
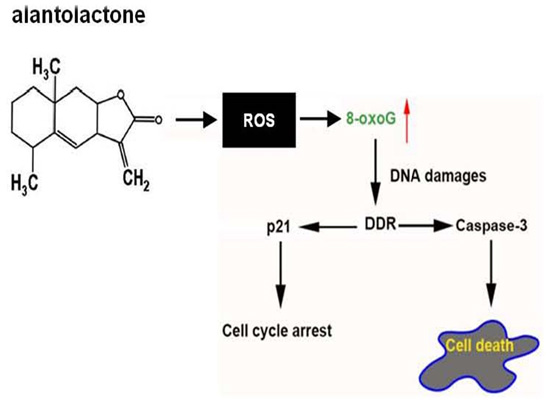
Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Cytotoxicity of Alantolactone (ATL) against SW480 and SW1116 Colorectal Cancer Cells
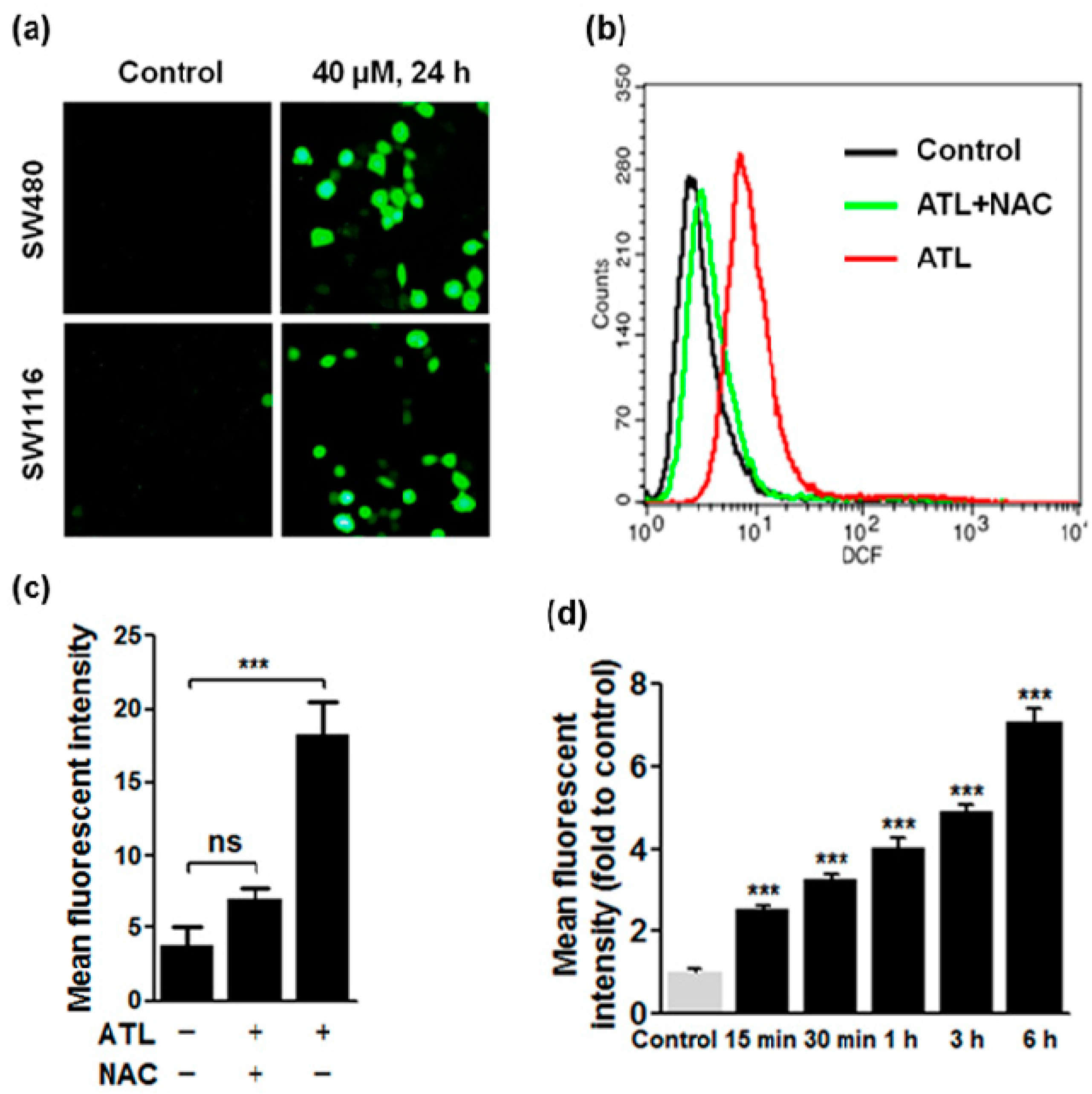
2.2. ATL Increased Reactive Oxygen Species (ROS) Levels in SW480 and SW1116 Colorectal Cancer Cells
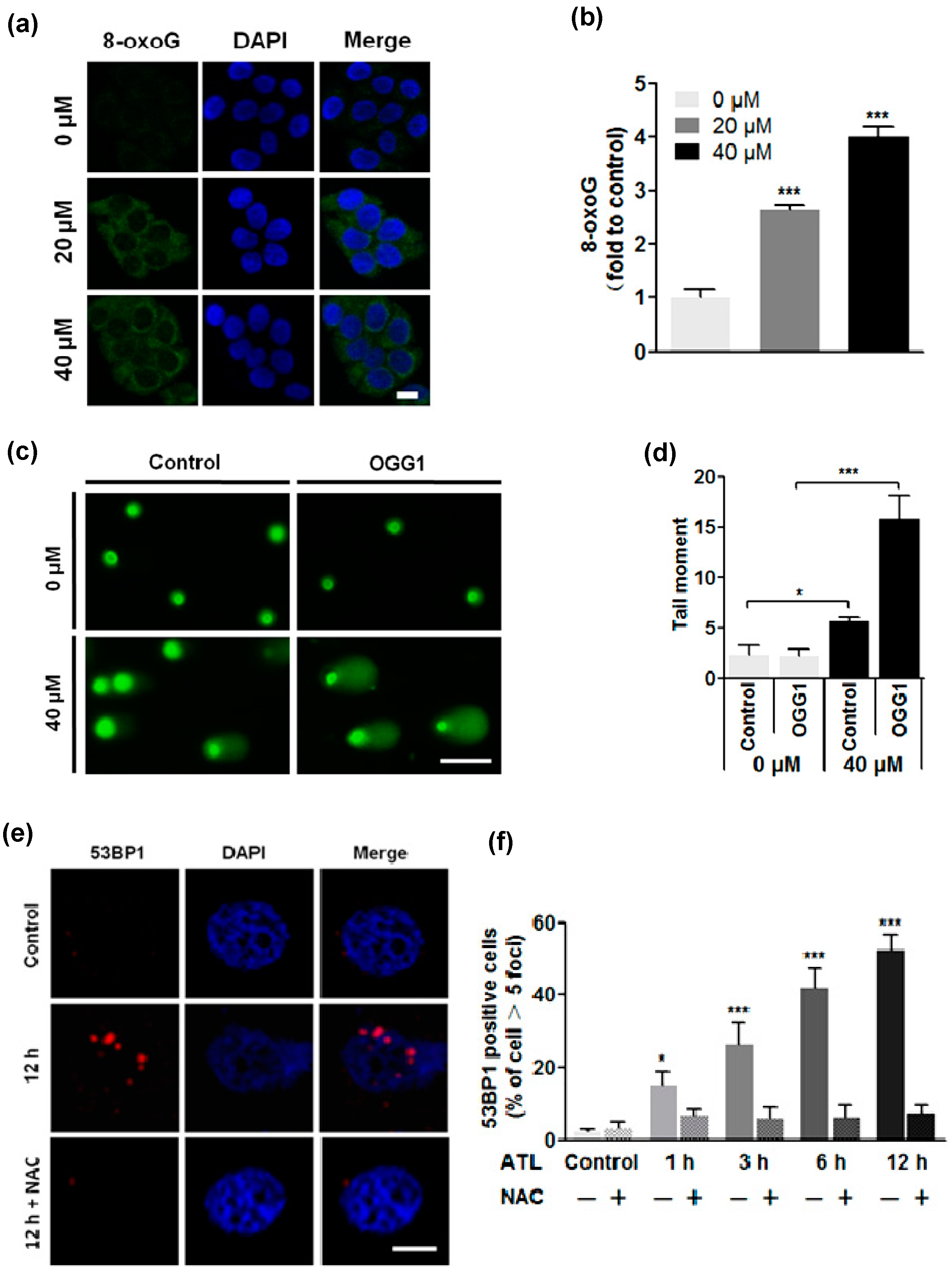
2.3. ROS Overload Resulted in Extensive DNA Damage
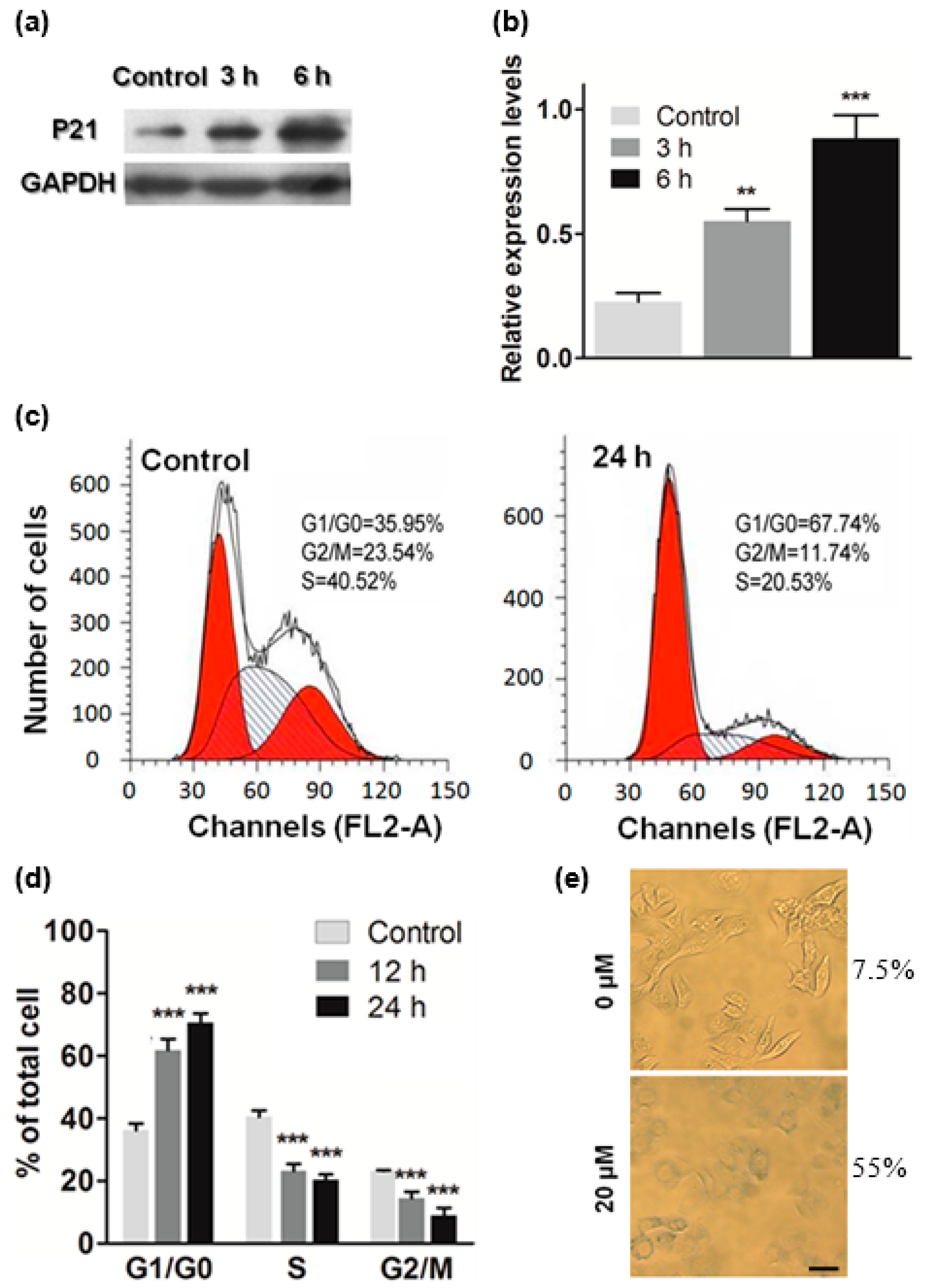
2.4. ATL Induced Cell Cycle Arrest at G1
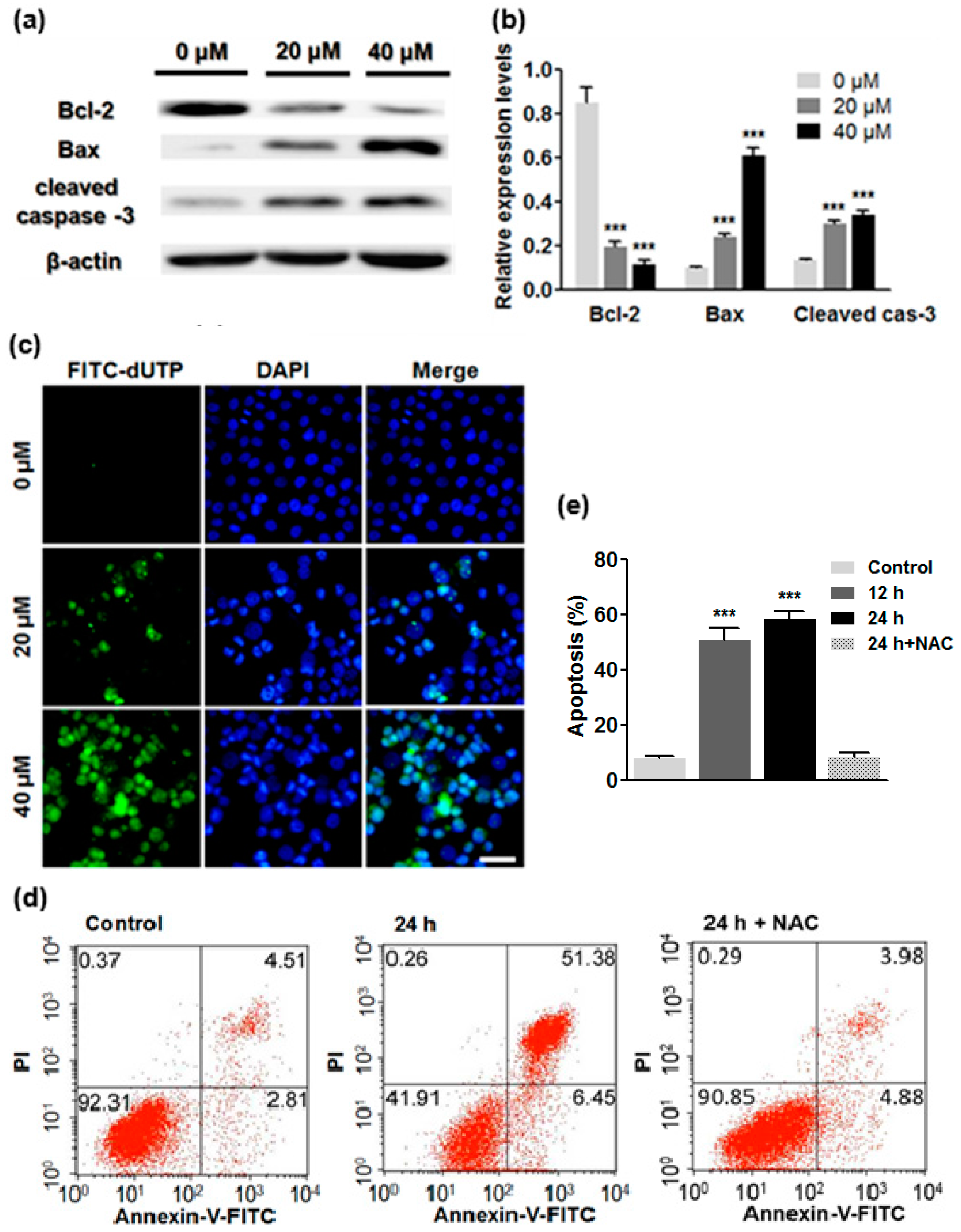
2.5. ATL Induced Apoptotic Cancer Cell Death
3. Materials and Methods
3.1. Reagents and Cells
3.2. MTT Assay
3.3. Colony Outgrowth Assay
3.4. Detection of Total Cellular ROS
3.5. Examination of Cellular 8-Oxoguanine (8-oxoG)
3.6. Comet Assay
3.7. Immunostaining
3.8. Western Blot
3.9. Cell Cycle Analysis
3.10. Detection of Cellular Senescence
3.11. Flow Cytometry Analysis of Apoptosis
3.12. TUNEL Assay
3.13. Determination of Mitochondrial Membrane Potential
3.14. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dancey, J.E.; Bedard, P.L.; Onetto, N.; Hudson, T.J. The genetic basis for cancer treatment decisions. Cell 2012, 148, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Block, K.I.; Gyllenhaal, C.; Lowe, L.; Amedei, A.; Amin, A.R.; Amin, A.; Aquilano, K.; Arbiser, J.; Arreola, A.; Arzumanyan, A.; et al. Designing a broad-spectrum integrative approach for cancer prevention and treatment. Semin. Cancer Biol. 2015, 35, S276–S304. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, Z. Understanding the genetic mechanisms of cancer drug resistance using genomic approaches. Trends Genet. 2016, 32, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Fece de la Cruz, F.; Gapp, B.V.; Nijman, S.M. Synthetic lethal vulnerabilities of cancer. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Cancer phenotypic lethality, exemplified by the non-essential MTH1 enzyme being required for cancer survival. Ann. Oncol. 2014, 25, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed]
- Tesori, V.; Piscaglia, A.C.; Samengo, D.; Barba, M.; Bernardini, C.; Scatena, R.; Pontoglio, A.; Castellini, L.; Spelbrink, J.N.; Maulucci, G.; et al. The multikinase inhibitor sorafenib enhances glycolysis and synergizes with glycolysis blockade for cancer cell killing. Sci. Rep. 2015, 5, 9149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Lu, Z. Pyruvate kinase M2 at a glance. J. Cell Sci. 2015, 128, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, Y.; Le, W.; Wang, K.; Kieffer, N.; Zhang, J. Redox control of the survival of healthy and diseased cells. Antioxid. Redox Signal. 