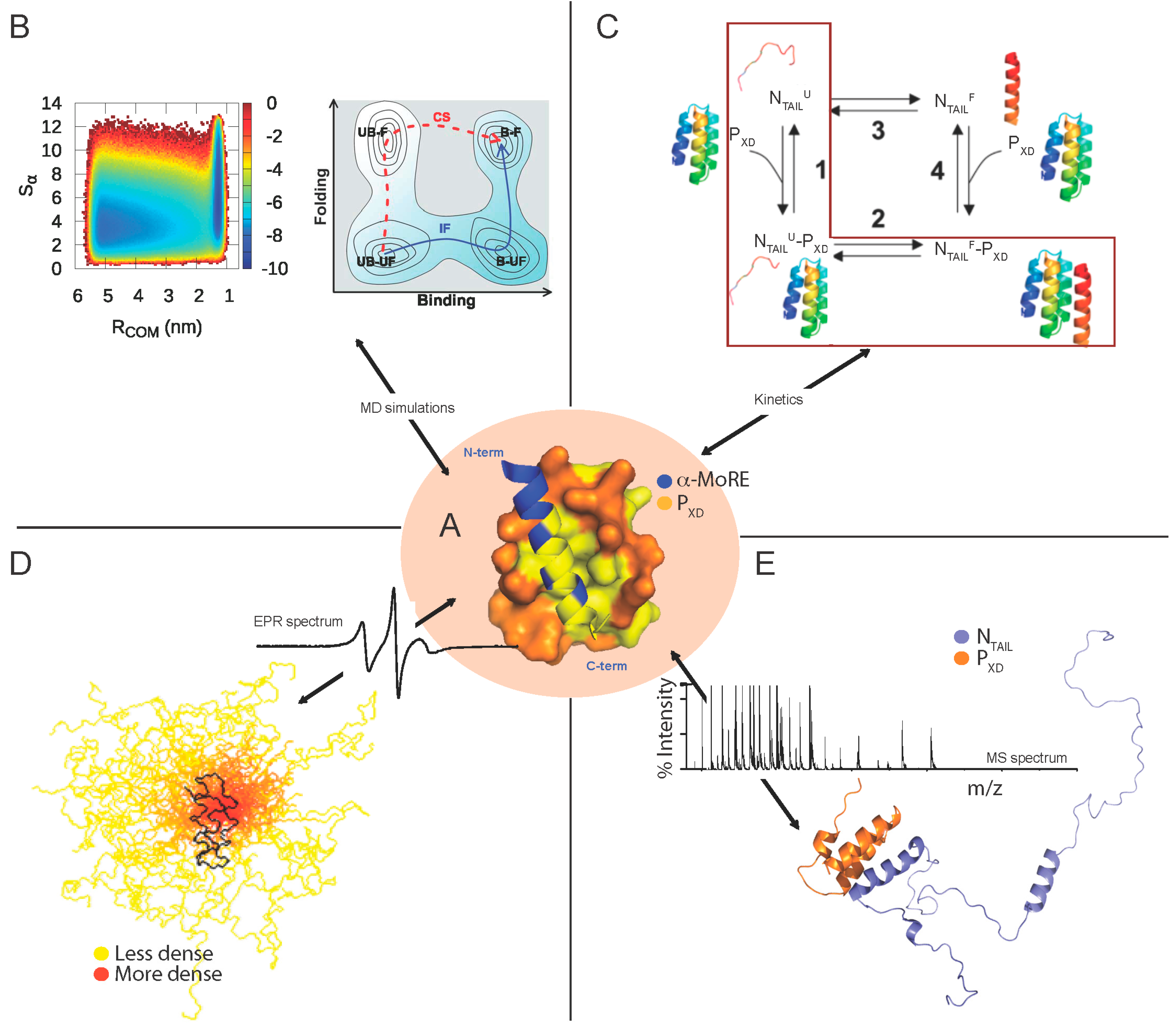
The dynamics of the MeV NTAIL–PXD complex in solution has been thoroughly investigated in SDSL EPR, SAXS and NMR studies [85,86,87,130]. Indeed, in SDSL EPR studies, 14 single-site MeV NTAIL cysteine variants were designed, purified and labeled and their EPR spectra were recorded in the presence or absence of PXD [85,86]. The mobility of the spin labels grafted within the 488–502 (i.e., Box2) and 505–522 regions was found to be severely and moderately reduced, respectively, upon addition of PXD [86]. The restrained motion of the 505–522 region upon binding to PXD was shown to be due to the α-helical transition occurring within the neighboring Box2 region and not to a direct interaction with PXD nor to gain of regular secondary structure [86]. ITC studies further supported the lack of contribution of Box3 in the interactions with PXD by showing that the removal of Box3 does not affect the binding strength of the NTAIL–PXD interaction [80].
The parallel orientation of the MoRE at the PXD surface is thus a conserved feature between MeV and HeV, arguing for a functional relevance. It is tempting to speculate that this could be related to the relative orientation of the whole P protein with respect to the NTAIL region protruding from the nucleocapsid. In its turn, this might be related to optimal positioning of the polymerase onto the nucleocapsid template and might impart directionality to the polymerase movement along the nucleocapsid.
Another common feature among MeV, HeV and, supposedly, NiV NTAIL–PXD complexes is the hydrophobic nature of their interface, in line with the findings by Meszaros and co-workers who reported that the binding interfaces of protein complexes involving IDPs are often enriched in hydrophobic residues [131]. Burying of hydrophobic residues of the MeV α-MoRE at the PXD surface is thought to provide the driving force of its induced folding. We therefore modeled the more hydrophobic side of the amphipathic α-MoRE of Henipavirus NTAIL at the hydrophobic surface delimited by helices α2 and α3 of PXD using the MeV NTAIL–PXD structure as a template. The two modeled complexes display a rather small interface area in agreement with previous reports indicating that the interfaces of complexes involving IDPs are generally smaller than those occurring in ordered complexes [70]. Strikingly, in the case of the SeV NTAIL–PXD complex, charged residues dominate the interface, thus giving a good illustration of how selection pressure allowed the C-terminal domains of N and P to evolve concomitantly within the Paramyxoviridae family leading to protein complexes having the same 3D fold and the same function, but with very limited sequence identity [34].
From a mechanistic point of view, two extreme mechanisms of IDPs-based interactions have been described in the literature (see [29] and references therein cited). According to these two mechanisms, IDP/Rs can fold either before or upon binding to the partner. In the first case, the partner binds to an already pre-folded MoRE through a conformational selection mechanism, thereby shifting the conformational ensemble to the folded form [132,133]. In the second mechanism, folding occurs upon binding, where the interaction with the partner induces the gain of structure within the MoRE, very often through hydrophobic contacts. However, a mixed mechanism involving both induced folding (i.e., folding after binding) and conformational selection (i.e., folding before binding) has been often reported [134].
In spite of the subtle differences distinguishing the Henipavirus and MeV NTAIL–PXD complexes, they share similar structural features, thus suggesting a possible common folding coupled to binding mechanism. Although the occurrence of a transiently populated α-helix even in the absence of the partner would suggest that the molecular mechanism governing the PXD-induced folding of NTAIL could rely on conformer selection (i.e., selection by the partner of a pre-existing conformation), quantitative analysis of NMR titration data obtained for the MeV NTAIL–PXD binding reaction [87] indicate that the binding reaction may also involve a binding intermediate in the form of a weak, non specific encounter complex hence implying also an induced folding mechanism [135]. A combined mechanism of conformational selection and induced folding was further supported by data obtained by MD simulations. Indeed, a synergistic mechanism in which the recognition event proceeds via (minor) conformational selection, followed by (major) induced folding has been proposed (Figure 5B) [84]. Interestingly, recent kinetic experiments on the MeV NTAIL–PXD binding reaction allowed the identification of at least two kinetic steps and revealed unambiguously that the reaction occurs via a folding after binding scenario, whereby the dominant pathway occurs via the accumulation of a partially folded intermediate followed by a subsequent monomolecular folding event (Figure 5C) [128].
