
3D-QSAR Studies on Barbituric Acid Derivatives as Urease Inhibitors and the Effect of Charges on the Quality of a Model




Abstract
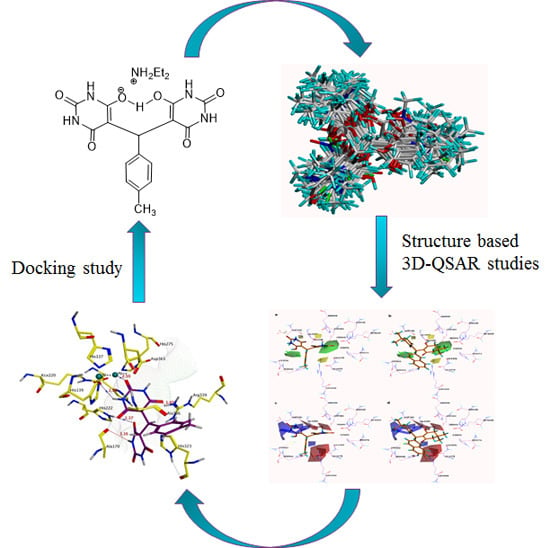
:
1. Introduction
2. Results
3. Discussion
3.1. Docking-Based Structural Alignment
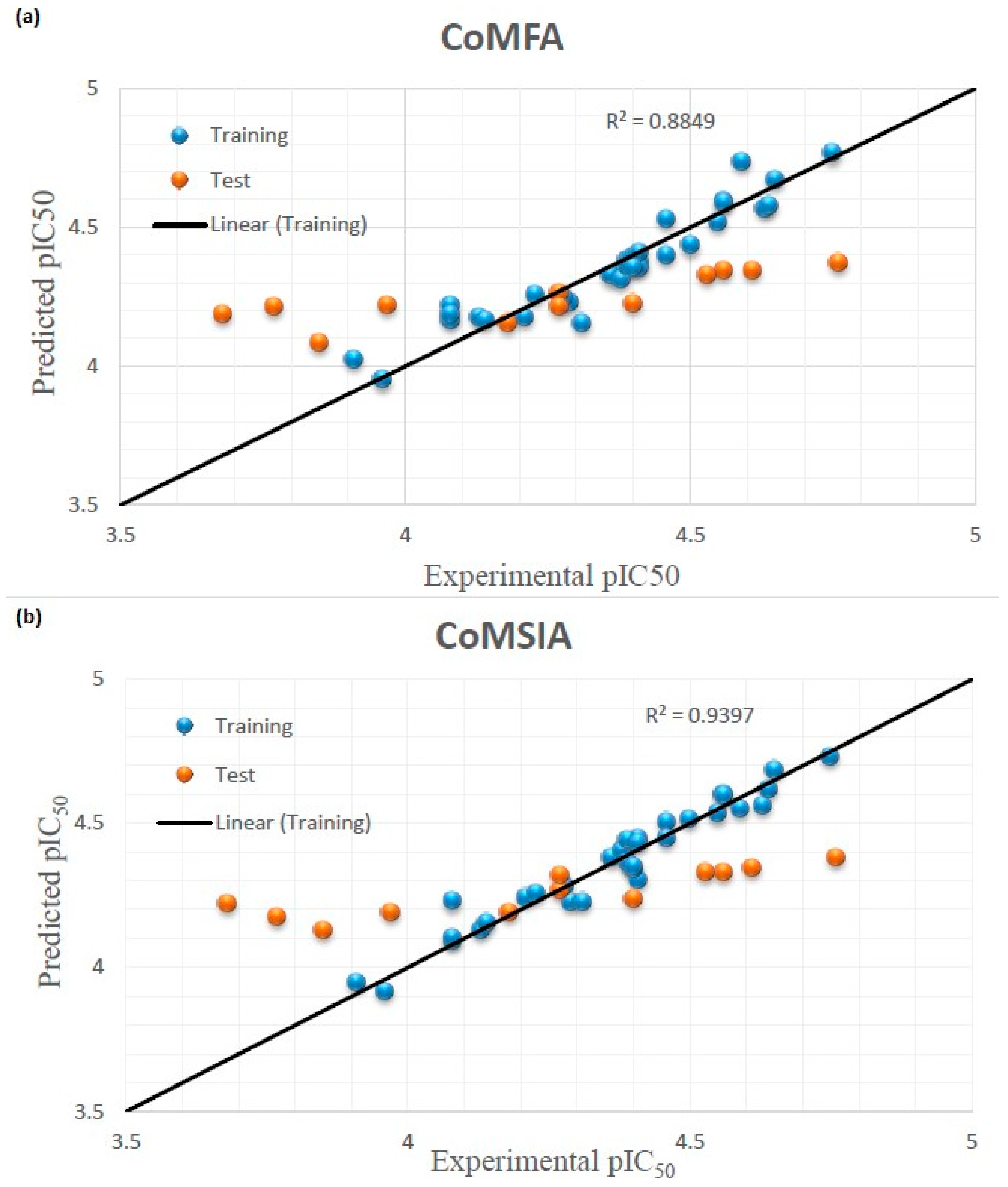
3.2. Effect of Charges on 3D-QSAR Model
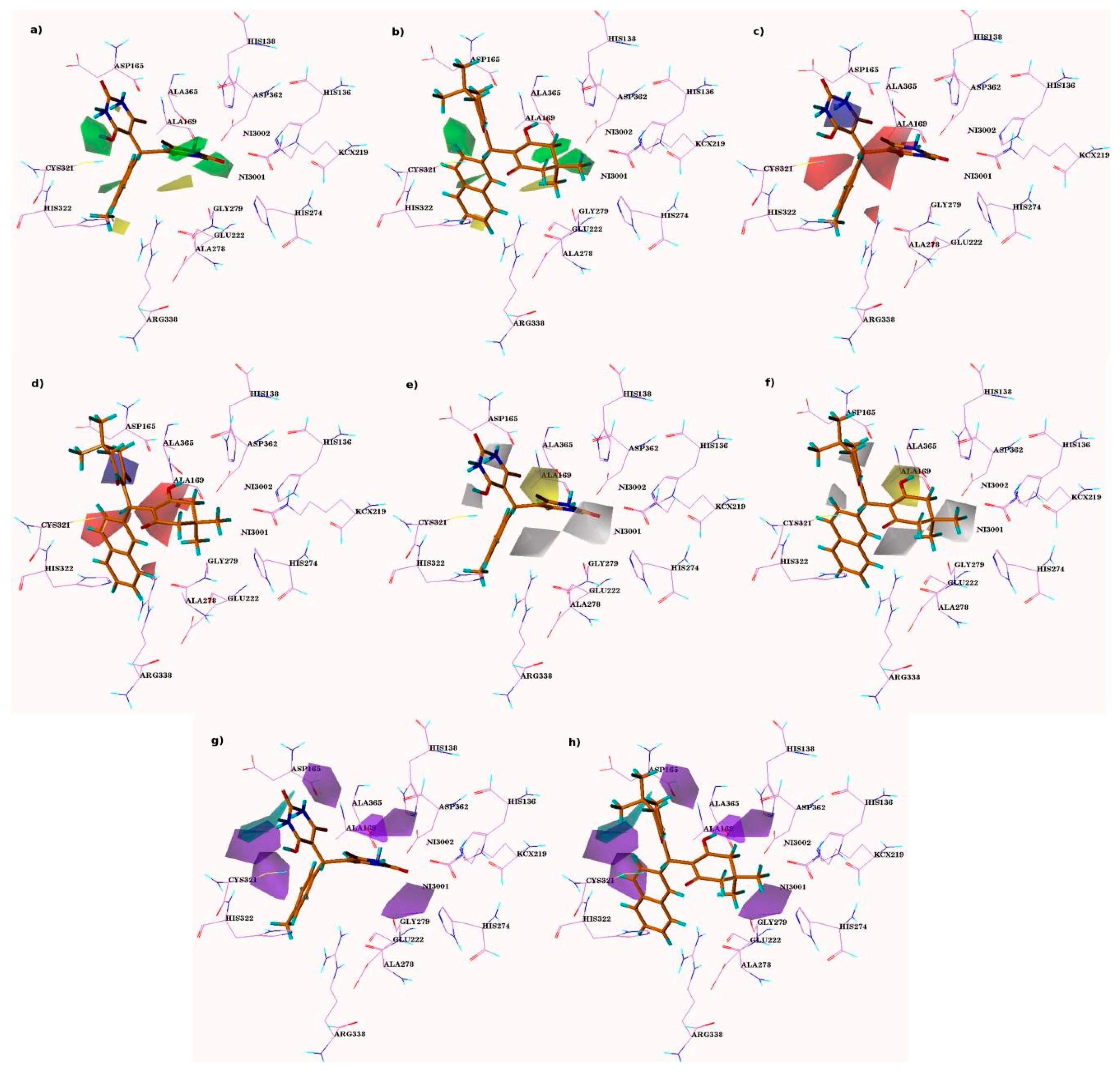
3.3. D-QSAR Contour Maps Analysis
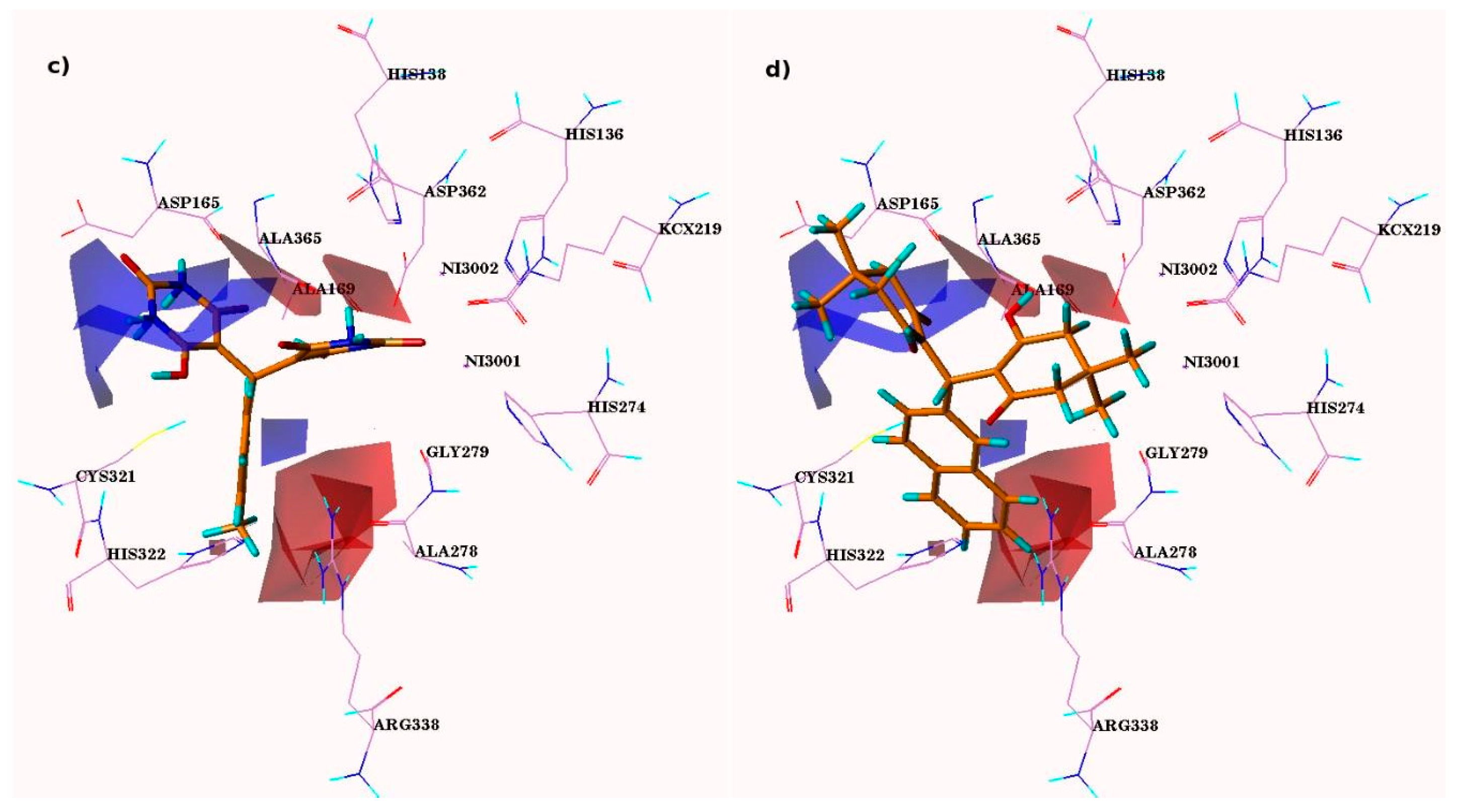
3.4. CoMFA Contour Analysis
3.5. CoMSIA Contour Maps Analysis
3.6. Steric and Electrostatic CoMSIA Contour Maps
3.7. CoMSIA Hydrophobic Contour Maps
3.8. CoMSIA Hydrogen Bond Donor Contours
3.9. Building of QSAR Model by MOE
4. Materials and Methods
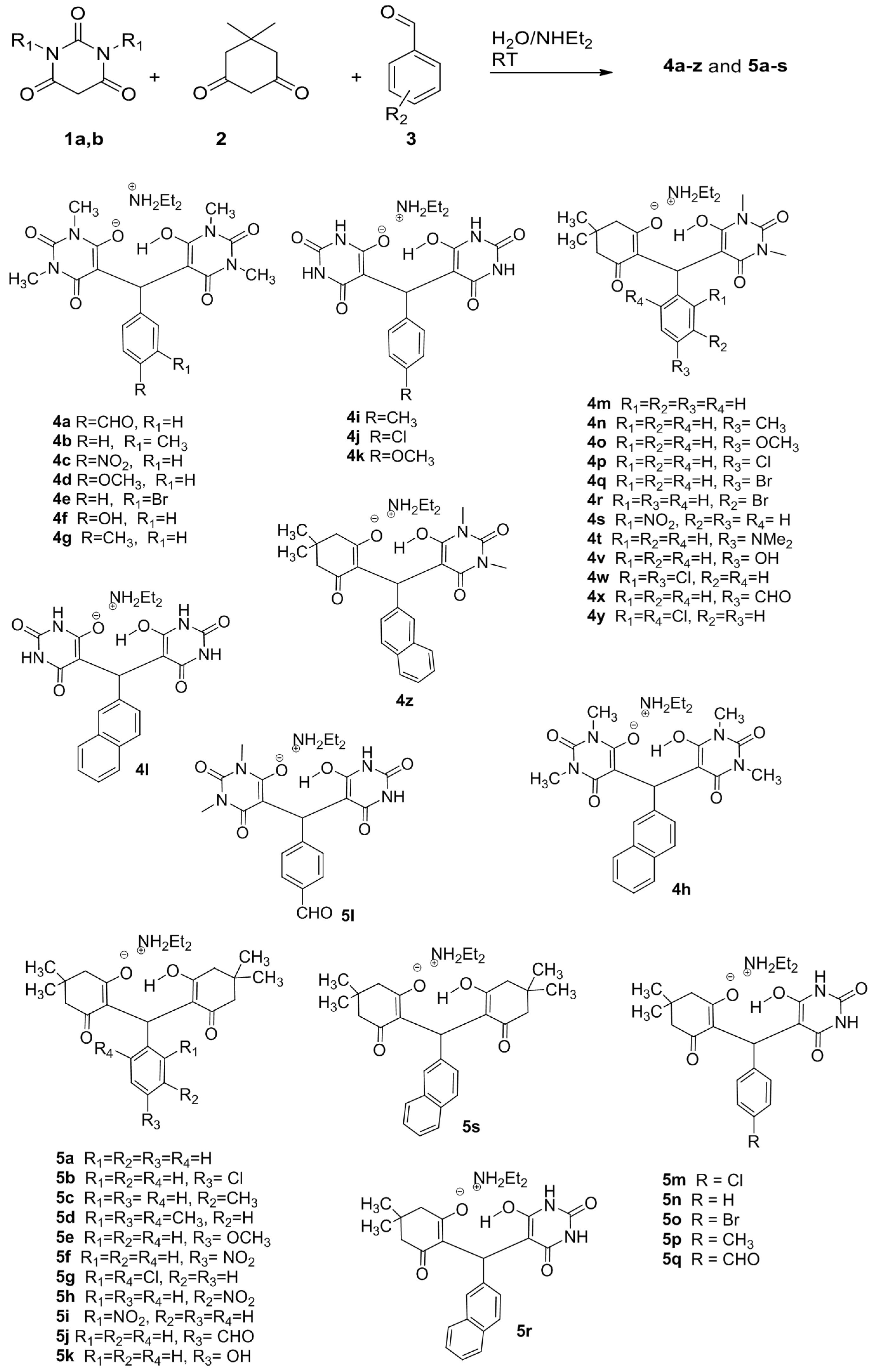
4.1. Dataset Preparation
4.2. Computational Modeling Tools
4.3. X-ray Crystal Structures of H. pylori
4.4. Alignment of Compounds Dataset
4.5. Partial Atomic Charges
4.6. CoMFA and CoMSIA Studies
4.7. Partial Least Squares (PLS)/Statistical Analysis
4.8. Molecular Descriptors
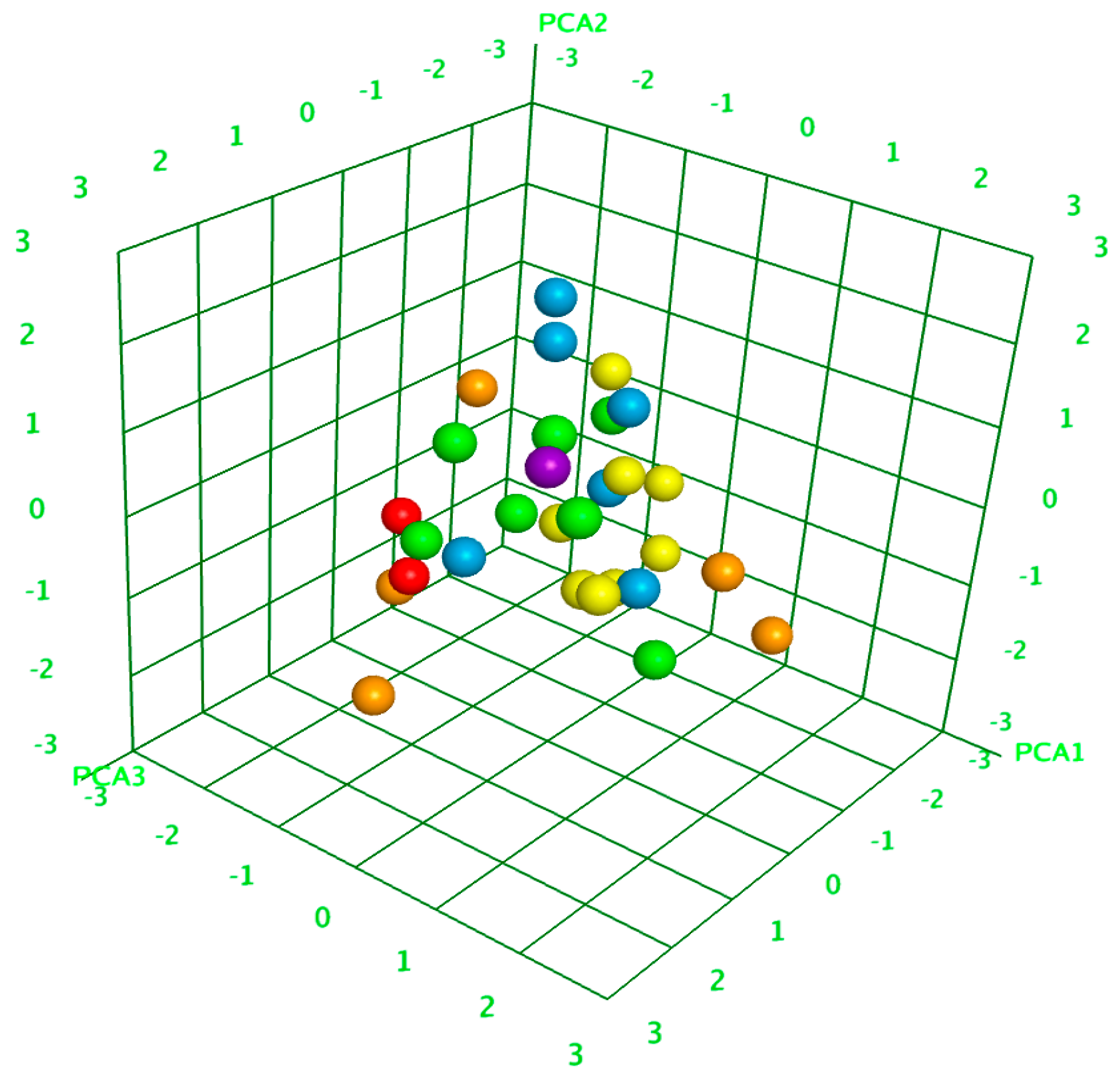
4.9. Principal Component Analysis (PCA)
4.10. Partial Least Squares (PLS) by MOE
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dixon, N.E.; Gazzola, C.; Blakeley, R.L.; Zerner, B. Jack bean urease (ec 3.5. 1.5). Metalloenzyme. Simple biological role for nickel. J. Am. Chem. Soc. 1975, 97, 4131–4133. [Google Scholar] [CrossRef] [PubMed]
- Sumner, J.B. The isolation and crystallization of the enzyme urease preliminary paper. J. Biol. Chem. 1926, 69, 435–441. [Google Scholar]
- Mobley, H.; Hausinger, R. Microbial ureases: Significance, regulation, and molecular characterization. Microbiol. Rev. 1989, 53, 85–108. [Google Scholar] [PubMed]
- Krajewska, B. Ureases I. Functional, catalytic and kinetic properties: A review. J. Mol. Catal. B Enzym. 2009, 59, 9–21. [Google Scholar] [CrossRef]
- Zonia, L.E.; Stebbins, N.E.; Polacco, J.C. Essential role of urease in germination of nitrogen-limited arabidopsis thaliana seeds. Plant Physiol. 1995, 107, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.; Island, M.D.; Hausinger, R.P. Molecular biology of microbial ureases. Microbiol. Rev. 1995, 59, 451–480. [Google Scholar] [PubMed]
