
Activin A-Smad Signaling Mediates Connective Tissue Growth Factor Synthesis in Liver Progenitor Cells

Abstract
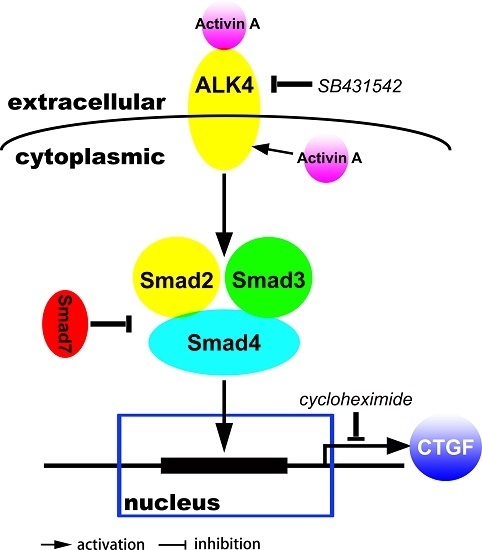
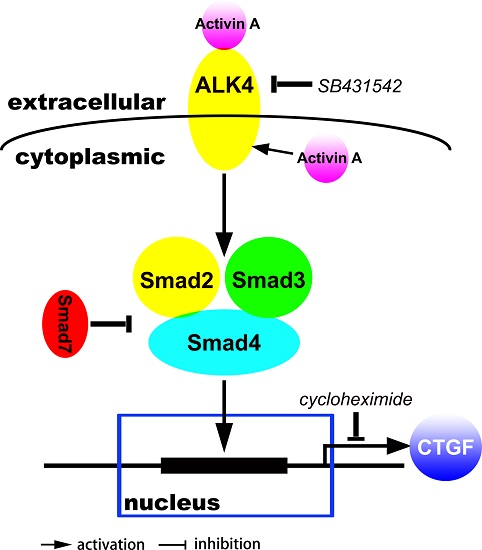
:
1. Introduction
2. Results
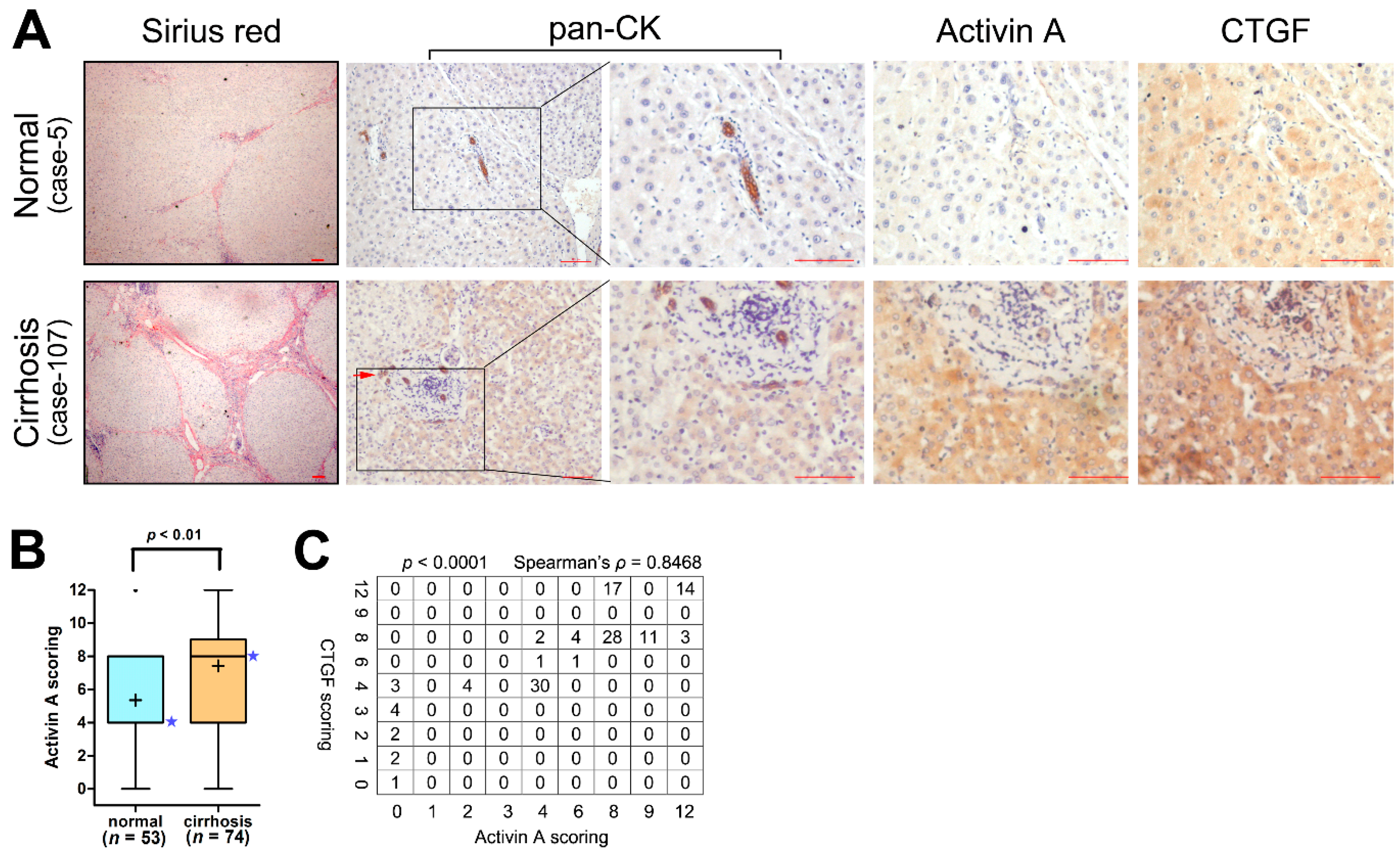
2.1. The Expression of Activin A and Connective Tissue Growth Factor (CTGF/CCN2) Are Elevated in the Cirrhotic Liver
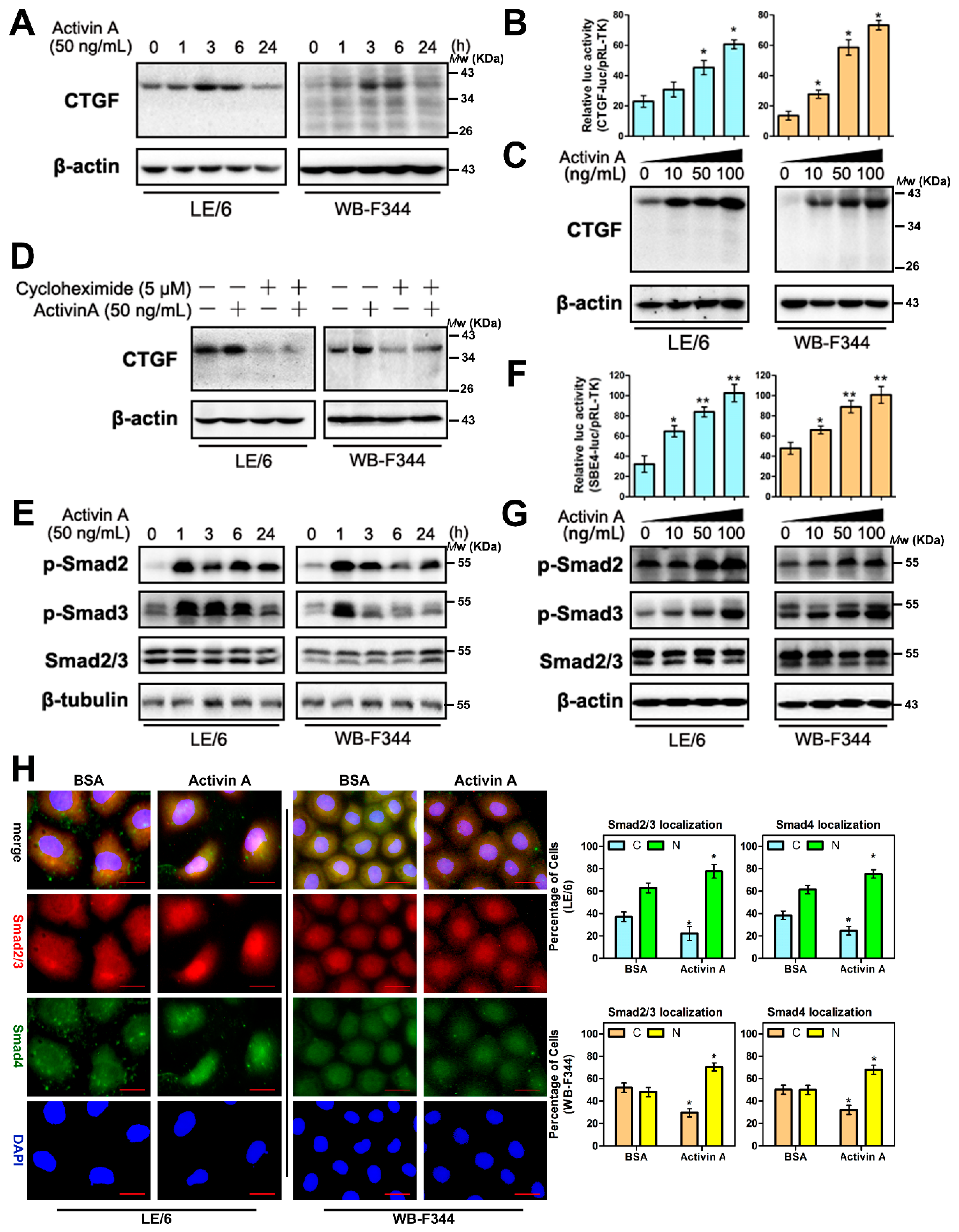
2.2 Activin A Induces CTGF/CCN2 Synthesis in Liver Progenitor Cells (LPCs)
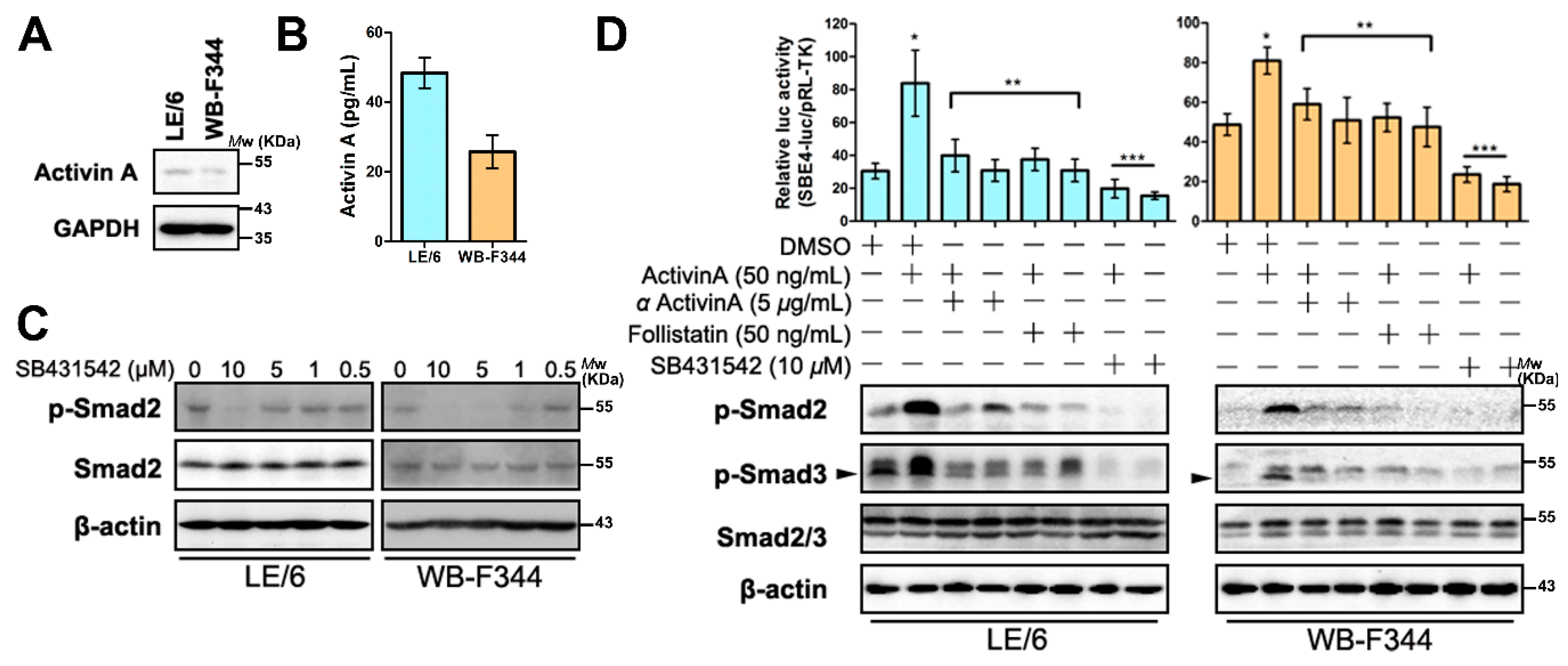
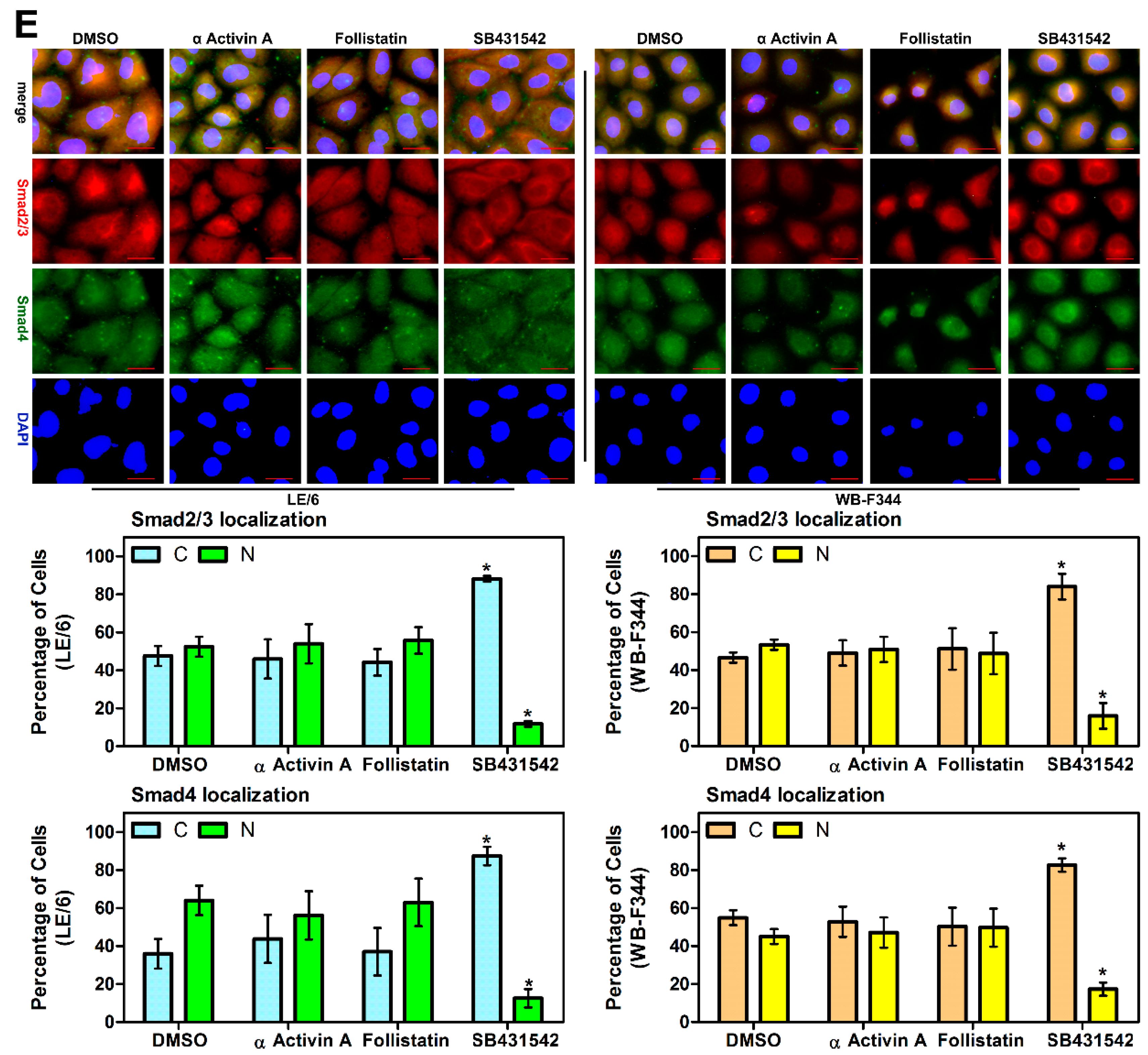
2.3. Intracrine Activin A Signaling Is Activated in LPCs
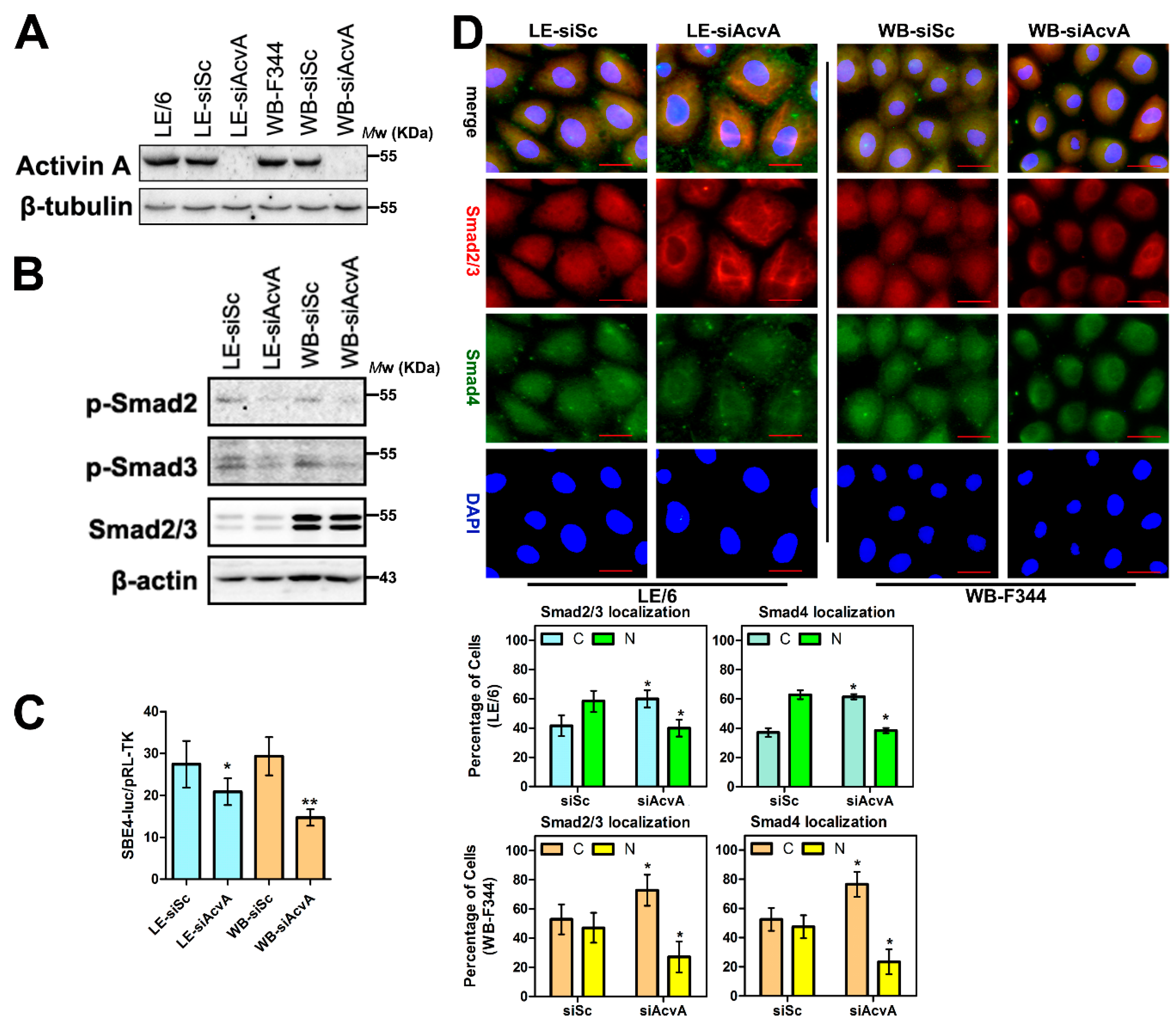
2.4. Knock Down of Activin A in LPCs Causes Reduced Activity of Smad Signaling
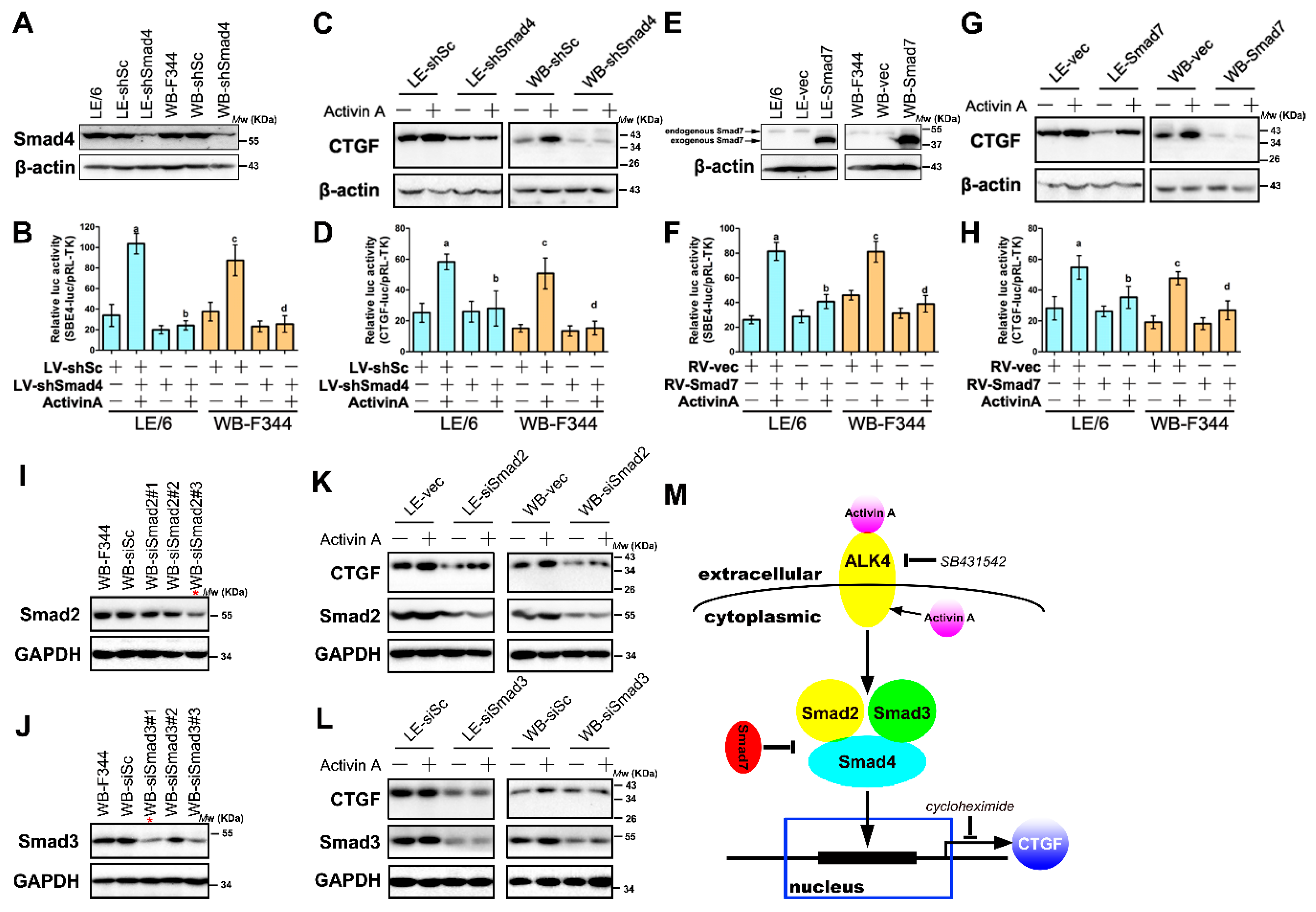
2.5. Intracrine Activin A Signaling Contributes to the CTGF/CCN2 Production in LPCs
2.6. Activin A Mediated the Production of CTGF/CCN2 through Smad Signaling in LPCs
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Liver Samples
4.3. Immunohistochemistry and Double Immunofluorescent Analyses
4.4. Cell Lines and Cell Culture
4.5. Plasmids
4.6. Virus Production, Cells Infection and Selection of Stable Cell Clones
4.7. Transient RNA Interference
4.8. Luciferase Reporter Analyses
4.9. Western Blot Analyses
4.10. Enzyme-Linked Immunosorbent Assay (ELISA) for Activin A
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CTGF/CCN2 | Connective tissue growth factor |
| LPCs | Liver progenitor cells |
| HCC | Hepatocellular carcinoma |
| CHX | Cycloheximide |
| shRNA | Small hairpin RNA |
| ALK4 | Activin receptor-like kinase 4 |
| HSCs | Hepatic stellate cells |
| TGF-β | Transforming growth factor β |
| TβRII | TGF-β receptor type II |
| RV | Retrovirus |
| LV | Lentivirus |
References
