
Rapid Diminution in the Level and Activity of DNA-Dependent Protein Kinase in Cancer Cells by a Reactive Nitro-Benzoxadiazole Compound


 and
and
Abstract
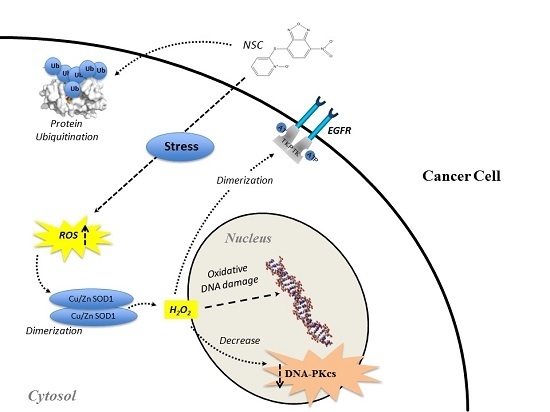
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
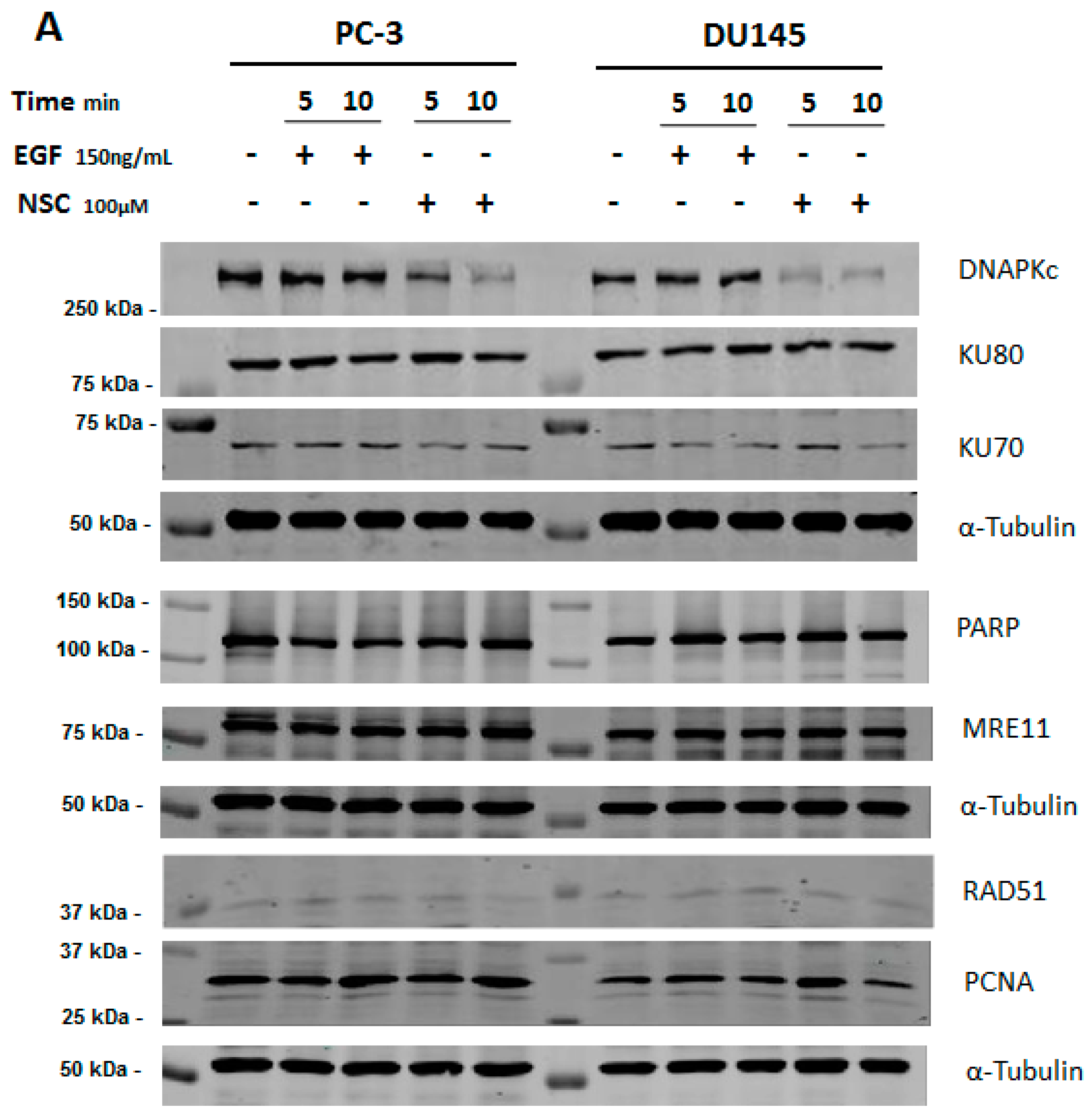
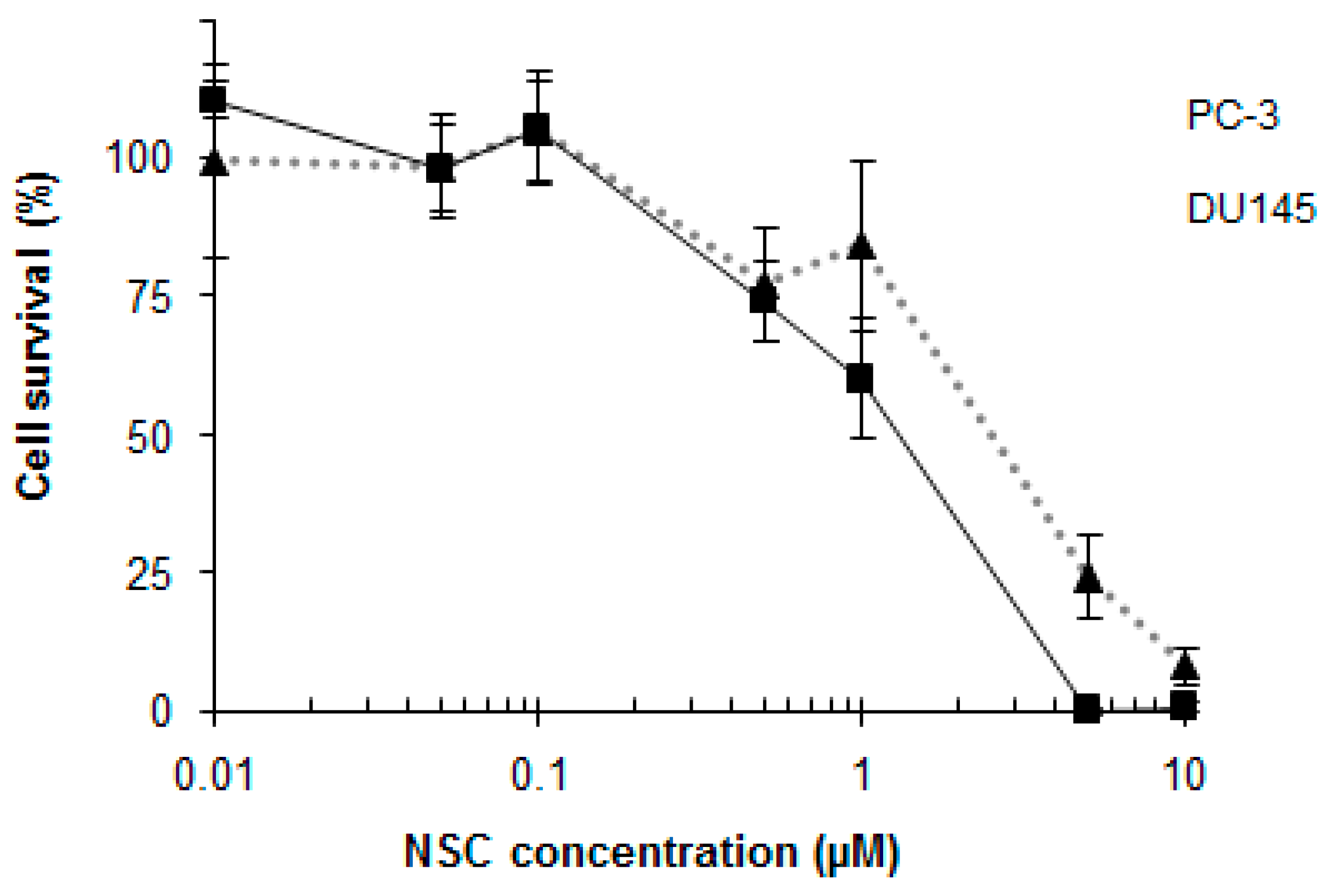
2.1. NSC Treatment Rapidly Affects the Amount of DNA-PKcs Protein
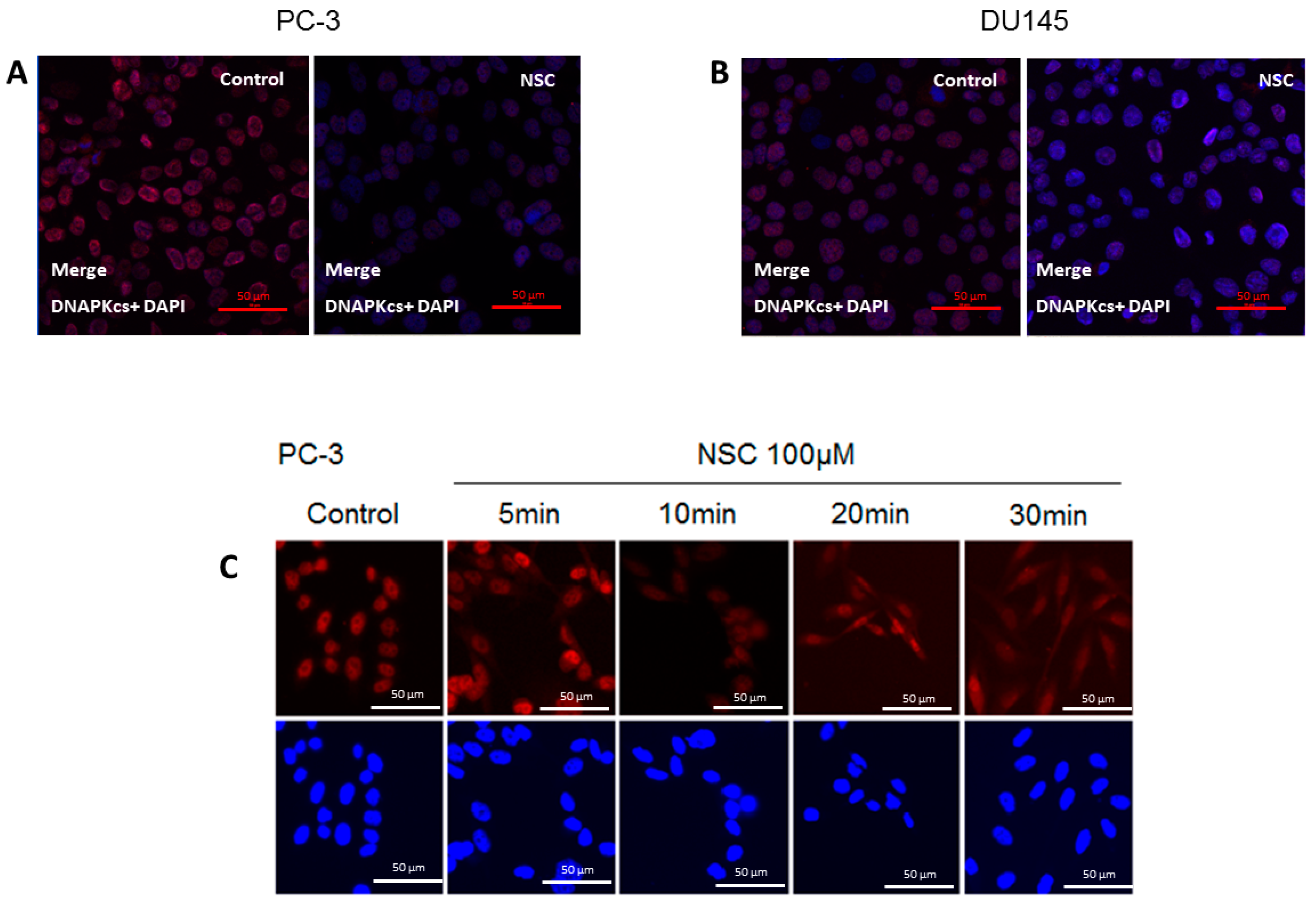
2.2. Subcellular Distribution of DNA-PKcs
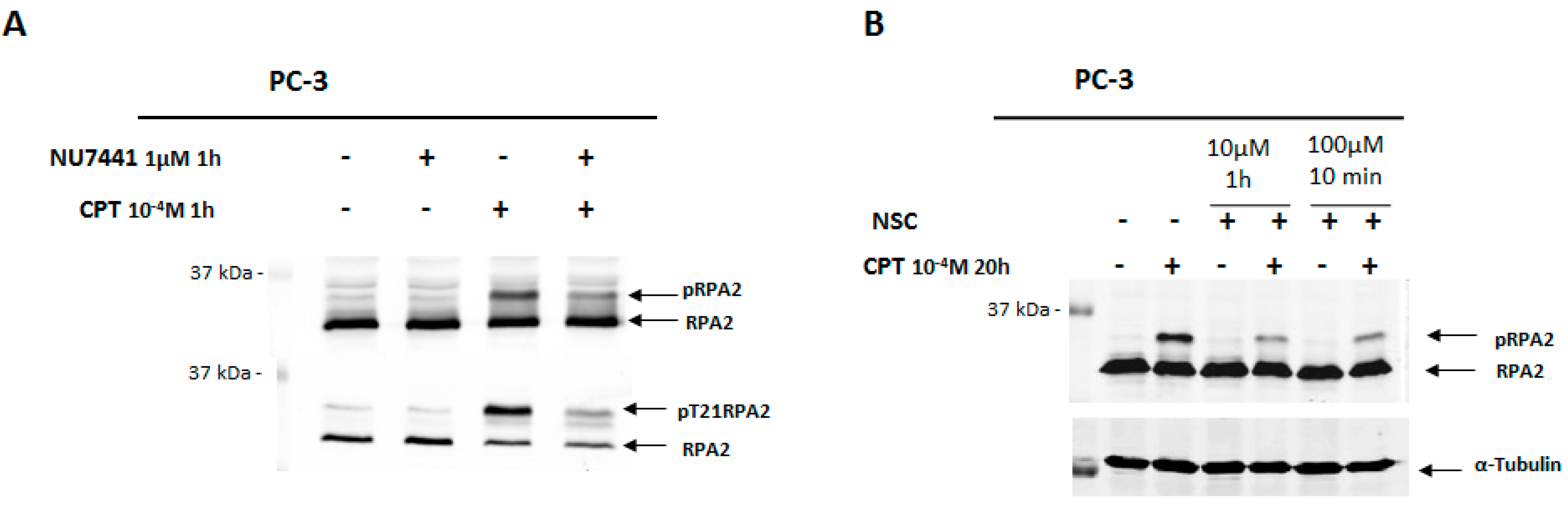