- Ermler, U.; Grabarse, W.; Shima, S.; Goubeaud, M.; Thauer, R.K. Active sites of transition-metal enzymes with a focus on nickel. Curr. Opin. Struct. Biol. 1998, 8, 749–758. [Google Scholar] [CrossRef]
- Lodhi, M.A.; Nawaz, S.A.; Iqbal, S.; Khan, K.M.; Rode, B.M.; Choudhary, M.I. 3D-QSAR CoMFA studies on bis-coumarine analogues as urease inhibitors: A strategic design in anti-urease agents. Bioorg. Med. Chem. 2008, 16, 3456–3461. [Google Scholar]
- Maroney, M.J.; Ciurli, S. Nonredox nickel enzymes. Chem. Rev. 2013, 114, 4206–4228. [Google Scholar] [CrossRef] [PubMed]
- Algood, H.M.S.; Cover, T.L. Helicobacter pylori persistence: An overview of interactions between H. Pylori and host immune defenses. Clin. Microbial. Rev. 2006, 19, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA A Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Follmer, C. Ureases as a target for the treatment of gastric and urinary infections. J. Clin. Pathol. 2010, 63, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Onoda, Y.; Magaribuchi, T.; Tamaki, H. Effects of the new anti-ulcer agent 12-sulfodehydroabietic acid monosodium salt on duodenal alkaline secretion in rats. Arzneimittel-Forschung 1990, 40, 576–578. [Google Scholar] [PubMed]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Yathindra, N. Structure of an enzyme revealed 80 years after it was crystallizeddifferential functional behaviour of plant and microbial ureases uncovered. Curr. Sci. 2010, 99, 566–568. [Google Scholar]
- Sirko, A.; Brodzik, R. Plant ureases: Roles and regulation. Acta Biochim. Pol. 1999, 47, 1189–1195. [Google Scholar]
- Sachs, G.; Weeks, D.L.; Wen, Y.; Marcus, E.A.; Scott, D.R.; Melchers, K. Acid acclimation by helicobacter pylori. Physiology 2005, 20, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Stingl, K.; Altendorf, K.; Bakker, E.P. Acid survival of helicobacter pylori: How does urease activity trigger cytoplasmic ph homeostasis? Trends Microbiol. 2002, 10, 70–74. [Google Scholar] [CrossRef]