Similarly to MeV NTAIL, NMR and SDSL EPR data [63,64,83] support the presence within Henipavirus NTAIL domains of a pre-formed α-MoRE in the free state. The extent to which this pre-configuration may presage the bound form and/or favor conformational selection is expectedly different between NiV and HeV given that the NiV α-MoRE is more populated and long-lived than its HeV counterpart [63,64,83]. In the case of HeV, quantitative analysis of peak intensities in the HSQC spectra of NTAIL at each PXD titration point showed that the signal intensity decreases faster for the residues located at the extremities of the α-MoRE and for which a smaller amount of residual helical structure is observed in the isolated state of NTAIL. This differential broadening suggests that PXD binds to a short, central helix within the α-MoRE, and that this helix is subsequently extended via helical folding of the adjacent residues. Data therefore indicate that NTAIL interacts with PXD via a folding-upon-binding mechanism, with the folding event occurring on the micro- to millisecond time scale [63]. Definite conclusions as to whether the higher helical sampling by the NiV NTAIL α-MoRE imparts a different mechanism of folding coupled to binding to PXD await direct experimental data that are so far missing.
From a functional point of view, the NTAIL–PXD interaction is thought to trigger the opening of the nucleocapsid to provide access of the polymerase to the viral RNA, which is tightly protected by the assembled N. In agreement, EM studies showed that addition of PXD triggers unwinding of MeV nucleocapsids (Bhella and Longhi, unpublished data). This dramatic conformational change is accompanied by an increased exposure of viral RNA to the solvent as indicated by its increased sensitivity to RNAse. In line with these observations, recent studies documented the ability of the MuV P protein to induce nucleocapsid uncoiling, with both N- and C-terminal P domains being involved [136]. In striking contrast with these findings, NMR studies have shown that addition of PXD to HeV nucleocapsids does not trigger any major nucleocapsid rearrangement, as judged from the fact that the only peaks that are affected by the addition of the partner are those of the residues belonging to the α-MoRE [63]. These findings provide evidence that the environment of NTAIL is identical in the free and the PXD-bound form within the nucleocapsids, thereby supporting the absence of major unwinding or rearrangements of the nucleocapsids [63]. The expectedly necessary nucleocapsid unwinding might require either the full-length P protein, or the P–L complex and/or cellular cofactors. One such a possible cellular cofactor could be hsp70, one of the experimentally confirmed MeV NTAIL partners [92,93,94]. The hypothesized ability of hsp70 to promote nucleocapsid conformational changes is corroborated by previous studies on the closely related canine distemper virus, where hsp70-nucleocapsid complexes were found to exhibit an expanded helical diameter, an increased fragility, and an enhanced exposure of the genomic RNA to nuclease degradation [137,138].
A tight N-P complex is predicted to hinder polymerase processivity according to the cartwheeling model. This model posits that contacts between NTAIL and PXD have to be dynamically made/broken to allow the polymerase to progress along the nucleocapsid template in order to allow transcription and replication to take place. In support of a relationship between affinity of the NTAIL–PXD and polymerase processivity, recent data obtained using a quantitative mammalian protein complementation assay and recombinant measles viruses allowing the conditional expression of wild-type or mutated P genes, showed that an increase in the MeV NTAIL–PXD binding strength resulting from PXD substitutions is associated with a slower transcript accumulation rate and that abolishing the interaction renders the polymerase non functional [139]. In striking contrast with these data, previous mutational studies that targeted the Box2 region of MeV NTAIL showed that a reduced binding strength has no impact on the polymerase rate [140]. Collectively, the results provided by these two independent studies suggest that while increasing the NTAIL–PXD binding strength has an effect on the polymerase rate, decreasing it (without abrogating it) has no impact. We can speculate that this tolerance of the polymerase to NTAIL substitutions leading to a reduced binding strength is probably true only in a certain range of affinities, where in spite of a pronounced drop in the affinity towards PXD, the NTAIL–PXD interaction remains strong enough to ensure recruitment of the polymerase. In all case, the results provided by the mutational study that targeted Box2 clearly point to the need of revisiting the accepted model whereby the NTAIL–PXD interaction has to be relatively weak to allow the polymerase to cartwheel on the nucleocapsid template. Indeed, a relatively labile complex can result either from an inherently lower affinity of the binding reaction, or from a tight complex whose strength is modulated by co-factors. Hsp70 is one such an experimentally confirmed modulator in the case of MeV. In fact, hsp70 binds to the same MeV NTAIL sites as PXD and thus competes out this latter [93]. It is, therefore, tempting to speculate that the progression of the MeV polymerase complex along the template could be ensured by hsp70. In this model hsp70 would promote successive cycles of binding and release thanks to its destabilizing effect on the NTAIL–PXD interaction.
4.2. Fuzziness of NTAIL–PXD Complexes
“Fuzziness” is a paradigm of protein structure and function that emerged from the observation that the ordering of an IDP upon binding to a target is often not complete, i.e., a significant residual structural disorder can persist in the complex [28]. In other words, an IDP could either sample a number of conformations at the surface of the partner (i.e., static fuzziness) or preserves polypeptide extensions in the disordered state (i.e., dynamic fuzziness). In this latter case, the flexible chains are usually not involved in complex formation but could serve for instance as tails for partner fishing with non-specific, transient contacts [141,142]. The abundance of such complexes in the literature supports the functional significance of residual structural disorder in macromolecular complexes.
The NTAIL–PXD complexes from paramyxoviruses provide illustrative examples of a combination of both static and dynamic fuzziness. Indeed, the induced α-MoRE within Henipavirus NTAIL domains was found to remain highly dynamic at the surface of PXD, i.e., to sample many sub-conformations reflecting a static fuzzy complex. On the other hand, the remainder of the NTAIL chain remains flexible within the three complexes thus supporting the formation of a dynamic fuzzy complex. These findings have been confirmed through several lines of evidences as described below (see also [143]).
For the three NTAIL domains, the majority of the peaks display chemical shifts that are nearly unaltered upon addition of the partner [63,64,87,90,91] indicating that the majority of NTAIL remains disordered in the bound form. In line with that, the experimentally determined RS of the NiV NTAIL–PXD complex (35.4 Å) suggests that binding to PXD does not imply formation of a compact complex (expected RS = 22.3 Å), with the resulting complex rather retaining a considerable flexibility [90]. In further support of the “fuzziness” within Henipavirus and MeV NTAIL–PXD complexes, EPR results pointed out lack of involvement of MeV Box1 and of Henipavirus Box1 and Box4 in complex formation, with the labels grafted within these Boxes retaining a high mobility and solvent accessibility [83,86]. In the same vein, intrinsic fluorescence spectroscopy data showed lack of PXD impact on the environment of a Trp residue introduced within Box4, thus further supporting a high conformational freedom and solvent exposure of the C-terminal region of Henipavirus NTAIL [90]. The NMR behavior of Henipavirus NTAIL, where some resonances disappear upon addition of PXD and never come back even at saturation [63,64,90], suggests that even when bound to PXD NTAIL remains dynamic, undergoing exchange between different conformers at the PXD surface [63]. SAXS studies of MeV NTAIL–PXD complex provided a low-resolution model of the NTAIL bound form, which showed that most of NTAIL (amino acids 401–488) remains disordered within the complex [91]. Furthermore, by combining SDSL EPR spectroscopy and modeling, an ensemble description of the structure of the MeV NTAIL region encompassing amino acids 489–525 bound to PXD could be obtained, which revealed that the region downstream the MoRE remains highly flexible (Figure 5D) [129]. Interestingly, a recent study combining ESI-IM-MS and modeling provided structural models of the MeV NTAIL–PXD complexes at the atomistic level (Figure 5E). These complexes are characterized by different levels of compaction, thus further supporting their structural heterogeneity [122]. Beyond documenting structural heterogeneity, those studies enabled to capture a collapsed form of the complex that had escaped detection in previous studies. Indeed, a bimodal charge state distribution was observed with a high-charge component (18+) and a low-charge (11+) component. While the former would correspond to an “open” conformation, in which the disordered arms of NTAIL flanking the α-MoRE fluctuate maintaining high solvent accessibility, the low-charge component likely represents a compact or “closed” conformation of the complex in which the NTAIL arms collapse onto the surface of the folded partner [122]. Computational modeling of the “open” complex in solution, using experimental chemical shifts as restraints, provided atomic-resolution structural models with calculated solvent accessible surface area (SASA) in good agreement with that experimentally determined by ESI-MS. In the resulting models, the intermolecular interactions are predominantly hydrophobic, not only in the ordered core of the complex, but also in the disordered regions.