2011, 15, 2867–2908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, K.; Lei, Y.; Li, Q.; Nice, E.C.; Huang, C. Redox signaling: Potential arbitrator of autophagy and apoptosis in therapeutic response. Free Radic. Biol. Med. 2015, 89, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, O.P.; Albanes, D. The effect of vitamin E and β carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar]
- Goodman, M.; Bostick, R.M.; Kucuk, O.; Jones, D.P. Clinical trials of antioxidants as cancer prevention agents: Past, present, and future. Free Radic. Biol. Med. 2011, 51, 1068–1084. [Google Scholar] [CrossRef] [PubMed]
- Kristal, A.R.; Darke, A.K.; Morris, J.S.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M.; Meyskens, F.L., Jr.; Goodman, G.E.; Minasian, L.M.; Parnes, H.L.; et al. Baseline selenium status and effects of selenium and vitamin E supplementation on prostate cancer risk. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.F. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Strom, C.E.; Svensson, L.M.; Schultz, N.; Lundback, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Gokturk, C.; Sanjiv, K.; Stromberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wang, H.; Niu, J.; Zou, M.; Wu, N.; Yu, D.; Wang, Y.; Zou, Z. Echinacoside induces apoptotic cancer cell death by inhibiting the nucleotide pool sanitizing enzyme MTH1. Onco Targets Ther. 2015, 8, 3649–3664. [Google Scholar] [PubMed]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Jutooru, I.; Guthrie, A.S.; Chadalapaka, G.; Pathi, S.; Kim, K.; Burghardt, R.; Jin, U.H.; Safe, S. Mechanism of action of phenethylisothiocyanate and other reactive oxygen species-inducing anticancer agents. Mol. Cell. Biol. 2014, 34, 2382–2395. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Zhang, J.; Xia, Y.; Kanchana, K.; Guo, G.; Chen, W.; Huang, Y.; Wang, Z.; Yang, S.; Liang, G. ROS generation mediates the anti-cancer effects of WZ35 via activating JNK and ER stress apoptotic pathways in gastric cancer. Oncotarget 2015, 6, 5860–5876. [Google Scholar] [CrossRef] [PubMed]
- Mileo, A.M.; Miccadei, S. Polyphenols as modulator of oxidative stress in cancer disease: New therapeutic strategies. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Demma, J.; El-Seedi, H.; Engidawork, E.; Aboye, T.L.; Goransson, U.; Hellman, B. An in vitro study on the DNA damaging effects of phytochemicals partially isolated from