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Gouw, A.S.; Clouston, A.D.; Theise, N.D. Ductular reactions in human liver: Diversity at the interface. Hepatology 2011, 54, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Miyajima, A. Liver regeneration by stem/progenitor cells. Hepatology 2014, 59, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, A.; Tanaka, M.; Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 2014, 14, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.Y.; Jin, G.N.; Liang, H.F.; Wang, W.; Chen, W.X.; Datta, P.K.; Zhang, M.Z.; Zhang, B.; Chen, X.P. Transforming growth factor β induces expression of connective tissue growth factor in hepatic progenitor cells through smad independent signaling. Cell. Signal. 2013, 25, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Clouston, A.D.; Forbes, S.J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 2014, 146, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Rodgarkia-Dara, C.; Vejda, S.; Erlach, N.; Losert, A.; Bursch, W.; Berger, W.; Schulte-Hermann, R.; Grusch, M. The activin axis in liver biology and disease. Mutat. Res. 2006, 613, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; de Guise, C.; Kim, C.; Lemay, S.; Wang, X.F.; Lebrun, J.J. Activin induces hepatocyte cell growth arrest through induction of the cyclin-dependent kinase inhibitor p15INK4B and Sp1. Cell. Signal. 2004, 16, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Schwall, R.H.; Robbins, K.; Jardieu, P.; Chang, L.; Lai, C.; Terrell, T.G. Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology 1993, 18, 347–356. [Google Scholar] [CrossRef]
- Oe, S.; Lemmer, E.R.; Conner, E.A.; Factor, V.M.; Leveen, P.; Larsson, J.; Karlsson, S.; Thorgeirsson, S.S. Intact signaling by transforming growth factor β is not required for termination of liver regeneration in mice. Hepatology 2004, 40, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, W.; Liang, H.F.; Zhou, Q.F.; Ding, Z.Y.; Yang, H.Q.; Liu, W.B.; Wu, Y.H.; Man, Q.; Zhang, B.X.; et al. Activin A induces growth arrest through a SMAD-dependent pathway in hepatic progenitor cells. Cell Commun. Signal. CCS 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Ichida, T.; Sato, T.; Ishikawa, T.; Matsuda, Y.; Asakura, H. Expression of activin A is increased in cirrhotic and fibrotic rat livers. Gastroenterology 1998, 114, 550–558. [Google Scholar] [CrossRef]
