
Effects of Non-Natural Amino Acid Incorporation into the Enzyme Core Region on Enzyme Structure and Function


Abstract
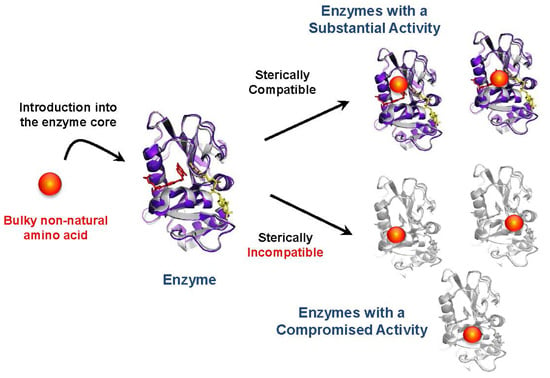
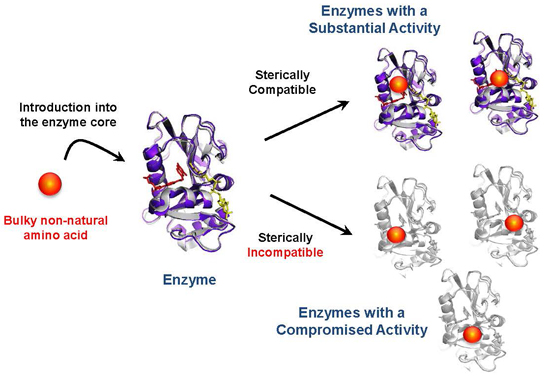
:
1. Introduction
2. Results and Discussion
2.1. Selection of Hydrophobic-Core Incorporation Sites
2.2. Expression and Purification of mDHFR Variants
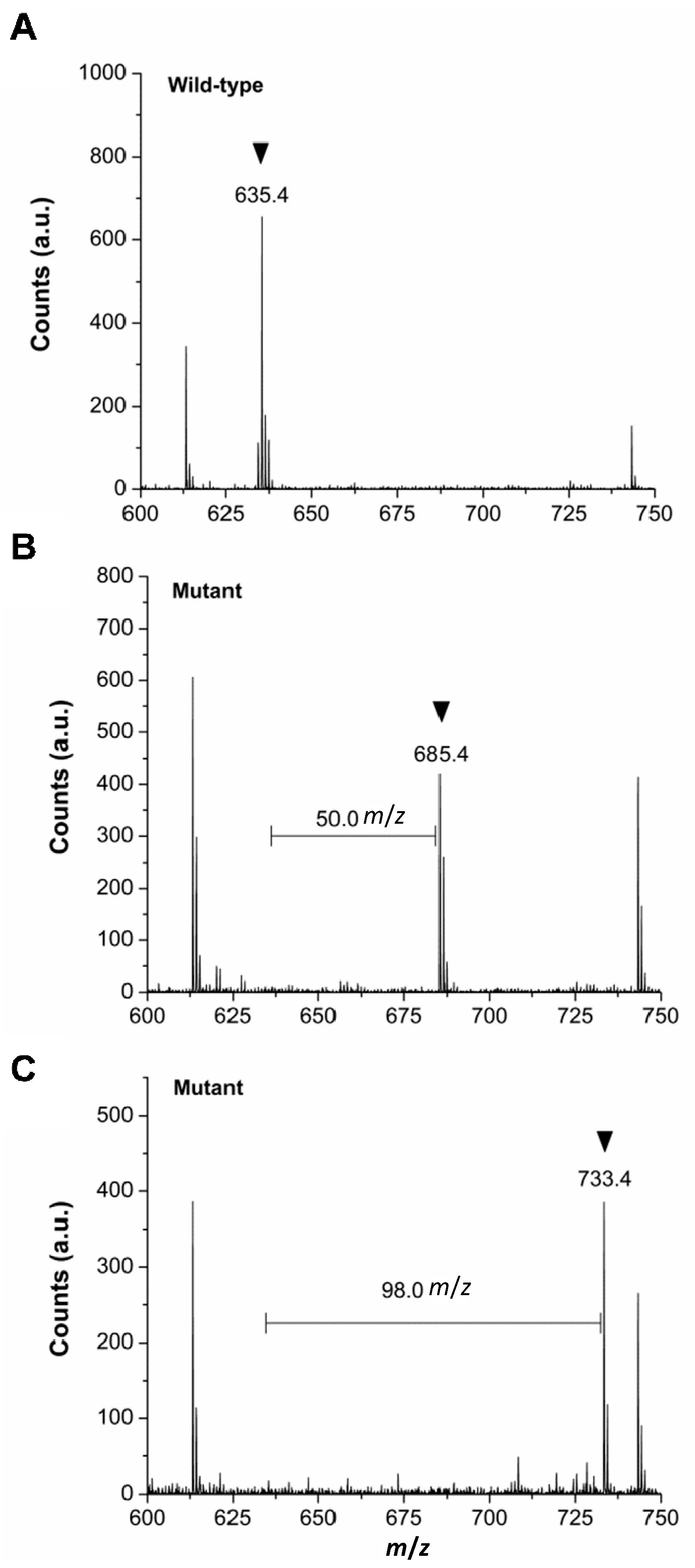
2.3. Verification of in Vivo 2Nal Incorporation by MALDI-TOF/MS

2.4. Computational Analysis and Circular Dichroism Spectroscopy of mDHFR Variants Containing 2Nal

{kind=link}
{kind=link}
| Variant | WT | W113Z | V112Z | F134Z | V135Z | I51Z | V50Z |
|---|---|---|---|---|---|---|---|
| fa_rep 1 (kcal/mol) | 14.1 ± 0.5 | 66.5 ± 0.7 | 121.3 ± 34.8 | 59.7 ± 2.3 | 77.9 ± 1.5 | 96.7 ± 0.6 | 121.4 ± 1.2 |
| DTS (Å) 2 | NA 3 | 9 | 12 | 6 | 7 | 8 | 8 |
2.5. Evaluating the Effects of 2Nal Incorporation on mDHFR Substrate Binding
| Variant | WT | W113Z | V112Z | F134Z | V135Z | I51Z | V50Z |
|---|---|---|---|---|---|---|---|
| kcat (s−1) | 3.66 ± 0.02 | 2.95 ± 0.002 | 0.029 ± 0.0001 | 1.46 ± 0.0002 | 0.32 ± 0.0004 | 0.028 ± 0.0002 | 0.020 ± 0.0002 |
| Km (μM) | 1.94 ± 0.08 | 2.10 ± 0.19 | 2.61 ± 0.12 | 1.87 ± 0.19 | 1.47 ± 0.08 | 3.01 ± 0.11 | 3.43 ± 0.24 |
| kcat/Km (μM/s) | 1.9 ± 0.1 | 1.4 ± 0.1 | 0.011 ± 0.001 | 0.78 ± 0.08 | 0.22 ± 0.01 | 0.0092 ± 0.001 | 0.0058 ± 0.0004 |
| Rel. kcat/Km (%) 2 | 100.0 | 74.5 ± 12.7 | 0.6 ± 0.05 | 41.4 ± 8.1 | 11.7 ± 1.2 | 0.5 ± 0.05 | 0.3 ± 0.04 |
| |ΔKm| (μM) 3 | NA | 0.16 | 0.67 | 0.07 | 0.47 | 1.07 | 1.49 |
2.6. Evaluating the Effect of 2Nal Incorporation on Catalytic Function
3. Experimental Section
3.1. Materials
3.2. Expression and Purification of Wild-Type mDHFR and Its Variants
3.3. Confirmation of in Vivo 2Nal Incorporation by MALDI-TOF/MS
3.4. Dihydrofolate Reduction Kinetics
3.5. Circular Dichroism Spectroscopy
3.6. Computational Analysis of Conformational Stability and Mutational Steric Compatibility
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zheng, S.; Kwon, I. Controlling enzyme inhibition using an expanded set of genetically encoded amino acids. Biotechnol. Bioeng. 2013, 110, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Cropp, T.A.; Anderson, J.C.; Mukherji, M.; Zhang, Z.W.; Schultz, P.G. An expanded eukaryotic genetic code. Science 2003, 301, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Noren, C.J.; Anthonycahill, S.J.; Griffith, M.C.; Schultz, P.G. A general-method for site-specific incorporation of unnatural amino-acids into proteins. Science 1989, 244, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-S.; Hohn, M.J.; Umehara, T.; Guo, L.-T.; Osborne, E.M.; Benner, J.; Noren, C.J.; Rinehart, J.; Soell, D. Expanding the genetic code of Escherichia coli with phosphoserine. Science 2011, 333, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.C.; Hammill, J.T.; Mehl, R.A. Site-specific incorporation of a 19F-amino acid into proteins as an NMR probe for characterizing protein structure and reactivity. J. Am. Chem. Soc. 2007, 129, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Prasher, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, J.M.; Deniz, A.A.; Schultz, P.G. Unnatural amino acid mutagenesis of green fluorescent protein. J. Org. Chem. 2003, 68, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Niu, W.; Guo, J.; Schultz, P.G. Unnatural amino acid mutagenesis of fluorescent proteins. Angew. Chem. Int. Ed. 2012, 51, 10132–10135. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Tirrell, D.A. Site-specific incorporation of tryptophan analogues into recombinant proteins in bacterial cells. J. Am. Chem. Soc. 2007, 129, 10431–10437. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.C.; Duffy, S.P.; Hess, K.R.; Mehl, R.A. Improving nature’s enzyme active site with genetically encoded unnatural amino acids. J. Am. Chem. Soc. 2006, 128, 11124–11127. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.B.; Carrico, I.S. New frontiers in protein bioconjugation editorial overview. Curr. Opin. Chem. Biol. 2010, 14, 771–773. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, A.J.; Kooijman, M.; Hennink, W.E.; Mastrobattista, E. Nonnatural amino acids for site-specific protein conjugation. Bioconj. Chem. 2009, 20, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef] [PubMed]
- Jewett, J.C.; Bertozzi, C.R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Liu, Y.; Liu, J.; Sun, R.; Chen, H.; Gao, X.; Yao, W. Designing and engineering of a site-specific incorporation of a keto group in uricase. Chem. Biol. Drug Design. 2011, 78, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Dozier, J.K.; Distefano, M.D. Enzymatic labeling of proteins: Techniques and approaches. Bioconj. Chem. 2013, 24, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Feldman, A.K.; Nugent, A.K.; Hawker, C.J.; Scheel, A.; Voit, B.; Pyun, J.; Frechet, J.M.J.; Sharpless, K.B.; Fokin, V.V. Efficiency and fidelity in a click-chemistry route to triazole dendrimers by the copper(I)-catalyzed ligation of azides and alkynes. Angew. Chem. Int. Ed. 2004, 43, 3928–3932. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.I.; Mizuta, Y.; Takasu, A.; Kim, Y.H.; Kwon, I. Site-specific bioconjugation of a murine dihydrofolate reductase enzyme by copper(I)-catalyzed azide-alkyne cycloaddition with retained activity. PLoS ONE 2014, 9, e98403. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lu, Y.; Fang, Z.; Liu, J.; Tian, H.; Gao, X.; Yao, W. High-level production of uricase containing keto functional groups for site-specific pegylation. Biochem. Eng. J. 2011, 58, 25–32. [Google Scholar] [CrossRef]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed]
- Kolev, J.N.; Zaengle, J.M.; Ravikumar, R.; Fasan, R. Enhancing the efficiency and regioselectivity of p450 oxidation catalysts by unnatural amino acid mutagenesis. ChemBioChem 2014, 15, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Kwon, I. Manipulation of enzyme properties by noncanonical amino acid incorporation. Biotechnol. J. 2012, 7, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.W.; Ayling, J.E. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc. Natl. Acad. Sci. USA 2009, 106, 15424–15429. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Appleman, J.R.; Delcamp, T.J.; Freisheim, J.H.; Blakley, R.L. Hydride transfer by dihydrofolate-reductase—Causes and consequences of the wide-range of rates exhibited by bacterial and vertebrate enzymes. J. Biol. Chem. 1989, 264, 9391–9399. [Google Scholar] [PubMed]