Many functional advantages can result from fuzziness, including interactions with alternative partners and simultaneous interactions with different partners. Indeed, the residual plasticity often allows adaptation of the same motif to different partners, or a variable arrangement of the recognition motifs, which can mediate interactions with alternative partners (i.e., promiscuity). Moreover, the disordered tails in complexes can serve for partner fishing via non-specific, transient contacts. Fuzzy parts of the complexes can harbor regulatory PTM sites. They can even directly or indirectly interfere with (promote or inhibit) binding of the part that undergoes folding transition. In addition, fuzziness provides a way to reduce the entropic penalty that accompanies the disorder-to-order transition, thereby affording enhanced affinity. Tuning fuzziness therefore provides an additional way to modulate the interaction strength.
In line with these expectations, the fuzzy Box3 region of MeV NTAIL was shown to serve as a binding site for hsp70 [93,94], where the latter is known to stimulate both viral transcription and replication [92,144,145]. Box3 constitutes however a low-affinity binding site for hsp70, with Box2 providing a high-affinity binding site (KD of 10 nM) [92,146]. Since hsp70 competitively inhibits PXD binding to NTAIL [93], hsp70 could enhance transcription and genome replication by reducing the stability of NTAIL–PXD complexes [91,93]. The hsp70-dependent reduction of the stability of P-NTAIL complexes would thus rely on competition between hsp70 and PXD for binding to the α-MoRE of NTAIL, with recruitment of hsp70 being ensured by both Box2 and Box3 [93].
As already mentioned, PTM is over-represented in IDPs [45,147], and disorder-to-order transitions occur in some cases after PTMs, such as phosphorylation, thus supporting a significant role of PTMs in regulating the functions of IDPs [45]. Interestingly, in the case of MeV, phosphorylation of the N protein has been shown to upregulate the transcriptional activity of minigenomic RNA and to regulate viral genomic RNA stability [148,149]. Likewise, in the case of NiV, phosphorylation of N was found to be involved in the regulation of viral RNA synthesis, with rapid turnover of phosphorylation being critical [150]. Two residues located within MeV NTAIL, namely S479 and S510, have been found to constitute the major phosphorylation sites within MeV N, while in the case of NiV only one phosphorylated residue, namely S451, was identified [150]. Interestingly, these residues are all located in fuzzy regions of NTAIL, which is in good agreement with the three structural requirements of PTMs: an appropriate local sequence, structural exposure, and flexibility of the site so that it can be productively accommodated by the active site of the modifying enzyme [45,147].
Finally, fuzzy regions flanking MoREs can also serve as natural modulators of the interactions established by IDPs. In fact, a recent random mutagenesis study of MeV NTAIL led to the identification of five regulatory regions that are located in the N-terminal fuzzy region of NTAIL and dampen the interaction [151]. This finding is consistent with recent observations based on mini-replicon studies that unveiled that the region upstream Box2 acts as a negative modulator for the binding of the polymerase complex [104]. In the same vein, MeV NTAIL variants devoid of Box3 were found to exhibit enhanced interaction with PXD, suggesting that Box3 would naturally serve as a dampener (see Section 4.3).
4.3. Molecular Determinants of the Affinity of the NTAIL–PXD Interaction as Unveiled by Random Mutagenesis
In the last decade, a wealth of bioinformatics and experimental studies showed that intrinsic disorder enhances protein interactivity, with IDPs and IDRs being able to bind several partners while maintaining both specificity and binding efficiencies. However, the molecular features of the binding efficiency of IDP/Rs (i.e., un/coupling between affinity and specificity) is far from being elucidated (see [152] for a review on this topic). In order to shed light onto these aspects, we applied a combinatorial experimental approach on the MeV NTAIL–PXD complex, which not only provided further perspicacity on the NTAIL–PXD complex, but also proved to be a valuable general approach to characterize complexes involving IDPs/IDRs.
This approach, termed “descriptive random mutagenesis”, is an unbiased method that relies on targeting the sequence of an IDP/IDR, in this case MeV NTAIL, by random mutagenesis thus allowing to assess how amino acid substitutions introduced at random affect partner recognition. Subsequent to the generation of a library of NTAIL random mutants, the interaction strength towards PXD was evaluated using a protein complementation assay based on green fluorescent protein (GFP) reassembly [153].
In this assay, each partner is fused to a GFP moiety and the interaction between the two proteins drives the re-assembly of the two GFP fragments thus producing a fluorescence signal that is directly related to the binding strength [154]. Besides confirming previous results on the crucial role of the α-MoRE in binding to PXD, the obtained results provided novel insights by dissecting the NTAIL region and identifying regulatory segments within the fuzzy parts of NTAIL [154].
In that study, 224 variants out of 300, which were randomly chosen without any selection pressure, were found to encode full-length forms of NTAIL with the substitutions providing a full coverage of the whole length of the NTAIL sequence (i.e., each amino acid of the sequence was found to be substituted at least once). Interestingly, most of these substitutions were shown to affect the NTAIL capacity to bind PXD. Substitutions within Box2 (i.e., the PXD-binding site) were found to lead to a reduced fluorescence, thus indicating that Box2 is poorly evolvable in terms of binding abilities towards PXD. The critical positions leading to the highest decrease in the fluorescence were found to correspond to residues with side chains oriented to the partner. The study led however also to the identification of Arg497 as an additional critical Box2 residue for stabilizing the NTAIL–PXD, whose role in complex formation had escaped detection in previous studies. Although the side chain of Arg497 points out of the binding surface, it is located at bonding distance from the hydroxyl group of Tyr480 of PXD. Through generation and characterization of a “mirror” PXD variant bearing the Y480F substitution, the crucial role of the Arg497-Tyr480 interaction in stabilizing the NTAIL–PXD complex was confirmed [154].
Beyond Box2, the study also allowed the identification of five regulatory regions, termed e-boxes (enhancer-boxes), located in the fuzzy region upstream Box2. Mutating these regions leads to enhanced interaction, indicating that e-boxes act as natural dampeners of the interaction. The precise mechanism by which they exert this role remains however unknown.
Random mutagenesis of NTAIL also resulted in the generation of truncated variants (i.e., 76 out of 300 variants) arising from the insertion of a stop codon. In line with expectations, variants devoid of Box2 showed a dramatic drop in the fluorescence, reflecting loss of interaction. Most interestingly, variants that are only devoid of Box3 were found to display an increased fluorescence, thus unveiling an inhibitory effect of Box3 on the interaction with PXD.
In conclusion, this study unveiled that most of the NTAIL sequence is sensitive to mutations and possesses a few regulatory sites located within fuzzy regions. The fuzziness of NTAIL may therefore not only serve as a way to capture other binding partners but also to modulate the strength of interactions established by NTAIL.
6. Conclusions
When we analyzed the modular organization of the P proteins within the Paramyxovirinae subfamily, we noticed that a larger PNT domain in Henipavirus P proteins accounts for the extra length of their P protein with respect to other paramyxoviruses [30]. This finding is consistent with the higher tolerance of disordered regions to insertions or major rearrangements as compared to ordered ones. Moreover, since the P-encoded proteins are believed to possess anti-interferon functions, the extension in Henipavirus P proteins might have evolved to better equip these viruses so as to enhance their capacity to overcome the cellular interferon response. Furthermore, the disordered nature of PNT and of the “spacer” region connecting PNT to PMD likely reflects a way of alleviating evolutionary constraints within overlapping reading frames. Indeed, PNT partially overlaps with the C protein (being encoded by the same RNA region), and the “spacer” region partially overlaps with the C-terminal domain of the V protein [30,31]. This observation is in agreement with previous reports pointing out a relationship between overlapping genes and structural disorder [31,170,171,172,173]. We thus reasoned that structural disorder, which is encoded by a much wider portion of sequence space as compared to order, can indeed represent a strategy by which genes encoding overlapping reading frames can lessen evolutionary constraints imposed on their sequence by the overlap, allowing the encoded overlapping protein products to sample a wider sequence space without losing function.