- Gressner, O.A.; Lahme, B.; Siluschek, M.; Rehbein, K.; Weiskirchen, R.; Gressner, A.M. Intracrine signalling of activin A in hepatocytes upregulates connective tissue growth factor (CTGF/CCN2) expression. Liver Int. 2008, 28, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Wada, W.; Kuwano, H.; Hasegawa, Y.; Kojima, I. The dependence of transforming growth factor-β-induced collagen production on autocrine factor activin A in hepatic stellate cells. Endocrinology 2004, 145, 2753–2759. [Google Scholar] [CrossRef] [PubMed]
- Date, M.; Matsuzaki, K.; Matsushita, M.; Tahashi, Y.; Sakitani, K.; Inoue, K. Differential regulation of activin A for hepatocyte growth and fibronectin synthesis in rat liver injury. J. Hepatol. 2000, 32, 251–260. [Google Scholar] [CrossRef]
- Gressner, O.A.; Gressner, A.M. Connective tissue growth factor: A fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008, 28, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Pi, L.; Robinson, P.M.; Jorgensen, M.; Oh, S.H.; Brown, A.R.; Weinreb, P.H.; Trinh, T.L.; Yianni, P.; Liu, C.; Leask, A.; et al. Connective tissue growth factor and integrin alphavβ6: A new pair of regulators critical for ductular reaction and biliary fibrosis in mice. Hepatology 2015, 61, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Lahme, B.; Siluschek, M.; Rehbein, K.; Weiskirchen, R.; Gressner, A.M. Connective tissue growth factor is a Smad2 regulated amplifier of transforming growth factor β actions in hepatocytes—But without modulating bone morphogenetic protein 7 signaling. Hepatology 2009, 49, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.L.; Ciuclan, L.; Liu, Y.; Hamzavi, J.; Godoy, P.; Gaitantzi, H.; Kanzler, S.; Heuchel, R.; Ueberham, U.; Gebhardt, R.; et al. Profibrogenic transforming growth factor-β/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology 2007, 46, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Lahme, B.; Demirci, I.; Gressner, A.M.; Weiskirchen, R. Differential effects of TGF-β on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J. Hepatol. 