- Chunduru, S.K.; Cody, V.; Luft, J.R.; Pangborn, W.; Appleman, J.R.; Blakley, R.L. Methotrexate-resistant variants of human dihydrofolate-reductase—Effects of Phe31 substitutions. J. Biol. Chem. 1994, 269, 9547–9555. [Google Scholar] [PubMed]
- Cody, V.; Luft, J.R.; Pangborn, W. Understanding the role of Leu22 variants in methotrexate resistance: Comparison of wild-type and Leu22Arg variant mouse and human dihydrofolate reductase ternary crystal complexes with methotrexate and NADPH. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.S.; Cody, V.; Galitsky, N.; Luft, J.R.; Pangborn, W.; Chunduru, S.K.; Spencer, H.T.; Appleman, J.R.; Blakley, R.L. Methotrexate-resistant variants of human dihydrofolate-reductase with substitutions of leucine-22. Kinetics, crystallography, and potential as selectable markers. J. Biol. Chem. 1995, 270, 5057–5064. [Google Scholar] [CrossRef] [PubMed]
- Tsay, J.T.; Appleman, J.R.; Beard, W.A.; Prendergast, N.J.; Delcamp, T.J.; Freisheim, J.H.; Blakley, R.L. Kinetic investigation of the functional-role of phenylalanine-31 of recombinant human dihydrofolate-reductase. Biochemistry 1990, 29, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Thillet, J.; Adams, J.A.; Benkovic, S.J. The kinetic mechanism of wild-type and mutant mouse dihydrofolate reductases. Biochemistry 1990, 29, 5195–5202. [Google Scholar] [CrossRef] [PubMed]
- Thillet, J.; Absil, J.; Stone, S.R.; Pictet, R. Site-directed mutagenesis of mouse dihydrofolate-reductase—Mutants with increased resistance to methotrexate and trimethoprim. J. Biol. Chem. 1988, 263, 12500–12508. [Google Scholar] [PubMed]
- Schweitzer, B.I.; Dicker, A.P.; Bertino, J.R. Dihydrofolate-reductase as a therapeutic target. FASEB J. 1990, 4, 2441–2452. [Google Scholar] [PubMed]
- Reyes, P.; Huennekens, F.M. Ion-dependent activation of dihydrofolate reductase from l1210 cells. Biochemistry 1967, 6, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.T.; Hanoian, P.; French, J.B.; Pringle, T.H.; Hammes-Schiffer, S.; Benkovic, S.J. Functional significance of evolving protein sequence in dihydrofolate reductase from bacteria to humans. Proc. Natl. Acad. Sci. USA 2013, 110, 10159–10164. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, E118–U185. [Google Scholar] [CrossRef] [PubMed]
- Bordo, D.; Argos, P. Suggestions for safe residue substitutions in site-directed mutagenesis. J. Mol. Biol. 1991, 217, 721–729. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Bowie, J.U.; Reidhaarolson, J.F.; Lim, W.A.; Sauer, R.T. Deciphering the message in protein sequences—Tolerance to amino-acid substitutions. Science 1990, 247, 1306–1310. [Google Scholar] [CrossRef] [PubMed]
- Ventura, S.; Vega, M.C.; Lacroix, E.; Angrand, I.; Spagnolo, L.; Serrano, L. Conformational strain in the hydrophobic core and its implications for protein folding and design. Nat. Struct. Biol. 2002, 9, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Vandenburg, B.; Dijkstra, B.W.; Vriend, G.; Vandervinne, B.; Venema, G.; Eijsink, V.G.H. Protein stabilization by hydrophobic interactions at the surface. Eur. J. Biochem. 1994, 220, 981–985. [Google Scholar]