Following our seminal studies that pointed out the abundance of disorder in Paramyxovirinae N and P proteins [31,32,57,174], several subsequent studies have documented the prevalence of disorder in viral proteins using both computational and experimental approaches (see [175] and references therein cited). Bioinformatics studies showed that viral proteins, and in particular proteins from RNA viruses, have a high disorder content [176,177]. In those studies, the authors propose that beyond affording a broad partnership, the wide occurrence of disordered regions in viral proteins could also be related to the typical high mutation rates of RNA viruses, i.e., it could represent a strategy for buffering the deleterious effects of mutations.
A detailed comparative examination of viral and non-viral proteins showed that, amongst several distinguishing characteristics, viral proteins possess (i) a larger fraction of residues that are not organized into regular secondary structural elements; (ii) conformational stabilities that are less affected by mutations; (iii) a high rate of mutations; (iv) enrichment in proteins encoded by overlapping reading frames; and (v) a higher content of polar residues. These features indicate that they have been shaped by evolution to be endowed with better adaptation to their hostile habitats and to rapid changes in their biological and physical environment. Indeed, one of the many noteworthy features of viruses is their ability to adapt to very harsh and hostile environments and to adjust themselves according to the biological and genetic features of the hosts, which in turn are often adapted to exist at extreme conditions (see [175] and references therein cited). The above-mentioned features are interestingly in intimate relationship with structural disorder, which indeed provides several advantages.
In fact, because viruses are obligate intracellular parasites, they have to interact with various components of the host, including membranes, nucleic acids, and proteins. The lack of a rigid 3D structure imparts to IDP/Rs the necessary plasticity to establish various interactions with several partners at once. In the course of evolution, viruses have “learned” to hijack and manipulate host proteins for their benefit, and to evade the host defense mechanisms. A recent study by Davey and co-workers showed that viruses have achieved this ability through broad mimicry of host protein short linear motifs (SLiMs) [178], where the latter are embedded in disordered regions and play a variety of roles, including targeting host proteins for proteosomal degradation, cell signaling, directing proteins to the correct subcellular localization, deregulating cell cycle checkpoints, and altering transcription of host proteins [179]. Importantly, binding to cell proteins through sites that mimic SLiMs also helps viral proteins in eluding the host cell’s immune system, by rendering viral epitopes poorly recognizable by the host immune system (see [175] and references therein cited).
Based on all these considerations, we proposed that the main advantage of the abundance of disorder within viral proteins would reside in pleiotropy and genetic compaction [175]. Indeed, disorder provides a solution to reduce both genome size and molecular crowding, where a single gene would (i) encode a single (regulatory) protein product that can establish multiple interactions via its disordered regions and hence exert multiple concomitant biological effects including evasion of the host immune response; and/or (ii) would encode more than one product by means of overlapping reading frames.
Acknowledgments
Sonia Longhi wishes to thank all the members of her lab and her co-workers for their critical contribution to the studies herein summarized. Within her group, she thanks David Karlin, François Ferron, Jean-Marie Bourhis, Kenth Johansson, Antoine Gruet, David Blocquel, Jenny Erales, Lorenzo Baronti, and Jennifer Roche (previous members) and Marion Dosnon, Matilde Beltrandi and Christophe Bignon (present members). Among her numerous past and present co-workers, she thanks Bruno Canard (AFMB, Marseille, France), Denis Gerlier (CIRI, Lyon, France), Michael Oglesbee (Ohio State University, Columbus, OH, USA), Hélène Valentin (CIRI, Lyon, France), Valerie Belle and Bruno Guigliarelli (BIP, Marseille, France), Janez Strancar (Jozef Stefan Institute, Ljubljana, Slovenia), Gary Daughdrill (University of South Florida, Tampa, FL, USA), Martin Blackledge and Malene Ringkjobin-Jensen (Institut de Biologie Structurale, Grenoble, France), Jin Wang (Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, China), Roberta Pierattelli and Isabella Felli (CERM, Florence, Italy), Rita Grandori (Universita’ degli Studi Milano-Bicocca, Milan, Italy), Paolo Carloni (Institute for Advanced Simulation IAS-5 and Institute of Neuroscience and Medicine INM-9, Jülich, Düren, Germany) and Stefano Gianni (La Sapienza, Universita’ di Roma, Rome, Italy). She is particularly grateful to Denis Gerlier (CIRI, Lyon, France) and to Vladimir Uversky (University of South Florida) for the numerous stimulating discussions and for their useful advice on various issues. The studies herein reviewed were carried out with the financial support of the Agence Nationale de la Recherche, specific programs “Physico-Chimie du Vivant”, ANR-08-PCVI-0020-01, and “ASTRID”, ANR-11-ASTR-003-01. They also benefited from support from the CNRS, the Direction Générale de l’Armement (DGA) and the Fondation pour la Recherche Médicale (FRM). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eaton, B.T.; Mackenzie, J.S.; Wang, L.F. Henipaviruses. In Fields Virology, 5th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2007; pp. 1587–1600. [Google Scholar]
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1450–1497. [Google Scholar]
- Wang, L.F.; Yu, M.; Hansson, E.; Pritchard, L.I.; Shiell, B.; Michalski, W.P.; Eaton, B.T. The exceptionally large genome of Hendra virus: Support for creation of a new genus within the family Paramyxoviridae. J. Virol. 2000, 74, 9972–9979. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Bankamp, B.; Harcourt, B.H.; Bellini, W.J.; Rota, P.A. Nipah virus conforms to the rule of six in a minigenome replication assay. J. Gen. Virol. 2004, 85, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Kolakofsky, D. Paramyxoviridae: The viruses and their replication. In Fields Virology, 4th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2001; pp. 1305–1340. [Google Scholar]
- Bhella, D.; Ralph, A.; Murphy, L.B.; Yeo, R.P. Significant differences in nucleocapsid morphology within the Paramyxoviridae. J. Gen. Virol. 2002, 83, 1831–1839. [Google Scholar] [PubMed]
- Tan, W.S.; Ong, S.T.; Eshaghi, M.; Foo, S.S.; Yusoff, K. Solubility, immunogenicity and physical properties of the nucleocapsid protein of nipah virus produced in Escherichia coli. J. Med. Virol. 2004, 73, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Cattaneo, R.; Spielhofer, P.; Orvell, C.; Norrby, E.; Messerli, M.; Perriard, J.C.; Billeter, M.A. Measles virus phosphoprotein retains the nucleocapsid protein in the cytoplasm. Virology 1991, 185, 299–308. [Google Scholar] [CrossRef]
- Spehner, D.; Drillien, R.; Howley, P.M. The assembly of the measles virus nucleoprotein into nucleocapsid-like particles is modulated by the phosphoprotein. Virology 1997, 232, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Albertini, A.A.V.; Schoehn, G.; Ruigrok, R.W. Structures impliquées dans la réplication et la transcription des virus à arn non segmentés de sens négatif. Virologie 2005, 9, 83–92. (In French) [Google Scholar]
- Blocquel, D.; Bourhis, J.M.; Eléouët, J.F.; Gerlier, D.; Habchi, J.; Jamin, M.; Longhi, S.; Yabukarski, F. Transcription et réplication des mononégavirales: Une machine moléculaire originale. Virologie 2012, 16, 225–257. (In French) [Google Scholar]
- Lamb, R.A.; Krug, R.M. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 4th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2001; pp. 1487–1531. [Google Scholar]
- Longhi, S.; Canard, B. Mécanismes de transcription et de réplication des Paramyxoviridae. Virologie 1999, 3, 227–240. (In French) [Google Scholar]
- Roux, L. Dans le génome des paramyxovirinae, les promoteurs et leurs activités sont façonnés par la «règle de six». Virologie 2005, 9, 19–34. (In French) [Google Scholar]