2007, 47, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Shi-Wen, X.; Leask, A.; Abraham, D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008, 19, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in p53 expression induce ctgf synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Investig. 2011, 121, 3343–3356. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.Y.; Liang, H.F.; Jin, G.N.; Chen, W.X.; Wang, W.; Datta, P.K.; Zhang, M.Z.; Zhang, B.; Chen, X.P. Smad6 suppresses the growth and self-renewal of hepatic progenitor cells. J. Cell. Physiol. 2014, 229, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A specific inhibitor of TGF-β receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Yovchev, M.I.; Xue, Y.; Shafritz, D.A.; Locker, J.; Oertel, M. Repopulation of the fibrotic/cirrhotic rat liver by transplanted hepatic stem/progenitor cells and mature hepatocytes. Hepatology 2014, 59, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Mavila, N.; James, D.; Shivakumar, P.; Nguyen, M.V.; Utley, S.; Mak, K.; Wu, A.; Zhou, S.; Wang, L.; Vendyres, C.; et al. Expansion of prominin-1-expressing cells in association with fibrosis of biliary atresia. Hepatology 2014, 60, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Patella, S.; Phillips, D.J.; Tchongue, J.; de Kretser, D.M.; Sievert, W. Follistatin attenuates early liver fibrosis: Effects on hepatic stellate cell activation and hepatocyte apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G137–G144. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Haukeland, J.W.; Dahl, T.B.; Bjoro, K.; Gladhaug, I.P.; Berge, C.; Damas, J.K.; Haaland, T.; Loberg, E.M.; Linnestad, P.; et al. A complex role of activin A in non-alcoholic fatty liver disease. Am. J. Gastroenterol. 2009, 104, 2196–2205. [Google Scholar] [CrossRef] [PubMed]
- Mashima, H.; Kanzaki, M.; Nobusawa, R.; Zhang, Y.Q.; Suzuki, M.; Mine, T.; Kojima, I. Derangements in the activin-follistatin system in hepatoma cells. Gastroenterology 1995, 108, 834–840. [Google Scholar] [CrossRef]