- Graham, D.Y.; Fischbach, L. Helicobacter pylori treatment in the era of increasing antibiotic resistance. Gut 2010, 2009, 192757. [Google Scholar] [CrossRef] [PubMed]
- Mégraud, F. The challenge of helicobacter pylori resistance to antibiotics: The comeback of bismuth-based quadruple therapy. Ther. Adv. Gastroenterol. 2012, 5, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Maneeruttanarungroj, C.; Nichols, S.E.; Lyons, T.M.; Tirado-Rives, J.; Jorgensen, W.L.; Yuthavong, Y.; Anderson, K.S. Exploiting structural analysis, in silico screening, and serendipity to identify novel inhibitors of drug-resistant falciparum malaria. ACS Chem. Biol. 2009, 4, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.; Yuan, H.; Petukhov, P.A.; Choudhary, M.I.; Madura, J.D. Receptor-based modeling and 3D-QSAR for a quantitative production of the butyrylcholinesterase inhibitors based on genetic algorithm. J. Chem. Inf. Model. 2008, 48, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Almajid, A.-M.; Lotfy, G.; Arshad, F.; Yousuf, S.; Choudhary, M.I.; Ashraf, S.; Ul-Haq, Z. Synthesis and dynamics studies of barbituric acid derivatives as urease inhibitors. Chem. Cent. J. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Al-Majid, A.M.; Barakat, A.; AL-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.-K. Tandem Aldol-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to bis-pyrimidine derivatives. Int. J. Mol. Sci. 2013, 14, 23762–23773. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Bruno, O.; Fossa, P. Docking-based comfa and comsia analyses of tetrahydro-β-carboline derivatives as type-5 phosphodiesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Buffa, L.; Fossa, P. 3,4,5-Trisubstituted-1,2,4–4h-Triazoles as Wt and Y188l mutant HIV-1 non-nucleoside reverse transcriptase inhibitors: Docking-based CoMFA and CoMSIA analyses. J. Mol. Model. 