- Lim, W.A.; Sauer, R.T. Alternative packing arrangements in the hydrophobic core of λrepressor. Nature 1989, 339, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Lazar, G.A.; Desjarlais, J.R.; Handel, T.M. De novo design of the hydrophobic core of ubiquitin. Protein Sci. 1997, 6, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Kellis, J.T.; Nyberg, K.; Fersht, A.R. Energetics of complementary side-chain packing in a protein hydrophobic core. Biochemistry 1989, 28, 4914–4922. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Moracci, M.; Elmasry, N.; Johnson, C.M.; Fersht, A.R. Effect of cavity-creating mutations in the hydrophobic core of chymotrypsin inhibitor-2. Biochemistry 1993, 32, 11259–11269. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.E.; Baase, W.A.; Zhang, X.J.; Heinz, D.W.; Blaber, M.; Baldwin, E.P.; Matthews, B.W. Response of a protein-structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 1992, 255, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Desjarlais, J.R.; Handel, T.M. De novo design of the hydrophobic cores of proteins. Protein Sci. 1995, 4, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Buckle, A.M.; Cramer, P.; Fersht, A.R. Structural and energetic responses to cavity-creating mutations in hydrophobic cores: Observation of a buried water molecule and the hydrophilic nature of such hydrophobic cavities. Biochemistry 1996, 35, 4298–4305. [Google Scholar] [CrossRef] [PubMed]
- Adamek, D.H.; Guerrero, L.; Blaber, M.; Caspar, D.L. Structural and energetic consequences of mutations in a solvated hydrophobic cavity. J. Mol. Biol. 2005, 346, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Schultz, P.G. Adding l-3-(2-naphthyl)alanine to the genetic code of E coli. J. Am. Chem. Soc. 2002, 124, 1836–1837. [Google Scholar] [CrossRef] [PubMed]
- Cornette, J.L.; Cease, K.B.; Margalit, H.; Spouge, J.L.; Berzofsky, J.A.; Delisi, C. Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. J. Mol. Biol. 1987, 195, 659–685. [Google Scholar] [CrossRef]
- Young, T.S.; Schultz, P.G. Beyond the canonical 20 amino acids: Expanding the genetic lexicon. J. Biol. Chem. 2010, 285, 11039–11044. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Wang, P.; Tirrell, D.A. Design of a bacterial host for site-specific incorporation of p-bromophenylalanine into recombinant proteins. J. Am. Chem. Soc. 2006, 128, 11778–11783. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Abraham, M.H.; Zissimos, A.M. Fast calculation of van der waals volume as a sum of atomic and bond contributions and its application to drug compounds. J. Org. Chem. 2003, 68, 7368–7373. [Google Scholar] [CrossRef] [PubMed]
- Gilis, D.; Rooman, M. Predicting protein stability changes upon mutation using database-derived potentials: Solvent accessibility determines the importance of local versus non-local interactions along the sequence. J. Mol. Biol. 1997, 272, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gromiha, M.; Fawareh, H.; Sarai, A. ASAview: Database and tool for solvent accessibility representation in proteins. BMC Bioinform. 2004, 5, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef] [PubMed]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [PubMed]