an extract of glinus lotoides. Phytother. Res. 2013, 27, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Rasul, A.; Khan, M.; Ali, M.; Li, J.; Li, X. Targeting apoptosis pathways in cancer with alantolactone and isoalantolactone. Sci. World J. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Yi, F.; Rasul, A.; Li, T.; Wang, N.; Gao, H.; Gao, R.; Ma, T. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life 2012, 64, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Li, T.; Ahmad Khan, M.K.; Rasul, A.; Nawaz, F.; Sun, M.; Zheng, Y.; Ma, T. Alantolactone induces apoptosis in HepG2 cells through GSH depletion, inhibition of STAT3 activation, and mitochondrial dysfunction. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Duan, D.; Yao, J.; Gao, K.; Fang, J. Inhibition of thioredoxin reductase by alantolactone prompts oxidative stress-mediated apoptosis of Hela cells. Biochem. Pharmacol. 2016, 102, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bao, Y.L.; Wu, Y.; Yu, C.L.; Huang, Y.X.; Sun, Y.; Zheng, L.H.; Li, Y.X. Alantolactone induces apoptosis in RKO cells through the generation of reactive oxygen species and the mitochondrial pathway. Mol. Med. Rep. 2013, 8, 967–972. [Google Scholar] [PubMed]
- Struthers, L.; Patel, R.; Clark, J.; Thomas, S. Direct detection of 8-oxodeoxyguanosine and 8-oxoguanine by avidin and its analogues. Anal. Biochem. 1998, 255, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y. Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 2014, 15, 12543–12557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Wang, H.; Niu, J.; Luo, M.; Gou, Y.; Miao, L.; Zou, Z.; Cheng, Y. Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells. Int. J. Mol. Sci. 2016, 17, 558. https://doi.org/10.3390/ijms17040558
Ding Y, Wang H, Niu J, Luo M, Gou Y, Miao L, Zou Z, Cheng Y. Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells. International Journal of Molecular Sciences. 2016; 17(4):558. https://doi.org/10.3390/ijms17040558
Chicago/Turabian StyleDing, Yushuang, Hongge Wang, Jiajing Niu, Manyu Luo, Yangmei Gou, Lining Miao, Zhihua Zou, and Ying Cheng. 2016. "Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells" International Journal of Molecular Sciences 17, no. 4: 558. https://doi.org/10.3390/ijms17040558
APA StyleDing, Y., Wang, H., Niu, J., Luo, M., Gou, Y., Miao, L., Zou, Z., & Cheng, Y. (2016). Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells. International Journal of Molecular Sciences, 17(4), 558. https://doi.org/10.3390/ijms17040558