2011, 17, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Cesarini, S.; Mosti, L.; Fossa, P. CoMFA and CoMSIA analyses on 1,2,3,4-tetrahydropyrrolo [3,4-b] indole and benzimidazole derivatives as selective CB2 receptor agonists. J. Mol. Model. 2010, 16, 1481–1498. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Fossa, P. Docking-based 3D-QSAR analyses of pyrazole derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Mol. Model. 2012, 18, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Al-Majid, A.M.; Islam, M.S.; Barakat, A.; Al-Qahtani, N.J.; Yousuf, S.; Choudhary, M.I. Tandem knoevenagel-michael reactions in aqueous diethylamine medium: A greener and efficient approach toward bis-dimedone derivatives. Arab. J. Chem. 2014. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Majid, A.M.; Al-Ghamdi, A.M.; Mabkhot, Y.N.; Siddiqui, M.R.H.; Ghabbour, H.A.; Fun, H.-K. Tandem Aldol–Michael reactions in aqueous diethylamine medium: A greener and efficient approach to dimedone-barbituric acid derivatives. Chem. Cent. J. 2014, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Al-Najjar, H.J.; Mabkhot, Y.N.; Javaid, S.; Yousuf, S.; Choudhary, M.I. Zwitterionic pyrimidinium adducts as antioxidants with therapeutic potential as nitric oxide scavenger. Eur. J. Med. Chem. 2014, 84, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wan, H.; Shi, Y.; Ouyang, P. Personal experience with four kinds of chemical structure drawing software: Review on chemdraw, chemwindow, ISIS/Draw, and ChemSketch. J. Chem. Inf. Comput. Sci. 2004, 44, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2015; Chemical Computing Group Inc.: Montreal, QC, Canada, 2015; H3A 2R7.
- SYBYL 7.3; Tripos International: St. Louis, MO, USA, 2007.
- Ha, N.-C.; Oh, S.-T.; Sung, J.Y.; Cha, K.A.; Lee, M.H.; Oh, B.-H. Supramolecular assembly and acid resistance of helicobacter pylori urease. Nat. Struct. Mol. Biol. 2001, 8, 505–509. [Google Scholar] [CrossRef] [PubMed]
- QUACPAC; Openeye Scientific Software, Inc.: Santa Fe, NM, USA, 2011.