- Eisenberg, D.; McLachlan, A.D. Solvation energy in protein folding and binding. Nature 1986, 319, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Mirny, L.A.; Shakhnovich, E.I. Universally conserved positions in protein folds: Reading evolutionary signals about stability, folding kinetics and function. J. Mol. Biol. 1999, 291, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Rausell, A.; Juan, D.; Pazos, F.; Valencia, A. Protein interactions and ligand binding: From protein subfamilies to functional specificity. Proc. Natl. Acad. Sci. USA 2010, 107, 1995–2000. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein structure prediction using rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar] [PubMed]
- Combs, S.A.; DeLuca, S.L.; DeLuca, S.H.; Lemmon, G.H.; Nannemann, D.P.; Nguyen, E.D.; Willis, J.R.; Sheehan, J.H.; Meiler, J. Small-molecule ligand docking into comparative models with rosetta. Nat. Protoc. 2013, 8, 1277–1298. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Farid, H.; Pistor, E.; Farid, R.S. A new approach to the design of uniquely folded thermally stable proteins. Protein Sci. 2000, 9, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Kussell, E.; Shimada, J.; Shakhnovich, E.I. Excluded volume in protein side-chain packing. J. Mol. Biol. 2001, 311, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Yoshidome, T.; Oshima, H.; Kodama, R.; Harano, Y.; Kinoshita, M. Effects of side-chain packing on the formation of secondary structures in protein folding. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoire, S.; Glitzos, K.; Kwon, I. Suppressing mutation-induced protein aggregation in mammalian cells by mutating residues significantly displaced upon the original mutation. Biochem. Eng. J. 2014, 91, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, S.; Zhang, S.; Costanzo, J.; Wilson, K.; Fernandez, E.J.; Kwon, I. Cis-suppression to arrest protein aggregation in mammalian cells. Biotechnol. Bioeng. 2014, 111, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.F.; Selzer, T.; Benkovic, S.J.; Hammes-Schiffer, S. Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 2005, 102, 6807–6812. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Goodey, N.M.; Benkovic, S.J.; Kohen, A. Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 2006, 103, 15753–15758. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Worth, C.L.; Gong, S.; Blundell, T.L. Structural and functional constraints in the evolution of protein families. Nat. Rev. Mol. Cell Biol. 2009, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, D. Binding, gating, affinity and efficacy: The interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 1998, 125, 924–947. [Google Scholar] [CrossRef] [PubMed]
- Luque, I.; Freire, E. Structural stability of binding sites: Consequences for binding affinity and allosteric effects. Proteins Struct. Funct. Genet. 2000, 41, 63–71. [Google Scholar] [CrossRef]
- Meiering, E.M.; Li, H.J.; Delcamp, T.J.; Freisheim, J.H.; Wagner, G. Contributions of tryptophan-24 and glutamate-30 to binding long-lived water-molecules in the ternary complex of human dihydrofolate-reductase with methotrexate and NADP studied by site-directed mutagenesis and nuclear-magnetic-resonance spectroscopy. J. Mol. Biol. 1995, 247, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, S.J.; Fierke, C.A.; Naylor, A.M. Insights into enzyme function from studies on mutants of dihydrofolate-reductase. Science 1988, 239, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Schnell, J.R.; McElheny, D.; Bae, S.-H.; Duggan, B.M.; Benkovic, S.J.; Dyson, H.J.; Wright, P.E. A distal mutation perturbs dynamic amino acid networks in dihydrofolate reductase. Biochemistry 2013, 52, 4605–4619. [Google Scholar] [CrossRef] [PubMed]