- Morin, B.; Rahmeh, A.A.; Whelan, S.P. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J. 2012, 31, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Banerjee, A.K. Phosphoprotein, P of human parainfluenza virus type 3 prevents self-association of RNA-dependent RNA polymerase, L. Virology 2009, 383, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, M.; Shaila, M.S. Recombinant L and P protein complex of Rinderpest virus catalyses mRNA synthesis in vitro. Virus Res. 2008, 135, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Kobayashi, M.; Iwama, M.; Mizumoto, K. Sendai virus RNA-dependent RNA polymerase L protein catalyzes cap methylation of virus-specific mRNA. J. Biol. Chem. 2005, 280, 4429–4435. [Google Scholar] [CrossRef] [PubMed]
- Noton, S.L.; Deflube, L.R.; Tremaglio, C.Z.; Fearns, R. The respiratory syncytial virus polymerase has multiple RNA synthesis activities at the promoter. PLoS Pathog. 2012, 8, e1002980. [Google Scholar] [CrossRef] [PubMed]
- Fix, J.; Galloux, M.; Blondot, M.L.; Eleouet, J.F. The insertion of fluorescent proteins in a variable region of respiratory syncytial virus L polymerase results in fluorescent and functional enzymes but with reduced activities. Open Virol. J. 2011, 5, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.M.; Longhi, S. Measles virus nucleoprotein: Structural organization and functional role of the intrinsically disordered C-terminal domain. In Measles Virus Nucleoprotein; Longhi, S., Ed.; Nova Publishers Inc.: Hauppage, NY, USA, 2007; pp. 1–35. [Google Scholar]
- Longhi, S. Measles Virus Nucleoprotein; Nova Publishers Inc.: Hauppage, NY, USA, 2007. [Google Scholar]
- Longhi, S. Nucleocapsid structure and function. Curr. Top. Microbiol. Immunol. 2009, 329, 103–128. [Google Scholar] [PubMed]
- Longhi, S.; Oglesbee, M. Structural disorder within the measles virus nucleoprotein and phosphoprotein. Protein Pept. Lett. 2010, 17, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S. Structural disorder within the measles virus nucleoprotein and phosphoprotein: Functional implications for transcription and replication. In Negative Strand RNA Virus; Luo, M., Ed.; World Scientific Publishing: Singapore, Singapore, 2011; pp. 95–125. [Google Scholar]
- Habchi, J.; Longhi, S. Structural disorder within paramyxovirus nucleoproteins and phosphoproteins. Mol. Biosyst. 2012, 8, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Mamelli, L.; Longhi, S. Structural disorder within the nucleoprotein and phosphoprotein from measles, nipah and hendra viruses. In Flexible Viruses: Structural Disorder in Viral Proteins; Uversky, V.N., Longhi, S., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 47–94. [Google Scholar]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein–protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Mamelli, L.; Darbon, H.; Longhi, S. Structural disorder within Henipavirus nucleoprotein and phosphoprotein: From predictions to experimental assessment. PLoS ONE 2010, 5, e11684. [Google Scholar] [CrossRef] [PubMed]
- Karlin, D.; Ferron, F.; Canard, B.; Longhi, S. Structural disorder and modular organization in Paramyxovirinae N and P. J. Gen. Virol. 2003, 84, 3239–3252. [Google Scholar] [CrossRef] [PubMed]
- Karlin, D.; Longhi, S.; Receveur, V.; Canard, B. The N-terminal domain of the phosphoprotein of morbilliviruses belongs to the natively unfolded class of proteins. Virology 2002, 296, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.; Johansson, K.; Receveur-Bréchot, V.; Oldfield, C.J.; Dunker, A.K.; Canard, B.; Longhi, S. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004, 99, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Houben, K.; Marion, D.; Tarbouriech, N.; Ruigrok, R.W.; Blanchard, L. Interaction of the C-terminal domains of sendai virus N and P proteins: Comparison of polymerase-nucleocapsid interactions within the Paramyxovirus family. J. Virol. 2007, 81, 6807–6816. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Bourhis, J.M.; Campanacci, V.; Cambillau, C.; Canard, B.; Longhi, S. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 2003, 278, 44567–44573. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.L.; Hamel, D.J.; Gay, L.S.; Dahlquist, F.W.; Matthews, B.W. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. USA 2004, 101, 8301–8306. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.L.; Walter, A.B.; Gay, L.S. Characterization of nucleocapsid binding by the measles and the mumps virus phosphoprotein. J. Virol. 2004, 78, 8630–8640. [Google Scholar] [CrossRef] [PubMed]
- Bernado, P.; Blanchard, L.; Timmins, P.; Marion, D.; Ruigrok, R.W.; Blackledge, M. A structural model for unfolded proteins from residual dipolar couplings and small-angle X-ray scattering. Proc. Natl. Acad. Sci. USA 2005, 102, 17002–17007. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, L.; Tarbouriech, N.; Blackledge, M.; Timmins, P.; Burmeister, W.P.; Ruigrok, R.W.; Marion, D. Structure and dynamics of the nucleocapsid-binding domain of the sendai virus phosphoprotein in solution. Virology 2004, 319, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Houben, K.; Blanchard, L.; Blackledge, M.; Marion, D. Intrinsic dynamics of the partly unstructured PX domain from the sendai virus RNA polymerase cofactor P. Biophys. J. 2007, 93, 2830–2844. [Google Scholar] [CrossRef] [PubMed]
- Communie, G.; Crepin, T.; Maurin, D.; Jensen, M.R.; Blackledge, M.; Ruigrok, R.W. Structure of the tetramerization domain of measles virus phosphoprotein. J. Virol. 2013, 87, 7166–7169. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.M.; Sun, D.; Schmitt, A.P.; He, B. Phosphorylation of paramyxovirus phosphoprotein and its role in viral gene expression. Future Microbiol. 2010, 5, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Gerard, F.C.; Ribeiro Ede, A., Jr.; Leyrat, C.; Ivanov, I.; Blondel, D.; Longhi, S.; Ruigrok, R.W.; Jamin, M. Modular organization of rabies virus phosphoprotein. J. Mol. Biol. 2009, 388, 978–996. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Gerard, F.C.; de Almeida Ribeiro, E., Jr.; Ivanov, I.; Ruigrok, R.W.; Jamin, M. Structural disorder in proteins of the rhabdoviridae replication complex. Protein Pept. Lett. 2010, 17, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Ringkjøbing Jensen, M.; Communie, G.; Ribeiro, E.D., Jr.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [Google Scholar]
- Ruigrok, R.W.; Crepin, T.; Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 2011, 14, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Warnes, A.; Fooks, A.R.; Dowsett, A.B.; Wilkinson, G.W.; Stephenson, J.R. Expression of the measles virus nucleoprotein gene in Escherichia coli and assembly of nucleocapsid-like structures. Gene 1995, 160, 173–178. [Google Scholar] [CrossRef]
- Bhella, D. Measles virus nucleocapsid structure, conformational flexibility and the rule of six. In Measles Virus Nucleoprotein; Longhi, S., Ed.; Nova Publishers Inc.: Hauppage, NY, USA, 2007. [Google Scholar]
- Eshaghi, M.; Tan, W.S.; Ong, S.T.; Yusoff, K. Purification and characterization of Nipah virus nucleocapsid protein produced in insect cells. J. Clin. Microbiol. 2005, 43, 3172–3177. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Bernado, P.; Houben, K.; Blanchard, L.; Marion, D.; Ruigrok, R.W.; Blackledge, M. Structural disorder within sendai virus nucleoprotein and phosphoprotein: Insight into the structural basis of molecular recognition. Protein Pept. Lett. 2010, 17, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Heggeness, M.H.; Scheid, A.; Choppin, P.W. Conformation of the helical nucleocapsids of paramyxoviruses and vesicular stomatitis virus: Reversible coiling and uncoiling induced by changes in salt concentration. Proc. Natl. Acad. Sci. USA 1980, 77, 2631–2635. [Google Scholar] [CrossRef] [PubMed]
- Heggeness, M.H.; Scheid, A.; Choppin, P.W. The relationship of conformational changes in the sendai virus nucleocapsid to proteolytic cleavage of the NP polypeptide. Virology 1981, 114, 555–562. [Google Scholar] [CrossRef]
- Karlin, D.; Longhi, S.; Canard, B. Substitution of two residues in the measles virus nucleoprotein results in an impaired self-association. Virology 2002, 302, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Bankamp, B.; Horikami, S.M.; Thompson, P.D.; Huber, M.; Billeter, M.; Moyer, S.A. Domains of the measles virus N protein required for binding to P protein and self-assembly. Virology 1996, 216, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Liston, P.; Batal, R.; DiFlumeri, C.; Briedis, D.J. Protein interaction domains of the measles virus nucleocapsid protein (NP). Arch. Virol. 1997, 142, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed]