- Cramer, R.D., 3rd; Patterson, D.E.; Bunce, J.D. Recent advances in comparative molecular field analysis (CoMFA). Prog. Clin. Biol. Res. 1989, 291, 161–165. [Google Scholar] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U. Comparative molecular similarity index analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J. Comput. Aided Mol. Des. 1999, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Bush, B.L.; Nachbar, R.B., Jr. Sample-distance partial least squares: PLS optimized for 510 many variables, with application to CoMFA. J. Comput. Aided Mol. 511 Des. 1993, 7, 587–619. [Google Scholar] [CrossRef]
- Vrontaki, E.; Melagraki, G.; Mavromoustakos, T.; Afantitis, A. Searching for anthranilic acid-based thumb pocket 2 HCV NS5B polymerase inhibitors through a combination of molecular docking, 3D-QSAR and virtual screening. J. Enzym. Inhib. Med. Chem. 2016, 31, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.H.; Reinders, J.H.; Tolboom, J.T.; Glennon, J.C.; Coolen, H.K.; Kruse, C.G. Principal component analysis differentiates the receptor binding profiles of three antipsychotic drug candidates from current antipsychotic drugs. J. Med. Chem. 2007, 50, 5103–5108. [Google Scholar] [CrossRef] [PubMed]
- Ringner, M. What is Principal Component Analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Charge | q2 | r2 | SEE | SEP | F | NOC | S | E |
|---|---|---|---|---|---|---|---|---|---|
| CoMFA-1 | AM1 | 0.590 | 0.996 | 0.014 | 0.150 | 124.56 | 6 | 0.510 | 0.490 |
| CoMFA-2 | AM1BCC | 0.522 | 0.965 | 0.042 | 0.155 | 184.465 | 4 | 0.568 | 0.432 |
| CoMFA-3 | GH⁄ | 0.507 | 0.970 | 0.039 | 0.158 | 220.940 | 4 | – | – |
| CoMFA-4 | GM⁄ | 0.510 | 0.935 | 0.056 | 0.155 | 135.194 | 3 | 0.494 | 0.506 |
| CoMFA-5 | MMFF94 | 0.597 | 0.897 | 0.071 | 0.140 | 81.306 | 3 | 0.481 | 0.519 |
| Combinations | q2 | r2 | r2 | F | C | SEE | SEP | 1% | 2% | 3% | 4% | 5% |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S + E | 0.555 | 0.808 | – | 39.401 | 3 | 0.097 | 0.147 | 21.4 | 78.6 | _ | _ | _ |
| S + H | 0.612 | 0.954 | – | 108.02 | 5 | 0.049 | 0.143 | 15 | 85 | _ | _ | _ |
| S + D | 0.621 | 0.925 | – | 51.758 | 6 | 0.064 | 0.144 | 25.7 | 74.3 | _ | _ | _ |