- Rod, T.H.; Radkiewicz, J.L.; Brooks, C.L. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 2003, 100, 6980–6985. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Johnson, K.; Matthews, R.; Benkovic, S.J. Effects of distal point-site mutations on the binding and catalysis of dihydrofolate-reductase from Escherichia coli. Biochemistry 1989, 28, 6611–6618. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Chen, J.T.; Taira, K.; Fierke, C.A.; Benkovic, S.J. Importance of a hydrophobic residue in binding and catalysis by dihydrofolate-reductase. Proc. Natl. Acad. Sci. USA 1986, 83, 7718–7720. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.A.; Fierke, C.A.; Benkovic, S.J. The function of amino-acid-residues contacting the nicotinamide ring of nadph in dihydrofolate-reductase from Escherichia coli. Biochemistry 1991, 30, 11046–11054. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.E.; Orengo, C.A.; Thornton, J.M. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 2001, 307, 1113–1143. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.W.; Lemmon, G.H.; DeLuca, S.L.; Sheehan, J.H.; Meiler, J. Practically useful: What the rosetta protein modeling suite can do for you. Biochemistry 2010, 49, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.N.; Campbell-Valois, F.X.; Michnick, S.W. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl. Acad. Sci. USA 1998, 95, 12141–12146. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Protein stability, flexibility and function. Biochim. Biophys. Acta 2011, 1814, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Eisenmesser, E.Z.; Millet, O.; Labeikovsky, W.; Korzhnev, D.M.; Wolf-Watz, M.; Bosco, D.A.; Skalicky, J.J.; Kay, L.E.; Kern, D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Francis, K.; Stojkovic, V.; Kohen, A. Preservation of protein dynamics in dihydrofolate reductase evolution. J. Biol. Chem. 2013, 288, 35961–35968. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Goodey, N.M.; Benkovic, S.J.; Kohen, A. The role of enzyme dynamics and tunnelling in catalysing hydride transfer: Studies of distal mutants of dihydrofolate reductase. Philos. Trans. R. Soc. Lond. Ser. B Biol. 2006, 361, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Baase, W.A.; Kuroki, R.; Matthews, B.W. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. USA 1995, 92, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Dima, R.I.; Thirumalai, D. Allosteric communication in dihydrofolate reductase: Signaling network and pathways for closed to occluded transition and back. J. Mol. Biol. 2007, 374, 250–266. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Benkovic, S.J. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry 1997, 36, 15792–15800. [Google Scholar] [CrossRef] [PubMed]