- Bhella, D.; Ralph, A.; Yeo, R.P. Conformational flexibility in recombinant measles virus nucleocapsids visualised by cryo-negative stain electron microscopy and real-space helical reconstruction. J. Mol. Biol. 2004, 340, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Schoehn, G.; Mavrakis, M.; Albertini, A.; Wade, R.; Hoenger, A.; Ruigrok, R.W. The 12Å structure of trypsin-treated measles virus N–RNA. J. Mol. Biol. 2004, 339, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagné, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. 3D structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Alayyoubi, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein-RNA complex. Proc. Natl. Acad. Sci. USA 2015, 112, E1792–E1799. [Google Scholar] [CrossRef] [PubMed]
- Desfosses, A.; Goret, G.; Farias Estrozi, L.; Ruigrok, R.W.; Gutsche, I. Nucleoprotein-RNA orientation in the measles virus nucleocapsid by three-dimensional electron microscopy. J. Virol. 2011, 85, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Communie, G.; Habchi, J.; Yabukarski, F.; Blocquel, D.; Schneider, R.; Tarbouriech, N.; Papageorgiou, N.; Ruigrok, R.W.; Jamin, M.; Ringkjøbing-Jensen, M.; et al. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of hendra virus. PLoS Pathog. 2013, 9, e1003631. [Google Scholar] [CrossRef] [PubMed]
- Baronti, L.; Erales, J.; Habchi, J.; Felli, I.C.; Pierattelli, R.; Longhi, S. Dynamics of the intrinsically disordered C-terminal domain of the nipah virus nucleoprotein and interaction with the X domain of the phosphoprotein as unveiled by NMR spectroscopy. ChemBioChem 2015, 16, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.; Sachse, C.; Schoehn, G. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Yabukarski, F.; Lawrence, P.; Tarbouriech, N.; Bourhis, J.M.; Delaforge, E.; Jensen, M.R.; Ruigrok, R.W.; Blackledge, M.; Volchkov, V.; Jamin, M. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat. Struct. Mol. Biol. 2014, 21, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Romero, P.; Dunker, A.K.; Brown, C.; Obradovic, Z. Predicting binding regions within disordered proteins. Genome Inform. 1999, 10, 41–50. [Google Scholar]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, morfs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Kawano, M.; Tsurudome, M.; Nishio, M.; Ito, M.; Ohgimoto, S.; Suga, S.; Komada, H.; Ito, Y. Binding of the V proteins to the nucleocapsid proteins of human parainfluenza type 2 virus. Med. Microbiol. Immunol. 1996, 185, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Karlin, D.; Belshaw, R. Detecting remote sequence homology in disordered proteins: Discovery of conserved motifs in the N-termini of Mononegavirales phosphoproteins. PLoS ONE 2012, 7, e31719. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Jensen, M.R.; Ribeiro, E.A., Jr.; Gerard, F.C.; Ruigrok, R.W.; Blackledge, M.; Jamin, M. The N0-binding region of the vesicular stomatitis virus phosphoprotein is globally disordered but contains transient α-helices. Protein Sci. 2011, 20, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Yabukarski, F.; Tarbouriech, N.; Ribeiro, E.A., Jr.; Jensen, M.R.; Blackledge, M.; Ruigrok, R.W.; Jamin, M. Structure of the vesicular stomatitis virus N0-P complex. PLoS Pathog. 2011, 7, e1002248. [Google Scholar] [CrossRef] [PubMed]
- Sweetman, D.A.; Miskin, J.; Baron, M.D. Rinderpest virus C and V proteins interact with the major (L) component of the viral polymerase. Virology 2001, 281, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Blocquel, D.; Beltrandi, M.; Erales, J.; Barbier, P.; Longhi, S. Biochemical and structural studies of the oligomerization domainof the Nipah virus phosphoprotein: evidence for an elongated coiled-coil homotrimer. Virology 2013, 446, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Beltrandi, M.; Blocquel, D.; Erales, J.; Barbier, P.; Cavalli, A.; Longhi, S. Insights into the coiled-coil organization of the Hendra virus phosphoprotein from combined biochemical and SAXS studies. Virology 2015, 477, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Blocquel, D.; Habchi, J.; Durand, E.; Sevajol, M.; Ferron, F.; Erales, J.; Papageorgiou, N.; Longhi, S. Coiled-coil deformations in crystal structures: The measles virus phosphoprotein multimerization domain as an illustrative example. Acta Cryst. D 2014, 70, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Blocquel, D.; Habchi, J.; Gruet, A.; Blangy, S.; Longhi, S. Compaction and binding properties of the intrinsically disordered C-terminal domain of henipavirus nucleoprotein as unveiled by deletion studies. Mol. Biosyst. 2012, 8, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Structure and Function of Intrinsically Disordered Proteins; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2010. [Google Scholar]
- Hazy, E.; Tompa, P. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem 2009, 10, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Martinho, M.; Habchi, J.; El Habre, Z.; Nesme, L.; Guigliarelli, B.; Belle, V.; Longhi, S. Assessing induced folding within the intrinsically disordered C-terminal domain of the Henipavirus nucleoproteins by site directed spin labeling EPR spectroscopy. J. Biomol. Struct. Dyn. 2013, 31, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chu, X.; Longhi, S.; Roche, P.; Han, W.; Wang, E.; Wang, J. Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein. Proc. Natl. Acad. Sci. USA 2013, 110, E3743–E3752. [Google Scholar] [CrossRef] [PubMed]
- Morin, B.; Bourhis, J.M.; Belle, V.; Woudstra, M.; Carrière, F.; Guigliarelli, B.; Fournel, A.; Longhi, S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling epr spectroscopy. J. Phys. Chem. B 2006, 110, 20596–20608. [Google Scholar] [CrossRef] [PubMed]
- Belle, V.; Rouger, S.; Costanzo, S.; Liquiere, E.; Strancar, J.; Guigliarelli, B.; Fournel, A.; Longhi, S. Mapping α-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins Struct. Funct. Bioinform. 2008, 73, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Gely, S.; Lowry, D.F.; Bernard, C.; Ringkjobing-Jensen, M.; Blackledge, M.; Costanzo, S.; Darbon, H.; Daughdrill, G.W.; Longhi, S. Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J. Mol. Recognit. 2010, 23, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Houben, K.; Lescop, E.; Blanchard, L.; Ruigrok, R.W.; Blackledge, M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: Application to the molecular recognition element of sendai virus nucleoprotein. J. Am. Chem. Soc. 2008, 130, 8055–8061. [Google Scholar] [CrossRef] [PubMed]
- Serrano, L.; Fersht, A.R. Capping and α-helix stability. Nature 1989, 342, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Blangy, S.; Mamelli, L.; Ringkjobing Jensen, M.; Blackledge, M.; Darbon, H.; Oglesbee, M.; Shu, Y.; Longhi, S. Characterization of the interactions between the nucleoprotein and the phosphoprotein of henipaviruses. J. Biol. Chem. 2011, 286, 13583–13602. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.M.; Receveur-Bréchot, V.; Oglesbee, M.; Zhang, X.; Buccellato, M.; Darbon, H.; Canard, B.; Finet, S.; Longhi, S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005, 14, 1975–1992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Glendening, C.; Linke, H.; Parks, C.L.; Brooks, C.; Udem, S.A.; Oglesbee, M. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 2002, 76, 8737–8746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bourhis, J.M.; Longhi, S.; Carsillo, T.; Buccellato, M.; Morin, B.; Canard, B.; Oglesbee, M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 2005, 337, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Couturier, M.; Buccellato, M.; Costanzo, S.; Bourhis, J.M.; Shu, Y.; Nicaise, M.; Desmadril, M.; Flaudrops, C.; Longhi, S.; Oglesbee, M. High affinity binding between Hsp70 and the C-terminal domain of the measles virus nucleoprotein requires an Hsp40 co-chaperone. J. Mol. Recognit. 2010, 23, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Tenoever, B.R.; Servant, M.J.; Grandvaux, N.; Lin, R.; Hiscott, J. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J. Virol. 2002, 76, 3659–3669. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Bourhis, J.M.; Chamontin, C.; Soriano, C.; Villet, S.; Costanzo, S.; Couturier, M.; Belle, V.; Fournel, A.; Darbon, H.; et al. The interaction between the measles virus nucleoprotein and the interferon regulator factor 3 relies on a specific cellular environment. Virol. J. 2009, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Laine, D.; Bourhis, J.; Longhi, S.; Flacher, M.; Cassard, L.; Canard, B.; Sautès-Fridman, C.; Rabourdin-Combe, C.; Valentin, H. Measles virus nucleoprotein induces cell proliferation arrest and apoptosis through NTAIL/NR and NCORE/FCγRIIB1 interactions, respectively. J. Gen. Virol. 2005, 86, 1771–1784. [Google Scholar] [CrossRef] [PubMed]