- Espanol-Suner, R.; Carpentier, R.; Van Hul, N.; Legry, V.; Achouri, Y.; Cordi, S.; Jacquemin, P.; Lemaigre, F.; Leclercq, I.A. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 2012, 143, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wong, P.P.; Sjeklocha, L.; Steer, C.J.; Sahin, M.B. Mature hepatocytes exhibit unexpected plasticity by direct dedifferentiation into liver progenitor cells in culture. Hepatology 2012, 55, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Jors, S.; Jeliazkova, P.; Ringelhan, M.; Thalhammer, J.; Durl, S.; Ferrer, J.; Sander, M.; Heikenwalder, M.; Schmid, R.M.; Siveke, J.T.; et al. Lineage fate of ductular reactions in liver injury and carcinogenesis. J. Clin. Investig. 2015, 125, 2445–2457. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo-Torres, D.; Affo, S.; Coll, M.; Morales-Ibanez, O.; Millan, C.; Blaya, D.; Alvarez-Guaita, A.; Rentero, C.; Lozano, J.J.; Maestro, M.A.; et al. The biliary epithelium gives rise to liver progenitor cells. Hepatology 2014, 60, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Kordes, C.; Sawitza, I.; Gotze, S.; Herebian, D.; Haussinger, D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J. Clin. Investig. 2014, 124, 5503–5515. [Google Scholar] [CrossRef] [PubMed]
- Hindley, C.J.; Mastrogiovanni, G.; Huch, M. The plastic liver: Differentiated cells, stem cells, every cell? J. Clin. Investig. 2014, 124, 5099–5102. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Furuya, M.H.; Wolfraim, L.A.; Nguyen, A.P.; Holdren, M.S.; Campbell, J.S.; Knight, B.; Yeoh, G.C.; Fausto, N.; Parks, W.T. Transforming growth factor-β differentially regulates oval cell and hepatocyte proliferation. Hepatology 2007, 45, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A. Intracrine signaling mechanisms of activin A and TGF-β. Vitam. Horm. 2011, 85, 59–77. [Google Scholar] [PubMed]