- Ohmae, E.; Iriyama, K.; Ichihara, S.; Gekko, K. Effects of point mutations at the flexible loop glycine-67 of Escherichia coli dihydrofolate reductase on its stability and function. J. Biochem. 1996, 119, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Gekko, K.; Kunori, Y.; Takeuchi, H.; Ichihara, S.; Kodama, M. Point mutations at glycine-121 of Escherichia coli dihydrofolate-reductase—Important roles of a flexible loop in the stability and function. J. Biochem. 1994, 116, 34–41. [Google Scholar] [PubMed]
- Mauldin, R.V.; Sapienza, P.J.; Petit, C.M.; Lee, A.L. Structure and dynamics of the g121v dihydrofolate reductase mutant: Lessons from a transition-state inhibitor complex. PLoS ONE 2012, 7, e33252. [Google Scholar] [CrossRef] [PubMed]
- Gauldie, J.; Marshall, L.; Hillcoat, B.L. Purification and properties of dihydrofolate reductase from cultured mammalian-cells. Biochem. J. 1973, 133, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Hillcoat, B.L.; Nixon, P.F.; Blakley, R.L. Effect of substrate decomposition on the spectrophotometric assay of dihydrofolate reductase. Anal. Biochem. 1967, 21, 178–189. [Google Scholar] [CrossRef]
- Haber, D.A.; Beverley, S.M.; Kiely, M.L.; Schimke, R.T. Properties of an altered dihydrofolate-reductase encoded by amplified genes in cultured mouse fibroblasts. J. Biol. Chem. 1981, 256, 9501–9510. [Google Scholar] [PubMed]
- Kuhlman, B.; Baker, D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. USA 2000, 97, 10383–10388. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, J.; Taylor, G.K.; Havranek, J.J.; Quadri, S.A.; Stoddard, B.L.; Baker, D. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 2010, 38, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, J.; Havranek, J.J.; Duarte, C.M.; Sussman, D.; Monnat, R.J.; Stoddard, B.L.; Baker, D. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature 2006, 441, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Cochran, F.V.; Wu, S.P.; Wang, W.; Nanda, V.; Saven, J.G.; Therien, M.J.; DeGrado, W.F. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J. Am. Chem. Soc. 2005, 127, 1346–1347. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Althoff, E.A.; Clemente, F.R.; Doyle, L.; Rothlisberger, D.; Zanghellini, A.; Gallaher, J.L.; Betker, J.L.; Tanaka, F.; Barbas, C.F., III; et al. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-S.; Love, J.J.; Mayo, S.L. A de novo designed protein-protein interface. Protein Sci. 2007, 16, 2770–2774. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, H.E.; Kwon, I. Effects of Non-Natural Amino Acid Incorporation into the Enzyme Core Region on Enzyme Structure and Function. Int. J. Mol. Sci. 2015, 16, 22735-22753. https://doi.org/10.3390/ijms160922735
Wong HE, Kwon I. Effects of Non-Natural Amino Acid Incorporation into the Enzyme Core Region on Enzyme Structure and Function. International Journal of Molecular Sciences. 2015; 16(9):22735-22753. https://doi.org/10.3390/ijms160922735
Chicago/Turabian StyleWong, H. Edward, and Inchan Kwon. 2015. "Effects of Non-Natural Amino Acid Incorporation into the Enzyme Core Region on Enzyme Structure and Function" International Journal of Molecular Sciences 16, no. 9: 22735-22753. https://doi.org/10.3390/ijms160922735
APA StyleWong, H. E., & Kwon, I. (2015). Effects of Non-Natural Amino Acid Incorporation into the Enzyme Core Region on Enzyme Structure and Function. International Journal of Molecular Sciences, 16(9), 22735-22753. https://doi.org/10.3390/ijms160922735