- Laine, D.; Trescol-Biémont, M.; Longhi, S.; Libeau, G.; Marie, J.; Vidalain, P.; Azocar, O.; Diallo, A.; Canard, B.; Rabourdin-Combe, C.; et al. Measles virus (MV) nucleoprotein binds to a novel cell surface receptor distinct from FcγRII via its C-terminal domain: Role in MV-induced immunosuppression. J. Virol. 2003, 77, 11332–11346. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Masuda, M.; Miura, R.; Yoneda, M.; Kai, C. Morbillivirus nucleoprotein possesses a novel nuclear localization signal and a CRM1-independent nuclear export signal. Virology 2006, 352, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Takeda, M.; Shirogane, Y.; Nakatsu, Y.; Nakamura, T.; Yanagi, Y. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J. Virol. 2009, 83, 10374–10383. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Yoneda, M.; Ikeda, F.; Sugai, A.; Sato, H.; Kai, C. Peroxiredoxin 1 is required for efficient transcription and replication of measles virus. J. Virol. 2011, 85, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- De, B.P.; Banerjee, A.K. Involvement of actin microfilaments in the transcription/replication of human parainfluenza virus type 3: Possible role of actin in other viruses. Microsc. Res. Tech. 1999, 47, 114–123. [Google Scholar] [CrossRef]
- Moyer, S.A.; Baker, S.C.; Horikami, S.M. Host cell proteins required for measles virus reproduction. J. Gen. Virol. 1990, 71, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Krumm, S.A.; Takeda, M.; Plemper, R.K. The measles virus nucleocapsid protein tail domain is dispensable for viral polymerase recruitment and activity. J. Biol. Chem. 2013, 288, 29943–29953. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cortay, J.C.; Gerlier, D. Measles virus protein interactions in yeast: New findings and caveats. Virus Res. 2003, 98, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Liston, P.; DiFlumeri, C.; Briedis, D.J. Protein interactions entered into by the measles virus P, V, and C proteins. Virus Res. 1995, 38, 241–259. [Google Scholar] [CrossRef]
- Curran, J.; Pelet, T.; Kolakofsky, D. An acidic activation-like domain of the sendai virus P protein is required for RNA synthesis and encapsidation. Virology 1994, 202, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.; Marq, J.B.; Kolakofsky, D. An N-terminal domain of the sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J. Virol. 1995, 69, 849–855. [Google Scholar] [PubMed]
- Tarbouriech, N.; Curran, J.; Ruigrok, R.W.; Burmeister, W.P. Tetrameric coiled coil domain of sendai virus phosphoprotein. Nat. Struct. Biol. 2000, 7, 777–781. [Google Scholar] [PubMed]
- Rahaman, A.; Srinivasan, N.; Shamala, N.; Shaila, M.S. Phosphoprotein of the rinderpest virus forms a tetramer through a coiled coil region important for biological function. A structural insight. J. Biol. Chem. 2004, 279, 23606–23614. [Google Scholar] [CrossRef] [PubMed]
- Llorente, M.T.; Barreno-Garcia, B.; Calero, M.; Camafeita, E.; Lopez, J.A.; Longhi, S.; Ferron, F.; Varela, P.F.; Melero, J.A. Structural analysis of the human respiratory syncitial virus phosphoprotein: Characterization of an α-helical domain involved in oligomerization. J. Gen. Virol. 2006, 87, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.; Green, T.J.; Purushotham, S.; Deivanayagam, C.; Bedwell, G.J.; Prevelige, P.E.; Luo, M. Structural and functional characterization of the mumps virus phosphoprotein. J. Virol. 2013, 87, 7558–7568. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Renner, M.; Harlos, K.; Grimes, J.M. Solution and crystallographic structures of the central region of the phosphoprotein from human metapneumovirus. PLoS ONE 2013, 8, e80371. [Google Scholar] [CrossRef] [PubMed]
- Bruhn-Johannsen, J.F.; Barnett, K.; Bibby, J.; Thomas, J.; Keegan, R.; Rigden, D.; Bornholdt, Z.A.; Saphire, E.O. Crystal structure of the Nipah virus phosphoprotein tetramerization domain. J. Virol. 2014, 88, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Dutta, K.; Alexandrov, A.; Huang, H.; Pascal, S.M. pH-induced folding of an apoptotic coiled coil. Protein Sci. 2001, 10, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A.N.; Gruber, M. The structure of α-helical coiled coils. Adv. Protein Chem. 2005, 70, 37–78. [Google Scholar] [PubMed]
- Oshaben, K.M.; Salari, R.; McCaslin, D.R.; Chong, L.T.; Horne, W.S. The native GCN4 leucine-zipper domain does not uniquely specify a dimeric oligomerization state. Biochemistry 2012, 51, 9581–9591. [Google Scholar] [CrossRef] [PubMed]
- Curran, J. A role for the sendai virus p protein trimer in RNA synthesis. J. Virol. 1998, 72, 4274–4280. [Google Scholar] [PubMed]
- Curran, J.; Boeck, R.; Lin-Marq, N.; Lupas, A.; Kolakofsky, D. Paramyxovirus phosphoproteins form homotrimers as determined by an epitope dilution assay, via predicted coiled coils. Virology 1995, 214, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.L.; Gay, L.S.; Baase, W.S.; Matthews, B.W. Structure of the nucleocapsid-binding domain from the mumps virus polymerase; an example of protein folding induced by crystallization. J. Mol. Biol. 2008, 379, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Yegambaram, K.; Bulloch, E.M.; Kingston, R.L. Protein domain definition should allow for conditional disorder. Protein Sci. 2013, 22, 1502–1518. [Google Scholar] [CrossRef] [PubMed]
- D’Urzo, A.; Konijnenberg, A.; Rossetti, G.; Habchi, J.; Li, J.; Carloni, P.; Sobott, F.; Longhi, S.; Grandori, R. Molecular basis for structural heterogeneity of an intrinsically disordered protein bound to a partner by combined ESI-IM-MS and modeling. J. Am. Soc. Mass Spectrom. 2015, 26, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Delano, W.L. The pymol molecular graphics system. Proteins Struct. Funct. Bioinform. 2002, 30, 442–454. [Google Scholar]
- Erales, J.; Beltrandi, M.; Roche, J.; Maté, M.; Longhi, S. Insights into the hendra virus NTAIL-XD complex: Evidence for a parallel organization of the helical more at the XD surface stabilized by a combination of hydrophobic and polar interactions. Biochim. Biophys. Acta 2015, 1854, 1038–1053. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G.; Haran, G.; Zhou, H.X. Fundamental aspects of protein-protein association kinetics. Chem. Rev. 2009, 109, 839–860. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Yuwen, T.; Zhu, F.; Skrynnikov, N.R. Role of electrostatic interactions in binding of peptides and intrinsically disordered proteins to their folded targets. 1. NMR and MD characterization of the complex between the c-Crk N-SH3 domain and the peptide sos. Biochemistry 2014, 53, 6473–6495. [Google Scholar] [CrossRef] [PubMed]
- Dosnon, M.; Bonetti, D.; Morrone, A.; Erales, J.; di Silvio, E.; Longhi, S.; Gianni, S. Demonstration of a folding after binding mechanism in the recognition between the measles virus NTAIL and X domains. ACS Chem. Biol. 2015, 10, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Kavalenka, A.; Urbancic, I.; Belle, V.; Rouger, S.; Costanzo, S.; Kure, S.; Fournel, A.; Longhi, S.; Guigliarelli, B.; Strancar, J. Conformational analysis of the partially disordered measles virus NTAIL-XD complex by SDSL EPR spectroscopy. Biophys. J. 2010, 98, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Belle, V.; Rouger, S.; Costanzo, S.; Longhi, S.; Fournel, A. Site-directed spin labeling EPR spectroscopy. In Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation; Uversky, V.N., Longhi, S., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Meszaros, B.; Tompa, P.; Simon, I.; Dosztanyi, Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.D.; Ma, B.; Kumar, S.; Wolfson, H.; Nussinov, R. Protein folding: Binding of conformationally fluctuating building blocks via population selection. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 399–433. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Ma, B.; Sham, Y.Y.; Kumar, S.; Nussinov, R. Structured disorder and conformational selection. Proteins Struct. Funct. Bioinform. 2001, 44, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M. Reconciling binding mechanisms of intrinsically disordered proteins. Biochem. Biophys. Res. Commun. 2009, 382, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.; Pickar, A.; Qiu, S.; Tsao, J.; Rodenburg, C.; Dokland, T.; Elson, A.; He, B.; Luo, M. Structural studies on the authentic mumps virus nucleocapsid showing uncoiling by the phosphoprotein. Proc. Natl. Acad. Sci. USA 2014, 111, 15208–15213. [Google Scholar] [CrossRef] [PubMed]