- Bauer, J.; Ozden, O.; Akagi, N.; Carroll, T.; Principe, D.R.; Staudacher, J.J.; Spehlmann, M.E.; Eckmann, L.; Grippo, P.J.; Jung, B. Activin and TGFβ use diverging mitogenic signaling in advanced colon cancer. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Pi, L.; Oh, S.H.; Shupe, T.; Petersen, B.E. Role of connective tissue growth factor in oval cell response during liver regeneration after 2-AAF/PHx in rats. Gastroenterology 2005, 128, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Pi, L.; Ding, X.; Jorgensen, M.; Pan, J.J.; Oh, S.H.; Pintilie, D.; Brown, A.; Song, W.Y.; Petersen, B.E. Connective tissue growth factor with a novel fibronectin binding site promotes cell adhesion and migration during rat oval cell activation. Hepatology 2008, 47, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, W.; Yan, H.X.; Luo, T.; Zhang, J.; Tang, L.; Wu, F.Q.; Zhang, H.L.; Yu, L.X.; Zheng, L.Y.; et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology 2012, 55, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor β gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Pirisi, M.; Fabris, C.; Luisi, S.; Santuz, M.; Toniutto, P.; Vitulli, D.; Federico, E.; Del Forno, M.; Mattiuzzo, M.; Branca, B.; et al. Evaluation of circulating Activin-A as a serum marker of hepatocellular carcinoma. Cancer Detect. Prev. 2000, 24, 150–155. [Google Scholar] [PubMed]
- Voumvouraki, A.; Notas, G.; Koulentaki, M.; Georgiadou, M.; Klironomos, S.; Kouroumalis, E. Increased serum Activin-A differentiates alcoholic from cirrhosis of other aetiologies. Eur. J. Clin. Investig. 2012, 42, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Fransvea, E.; Dituri, F.; Lupo, L.; Antonaci, S.; Giannelli, G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor β blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010, 51, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, R.; Latasa, M.U.; Demartis, M.I.; Balzani, S.; Goni, S.; Garcia-Irigoyen, O.; Elizalde, M.; Azcona, M.; Pascale, R.M.; Feo, F.; et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: Oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 2011, 54, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Yang, S.L.; Liang, H.F.; Li, C.H. HBx protein promotes oval cell proliferation by up-regulation of cyclin D1 via activation of the MEK/ERK and PI3K/Akt pathways. Int. J. Mol. Sci. 2014, 15, 3507–3518. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.Y.; Jin, G.N.; Wang, W.; Chen, W.X.; Wu, Y.H.; Ai, X.; Chen, L.; Zhang, W.G.; Liang, H.F.; Laurence, A.; et al. Reduced expression of transcriptional intermediary factor 1 γ promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology 2014, 60, 1620–1636. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, A.; Vaughn, D.A.; Choi, Y.; Nigam, S.K. Activin A is an endogenous inhibitor of ureteric bud outgrowth from the wolffian duct. Dev. Biol. 2006, 295, 473–485. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemicals | Manufacturers |
|---|---|
| Recombinant human Activin A | 120-14E, PeproTech, Rocky Hill, NJ, USA |
| Recombinant human Follistatin 315 aa 30-344 | 4889-FN/CF, R&D Systems, Minneapolis, MN, USA |
| SB431542 | 301836-41-9, Cayman, Ann Arbor, MI, USA |
| LY364947 | 616451, Merck Calbiochem, Darmstadt, Germany |
| Cycloheximide | SI005, Beyotime Institute of Biotechnology, Haimen, Jiangsu, China |
| Antigens | Manufacturers | Application |
|---|---|---|
| CTGF/CCN2 | sc-14939, Santa Cruz Biotechnology, Santa Cruz, CA, USA | 1:200 for WB, I:50 for IHC |
| Pan-cytokeratin | IR053, Dako, Glostrup, Denmark. | Ready-to-Use for IHC |
| Activin A | AF338, R&D Systems, Minneapolis, MN, USA | 1:50 (5 μg/mL) for IHC |
| Activin A | MAB3381, R&D Systems, Minneapolis, MN, USA | neutralization |
| Activin A | 5624-1, Epitomics, Burlingame, CA, USA | 1:500 for WB |
| Phospho-Smad2 (Ser465/467) | #3108, Cell Signaling Technology, Beverly, MA, USA | 1:2000 for WB |
| Phospho-Smad3 (Ser423/425) | 1880-1, Epitomics, Burlingame, CA, USA | 1:2000 for WB |
| Smad2/3 | sc-133098, Santa Cruz Biotechnology, Santa Cruz, CA, USA | 1:500 for WB, 1:50 for IF |
| Smad4 | 1676-1, Epitomics, Burlingame, CA, USA | 1:2000 for WB, 1:100 for IF |
| Smad7 | 3894-1, Epitomics, Burlingame, CA, USA | 1:1000 for WB |
| β-Actin | sc-47778, Santa Cruz Biotechnology, Santa Cruz, CA, USA | 1:10000 for WB |
| β-Tubulin | M30109, Abmart, Shanghai, China. | 1:5000 for WB |