| H + D | 0.610 | 0.973 | – | 148.944 | 6 | 0.696 | 0.146 | 56.4 | 43.6 | _ | _ | _ |
| S + E + H | 0.572 | 0.953 | – | 105.061 | 5 | 0.05 | 0.15 | 11.9 | 35.6 | 52.5 | _ | _ |
| S + E + D | 0.599 | 0.947 | – | 93.825 | 5 | 0.053 | 0.145 | 13.1 | 40.1 | 46.8 | _ | _ |
| S + H + D | 0.634 | 0.978 | – | 184.087 | 6 | 0.035 | 0.141 | 08 | 50.7 | 40.4 | _ | _ |
| E + H + D | 0.552 | 0.978 | – | 183.48 | 6 | 0.035 | 0.156 | 25.2 | 42 | 32.8 | _ | _ |
| S + E + H + A | 0.519 | 0.925 | – | 82.96 | 4 | 0.062 | 0.156 | 8.2 | 30.2 | 40.6 | 21.0 | _ |
| S + E + H + D | 0.602 | 0.98 | 0.84 | 209.259 | 6 | 0.033 | 0.147 | 7.3 | 24 | 38.5 | 30.2 | _ |
| S + H + D + A | 0.509 | 0.962 | – | 131.282 | 5 | 0.045 | 0.161 | 6.5 | 40.8 | 31.7 | 20.9 | _ |
| S + E + H + D + A | 0.533 | 0.945 | – | 115.483 | 4 | 0.053 | 0.154 | 5.4 | 20.3 | 32.3 | 24.6 | 17.4 |
| Compounds | pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Experimental | Predicted | Residual | Predicted | Residual | |
| 4a | 4.41 | 4.374 | 0.03 | 4.396 | 0.01 |
| 4b | 4.46 | 4.399 | 0.06 | 4.478 | −0.01 |
| 4c * | 4.26 | 4.263 | 0 | 4.265 | 0 |
| 4d | 4.5 | 4.435 | 0.07 | 4.489 | 0.01 |
| 4e | 4.56 | 4.592 | −0.03 | 4.569 | −0.01 |
| 4f * | 4.26 | 4.215 | 0.05 | 4.318 | −0.05 |
| 4g | 4.54 | 4.517 | 0.03 | 4.519 | 0.03 |
| 4h | 4.39 | 4.382 | 0.01 | 4.389 | 0.01 |
| 4i | 4.75 | 4.766 | −0.01 | 4.773 | −0.02 |
| 4j | 4.65 | 4.67 | −0.02 | 4.654 | 0 |
| 4k | 4.58 | 4.735 | −0.15 | 4.592 | 0 |
| 4l | 4.64 | 4.579 | 0.07 | 4.624 | 0.02 |
| 4m | 4.4 | 4.355 | 0.05 | 4.326 | 0.08 |
| 4n | 4.38 | 4.362 | 0.02 | 4.36 | 0.02 |
| 4o | 4.08 | 4.166 | −0.09 | 4.151 | −0.07 |
| 4p | 4.4 | 4.393 | 0.01 | 4.403 | 0 |
| 4q * | 4.61 | 4.344 | 0.27 | 4.343 | 0.27 |
| 4r | 4.56 | 4.59 | −0.03 | 4.607 | −0.05 |
| 4s | 3.96 | 3.951 | 0.01 | 3.974 | −0.01 |
| 4t * | 3.84 | 4.083 | −0.24 | 4.128 | −0.28 |
| 4v | 4.28 | 4.248 | 0.03 | 4.27 | 0.01 |
| 4w | 4.22 | 4.256 | −0.03 | 4.246 | −0.02 |
| x | 4.41 | 4.41 | 0 | 4.418 | 0 |
| 4y | 4.07 | 4.186 | −0.11 | 4.074 | 0 |