2.3. Exposure to NSC Alters the Activity of DNA-PKcs
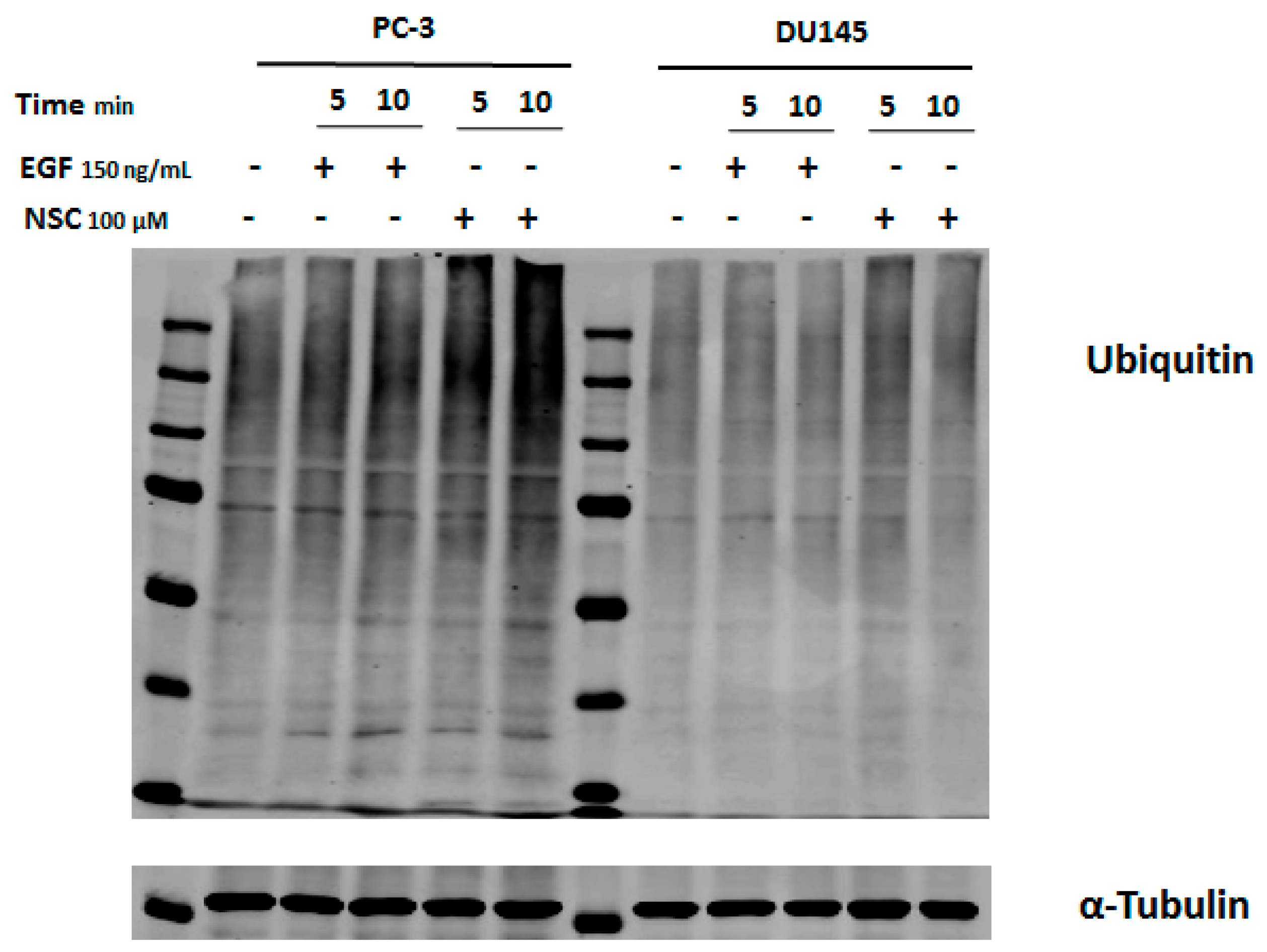
2.4. Exposure to NSC Promotes Protein Ubiquitination
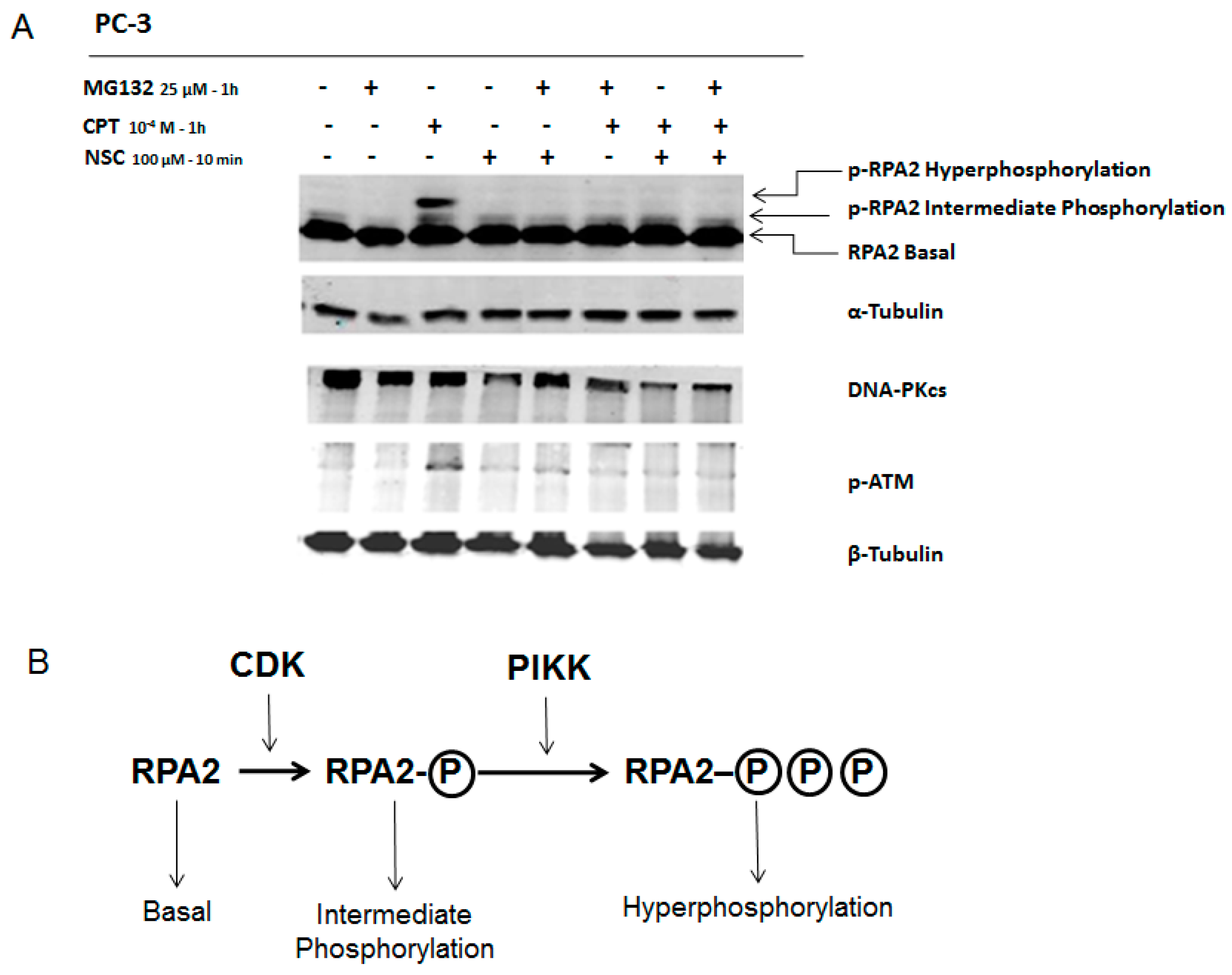
2.5. NSC Inhibits DNA-PK Activity Differently from a Proteasome Inhibitor
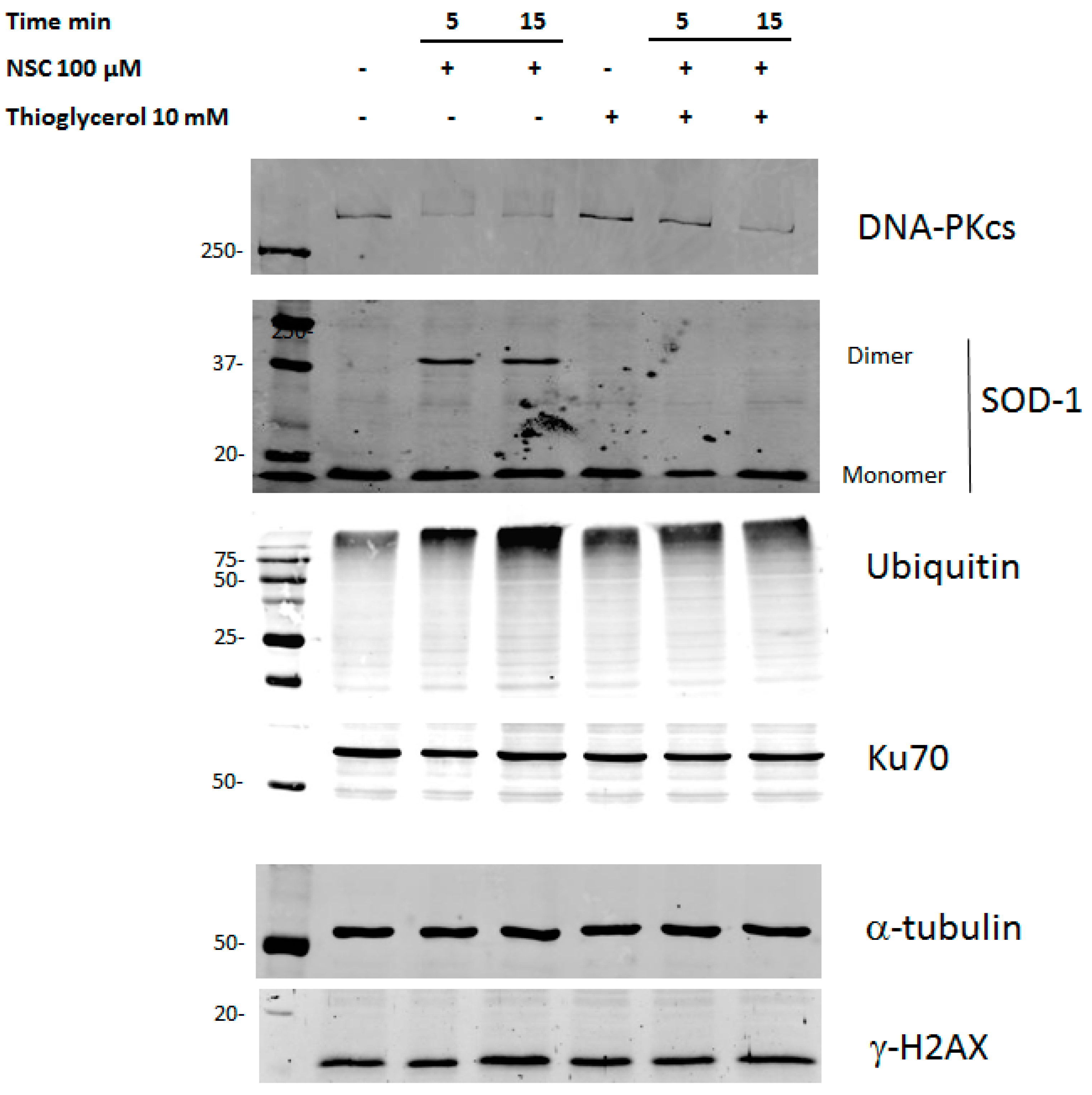
2.6. Thioglycerol Prevents the Action of NSC on DNA-PKcs
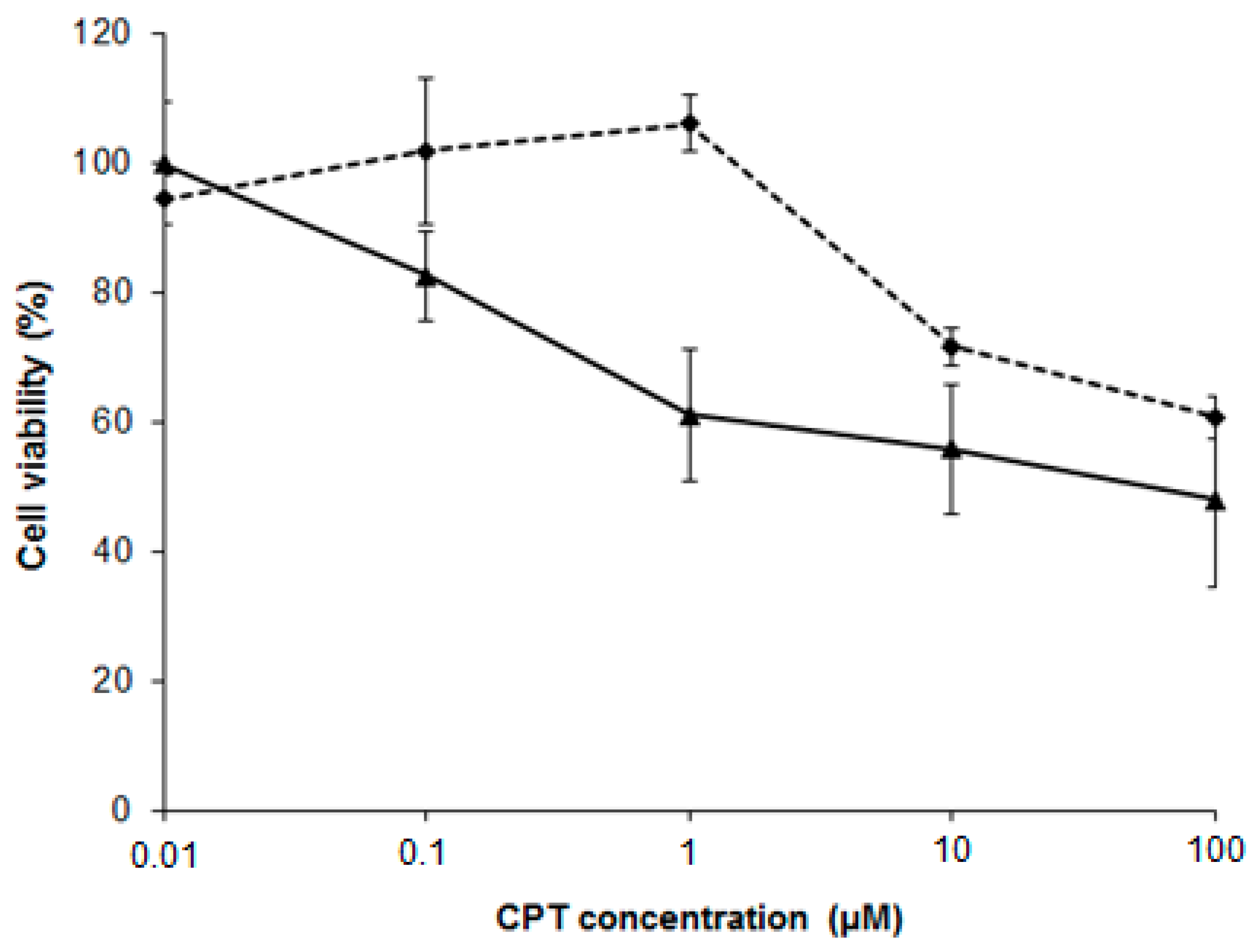
2.7. NSC Can Sensitize Cancer Cell Lines to Camptothecin-Mediated DNA Damage
3. Discussion