- Oglesbee, M.; Ringler, S.; Krakowka, S. Interaction of canine distemper virus nucleocapsid variants with 70K heat-shock proteins. J. Gen. Virol. 1990, 71, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Oglesbee, M.; Tatalick, L.; Rice, J.; Krakowka, S. Isolation and characterization of canine distemper virus nucleocapsid variants. J. Gen. Virol. 1989, 70, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.; Chopy, D.; Dosnon, M.; Bloyet, L.M.; Devaux, P.; Urzua, E.; Cattaneo, R.; Longhi, S.; Gerlier, D. Sequence of events in measles virus replication: Role of phosphoprotein-nucleocapsid interactions. J. Virol. 2014, 88, 10851–10863. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Habchi, J.; Costanzo, S.; Padilla, A.; Brunel, J.; Gerlier, D.; Oglesbee, M.; Longhi, S. Plasticity in structural and functional interactions between the phosphoprotein and nucleoprotein of measles virus. J. Biol. Chem. 2012, 287, 11951–11967. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P. Fuzzy interactome: The limitations of models in molecular biology. Trends Biochem. Sci. 2009, 34, 3. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzziness: Linking regulation to protein dynamics. Mol. Biosyst. 2012, 8, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S. The measles virus NTAIL-XD complex: An illustrative example of fuzziness. Adv. Exp. Med. Biol. 2012, 725, 126–141. [Google Scholar] [PubMed]
- Carsillo, T.; Zhang, X.; Vasconcelos, D.; Niewiesk, S.; Oglesbee, M. A single codon in the nucleocapsid protein C terminus contributes to in vitro and in vivo fitness of edmonston measles virus. J. Virol. 2006, 80, 2904–2912. [Google Scholar] [CrossRef] [PubMed]
- Oglesbee, M. Nucleocapsid protein interactions with the major inducible heat shock protein. In Measles Virus Nucleoprotein; Longhi, S., Ed.; Nova Publishers Inc.: Hauppage, NY, USA, 2007; pp. 53–98. [Google Scholar]
- Carsillo, T.; Traylor, Z.; Choi, C.; Niewiesk, S.; Oglesbee, M. Hsp72, a host determinant of measles virus neurovirulence. J. Virol. 2006, 80, 11031–11039. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Sato, H.; Inoue, Y.; Watanabe, A.; Yoneda, M.; Ikeda, F.; Fujita, K.; Fukuda, H.; Takamura, C.; Kozuka-Hata, H.; et al. Phosphorylation of measles virus nucleoprotein upregulates the transcriptional activity of minigenomic RNA. Proteomics 2008, 8, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Sugai, A.; Sato, H.; Yoneda, M.; Kai, C. Phosphorylation of measles virus nucleoprotein affects viral growth by changing gene expression and genomic RNA stability. J. Virol. 2013, 87, 11684–11692. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Sato, H.; Hagiwara, K.; Watanabe, A.; Sugai, A.; Ikeda, F.; Kozuka-Hata, H.; Oyama, M.; Yoneda, M.; Kai, C. Determination of a phosphorylation site in Nipah virus nucleoprotein and its involvement in virus transcription. J. Gen. Virol. 2011, 92, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Gruet, A.; Longhi, S.; Bignon, C. One-step generation of error-prone PCR libraries using Gateway® technology. Microb. Cell Fact. 2012, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Fuxreiter, M. Interactions via intrinsically disordered regions: What kind of motifs? IUBMB Life 2012, 64, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.G.; Magliery, T.J.; Regan, L. Detecting protein–protein interactions with GFP-fragment reassembly. Nat. Methods 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gruet, A.; Dosnon, M.; Vassena, A.; Lombard, V.; Gerlier, D.; Bignon, C.; Longhi, S. Dissecting partner recognition by an intrinsically disordered protein using descriptive random mutagenesis. J. Mol. Biol. 2013, 425, 3495–3509. [Google Scholar] [CrossRef] [PubMed]
- Sebolt-Leopold, J.S.; English, J.M. Mechanisms of drug inhibition of signalling molecules. Nature 2006, 441, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Lagerstrom, M.C.; Schioth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Betzi, S.; Restouin, A.; Opi, S.; Arold, S.T.; Parrot, I.; Guerlesquin, F.; Morelli, X.; Collette, Y. Protein protein interaction inhibition (2P2I) combining high throughput and virtual screening: Application to the HIV-1 Nef protein. Proc. Natl. Acad. Sci. USA 2007, 104, 19256–19261. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Gabiane, G.; Sourimant, J.; Richard, C.A.; England, P.; Moudjou, M.; Aumont-Nicaise, M.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Identification and characterization of the binding site of the respiratory syncytial virus phosphoprotein to RNA-free nucleoprotein. J. Virol. 2015, 89, 3484–3496. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Legall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 24, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: Another illustration of the D2 concept. Expert Rev. Proteomics 2010, 7, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Uversky, V.N. Drugs for “protein clouds”: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010, 10, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug Discov. 2012, 7, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Tsai, C.J.; Nussinov, R. Analysis of ordered and disordered protein complexes reveals structural features discriminating between stable and unstable monomers. J. Mol. Biol. 2004, 341, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Vassilev, L.T. Targeting the p53-MDM2 interaction to treat cancer. Br. J. Cancer 2004, 91, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T. Small-molecule antagonists of p53-MDM2 binding: Research tools and potential therapeutics. Cell Cycle 2004, 3, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [PubMed]
- Dey, S.; Pal, A.; Chakrabarti, P.; Janin, J. The subunit interfaces of weakly associated homodimeric proteins. J. Mol. Biol. 2010, 398, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Bourgeas, R.; Basse, M.J.; Morelli, X.; Roche, P. Atomic analysis of protein–protein interfaces with known inhibitors: The 2P2I database. PLoS ONE 2010, 5, e9598. [Google Scholar] [CrossRef] [PubMed]
- Jordan, I.K.; Sutter, B.A.; McClure, M.A. Molecular evolution of the Paramyxoviridae and Rhabdoviridae multiple-protein-encoding P gene. Mol. Biol. Evol. 2000, 17, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Narechania, A.; Terai, M.; Burk, R.D. Overlapping reading frames in closely related human papillomaviruses result in modular rates of selection within E2. J. Gen. Virol. 2005, 86, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Rancurel, C.; Khosravi, M.; Dunker, K.A.; Romero, P.R.; Karlin, D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J. Virol. 2009, 83, 10719–10736. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Tompa, P.; Liliom, K.; Kalmar, L. Dual coding in alternative reading frames correlates with intrinsic protein disorder. Proc. Natl. Acad. Sci. USA 2010, 107, 5429–5434. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.M.; Canard, B.; Longhi, S. Structural disorder within the replicative complex of measles virus: Functional implications. Virology 2006, 344, 94–110. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Blocquel, D.; Habchi, J.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. Structural disorder in viral proteins. Chem. Rev. 2014, 114, 6880–6911. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Oldfield, C.J.; Uversky, V.N.; Berezovsky, I.N.; Tawfik, D.S. Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 2009, 34, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Goh, G.K.; Dunker, A.K.; Uversky, V.N. Viral disorder or disordered viruses: Do viral proteins possess unique features? Protein Pept. Lett. 2010, 17, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Trave, G.; Gibson, T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011, 36, 159–169. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).