| 4z | 4.4 | 4.354 | 0.05 | 4.336 | 0.06 |
| 5a | 4.13 | 4.175 | −0.05 | 4.115 | 0.01 |
| 5b * | 4.53 | 4.326 | 0.2 | 4.331 | 0.2 |
| 5c | 4.21 | 4.177 | 0.03 | 4.231 | −0.02 |
| 5d | 4.29 | 4.231 | 0.06 | 4.263 | 0.03 |
| 5e * | 4.4 | 4.226 | 0.17 | 4.238 | 0.16 |
| 5f * | 3.97 | 4.219 | −0.25 | 4.189 | −0.22 |
| 5g * | 3.77 | 4.214 | −0.45 | 4.175 | −0.41 |
| 5h | 4.31 | 4.156 | 0.15 | 4.269 | 0.04 |
| 5i * | 3.68 | 4.129 | −0.45 | 4.322 | −0.65 |
| 5j | 4.14 | 4.164 | −0.02 | 4.154 | −0.01 |
| 5k | 4.35 | 4.325 | 0.03 | 4.399 | −0.04 |
| 5l * | 4.76 | 4.372 | 0.39 | 4.372 | 0.39 |
| 5m * | 4.18 | 4.229 | −0.05 | 4.229 | −0.05 |
| 5n | 4.62 | 4.569 | 0.06 | 4.634 | −0.01 |
| 5o | 4.46 | 4.529 | −0.07 | 4.485 | −0.02 |
| 5p * | 4.56 | 4.343 | 0.22 | 4.343 | 0.22 |
| 5q | 4.38 | 4.313 | 0.07 | 4.39 | −0.01 |
| 5r | 4.08 | 4.219 | −0.14 | 4.09 | −0.01 |
| 5s | 3.91 | 4.022 | −0.11 | 3.922 | −0.01 |
| Compounds Name | IC50 (μM) | pIC50 | Compounds Name | IC50 (μM) | pIC50 |
|---|---|---|---|---|---|
| 4a | 39.3 ± 0.36 | 4.41 | 4x | 38.5 ± 0.28 | 4.41 |
| 4b | 34.4 ± 1.57 | 4.46 | 4y | 83.4 ± 1.00 | 4.07 |
| 4c * | 54.2 ± 0.47 | 4.26 | 4z | 39.8 ± 1.38 | 4.4 |
| 4d | 31.6 ± 0.79 | 4.5 | 5a | 74.5 ± 0.88 | 4.13 |
| 4e | 27.5 ± 0.12 | 4.56 | 5b * | 29.7 ± 0.67 | 4.53 |
| 4f * | 54.2 ± 0.83 | 4.26 | 5c | 61.4 ± 1.12 | 4.21 |
| 4g | 28.5 ± 0.41 | 4.54 | 5d | 51.3 ± 0.45 | 4.29 |
| 4h | 40.3 ± 0.32 | 4.39 | 5e * | 39.8 ± 0.75 | 4.4 |
| 4i | 17.6 ± 0.23 | 4.75 | 5f * | 106.4 ± 1.49 | 3.97 |
| 4j | 22.3 ± 0.73 | 4.65 | 5g * | 170.7 ± 1.55 | 3.77 |
| 4k | 25.8 ± 0.23 | 4.58 | 5h | 49.0 ± 0.55 | 4.31 |
| 4l | 22.7 ± 0.20 | 4.64 | 5i * | 210.1 ± 0.29 | 3.68 |
| 4m | 39.3 ± 0.79 | 4.4 | 5j | 72.6 ± 0.59 | 4.14 |
| 4n | 41.2 ± 0.58 | 4.38 | 5k | 43.8 ± 0.33 | 4.35 |
| 4o | 83.0 ± 0.66 | 4.08 | 5l * | 17.2 ± 0.44 | 4.76 |
| 4p | 39.7 ± 0.70 | 4.4 | 5m * | 65.9 ± 0.61 | 4.18 |
| 4q * | 24.6 ± 0.42 | 4.61 | 5n | 23.7 ± 0.57 | 4.62 |
| 4r | 27.5 ± 0.19 | 4.56 | 5o | 34.6 ± 0.79 | 4.46 |
| 4s | 109.7 ± 1.10 | 3.96 | 5p * | 27.4 ± 0.54 | 4.56 |