| GAPDH | KC-5G4, KangChen Bio-tech, Shanghai, China. | 1:20000 for WB |
| Alexa Flour 488-conjugated anti-rabbit IgG | Beyotime Institute of Biotechnology, Haimen, Jiangsu, China | 1:500 for IF |
| Alexa Flour 555-conjugated anti-mouse IgG | Beyotime Institute of Biotechnology, Haimen, Jiangsu, China | 1:500 for IF |
| Horseradish peroxidase (HRP) conjugated anti-rabbit IgG | Jackson ImmunoResearch Laboratories, Inc. West Grove, PA, USA | 1:5000 for WB |
| HRP conjugated anti-mouse IgG | Jackson ImmunoResearch Laboratories, Inc. West Grove, PA, USA | 1:5000 for WB |
| HRP conjugated anti-goat IgG | Jackson ImmunoResearch Laboratories, Inc. West Grove, PA, USA | 1:5000 for WB |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Z.-Y.; Jin, G.-N.; Wang, W.; Sun, Y.-M.; Chen, W.-X.; Chen, L.; Liang, H.-F.; Datta, P.K.; Zhang, M.-Z.; Zhang, B.; et al. Activin A-Smad Signaling Mediates Connective Tissue Growth Factor Synthesis in Liver Progenitor Cells. Int. J. Mol. Sci. 2016, 17, 408. https://doi.org/10.3390/ijms17030408
Ding Z-Y, Jin G-N, Wang W, Sun Y-M, Chen W-X, Chen L, Liang H-F, Datta PK, Zhang M-Z, Zhang B, et al. Activin A-Smad Signaling Mediates Connective Tissue Growth Factor Synthesis in Liver Progenitor Cells. International Journal of Molecular Sciences. 2016; 17(3):408. https://doi.org/10.3390/ijms17030408
Chicago/Turabian StyleDing, Ze-Yang, Guan-Nan Jin, Wei Wang, Yi-Min Sun, Wei-Xun Chen, Lin Chen, Hui-Fang Liang, Pran K. Datta, Ming-Zhi Zhang, Bixiang Zhang, and et al. 2016. "Activin A-Smad Signaling Mediates Connective Tissue Growth Factor Synthesis in Liver Progenitor Cells" International Journal of Molecular Sciences 17, no. 3: 408. https://doi.org/10.3390/ijms17030408
APA StyleDing, Z.-Y., Jin, G.-N., Wang, W., Sun, Y.-M., Chen, W.-X., Chen, L., Liang, H.-F., Datta, P. K., Zhang, M.-Z., Zhang, B., & Chen, X.-P. (2016). Activin A-Smad Signaling Mediates Connective Tissue Growth Factor Synthesis in Liver Progenitor Cells. International Journal of Molecular Sciences, 17(3), 408. https://doi.org/10.3390/ijms17030408