| 4t * | 142.1 ± 0.64 | 3.84 | 5q | 41.6 ± 0.41 | 4.38 |
| 4v | 52.2 ± 1.26 | 4.28 | 5r | 82.8 ± 0.72 | 4.08 |
| 4w | 59.4 ± 0.98 | 4.22 | 5s | 123.2 ± 0.37 | 3.91 |
| Code | Description |
|---|---|
| a_acc | Number of hydrogen bond acceptor atoms |
| a_don | Number of hydrogen bond donor atoms |
| a_hyd | Number of hydrophobic atoms. |
| vsa_acc | Approximation to the sum of VDW surface areas (Å2) of pure hydrogen bond acceptors |
| Q_PC+PEOE_PC+ | Total positive partial charge |
| Q_PC-PEOE_PC− | Total negative partial charge |
| i3D | Dipole moment |
| 1K | Kier first shape index |
| 2K | Kier second shape index |
| 3K | Kier third shape index |
| Weight | Molecular weight (including implicit hydrogens) in atomic mass units with atomic weights |
| SlogP | Log of the octanol/water partition coefficient. |
| TPSA | Polar surface area (Å2) |
| a_nN | Number of nitrogen atoms |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ul-Haq, Z.; Ashraf, S.; Al-Majid, A.M.; Barakat, A. 3D-QSAR Studies on Barbituric Acid Derivatives as Urease Inhibitors and the Effect of Charges on the Quality of a Model. Int. J. Mol. Sci. 2016, 17, 657. https://doi.org/10.3390/ijms17050657
Ul-Haq Z, Ashraf S, Al-Majid AM, Barakat A. 3D-QSAR Studies on Barbituric Acid Derivatives as Urease Inhibitors and the Effect of Charges on the Quality of a Model. International Journal of Molecular Sciences. 2016; 17(5):657. https://doi.org/10.3390/ijms17050657
Chicago/Turabian StyleUl-Haq, Zaheer, Sajda Ashraf, Abdullah Mohammed Al-Majid, and Assem Barakat. 2016. "3D-QSAR Studies on Barbituric Acid Derivatives as Urease Inhibitors and the Effect of Charges on the Quality of a Model" International Journal of Molecular Sciences 17, no. 5: 657. https://doi.org/10.3390/ijms17050657
APA StyleUl-Haq, Z., Ashraf, S., Al-Majid, A. M., & Barakat, A. (2016). 3D-QSAR Studies on Barbituric Acid Derivatives as Urease Inhibitors and the Effect of Charges on the Quality of a Model. International Journal of Molecular Sciences, 17(5), 657. https://doi.org/10.3390/ijms17050657