4. Experimental Section
4.1. Chemicals and Antibodies
4.2. Cell Culture
4.3. Cell Viability Assay and Chemical Treatments
4.4. Western Blotting
4.5. Immunofluorescence Microscopy
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATM | Ataxia Telangiectasia Mutated |
| ATR | ATM-Rad3 Related |
| CDKs | Cyclin-dependent kinases |
| CPT | Camptothecin |
| DNA-PKcs | DNA-dependent Protein Kinase catalytic subunit |
| DSBs | DNA double-strand breaks |
| EGFR | Epidermal Growth Factor Receptor |
| HR | Homologous Recombination |
| NBD | Nitro-BenzoxaDiazole |
| NHEJ | Non-Homologous End Joining |
| NSC | 4-Nitro-7-[(1-oxidopyridin-2-yl)sulfanyl]-2,1,3-benzoxadiazole |
| PIKK | Phosphatidyl inositol 3′ kinase-related kinases |
| PTP-1B | Protein Tyrosine Phosphatase 1B |
| RPA | Replication Protein A |
| ROS | Reactive Oxygen Species |
| SOD1 | Superoxide Dismutase 1 |
References
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair 2004, 3, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The life and death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Mody, C.H.; Ting, N.S.; Lees-Miller, S.P. Purification and characterization of the double-stranded DNA-activated protein kinase, DNA-PK, from human placenta. Biochem. Cell Biol. 1996, 74, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. PI 3-kinase related kinases: “Big” players in stress-induced signaling pathways. DNA Repair 2004, 3, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. Initiating cellular stress responses. Cell 2004, 118, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Shen, Y.; Jiang, N.; Fei, X.; Mi, J. Emerging roles of DNA-PK besides DNA repair. Cell Signal. 2011, 23, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Mitchell, R.A.; Cheng, A.; Hendrickson, E.A. Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol. Cell. Biol. 1997, 17, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Lucero, H.; Gae, D.; Taccioli, G.E. Novel localization of the DNA-PK complex in lipid rafts: A putative role in the signal transduction pathway of the ionizing radiation response. J. Biol. Chem. 2003, 278, 22136–22143. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Chen, B.P.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Yu, Y.; Gupta, S.; Cho, Y.M.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell. Biol. 2005, 25, 10842–10852. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Reddy, Y.V.; Wang, W.; Woods, T.; Douglas, P.; Ramsden, D.A.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 2003, 23, 5836–5848. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.; Moorhead, G.B.; Ye, R.; Lees-Miller, S.P. Protein phosphatases regulate DNA-dependent protein kinase activity. J. Biol. Chem. 2001, 276, 18992–18998. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Nirodi, C.S. The epidermal growth factor receptor: A role in repair of radiation-induced DNA damage. Clin. Cancer Res. 2007, 13, 6555–6560. [Google Scholar] [CrossRef] [PubMed]
- Kriegs, M.; Kasten-Pisula, U.; Rieckmann, T.; Holst, K.; Saker, J.; Dahm-Daphi, J.; Dikomey, E. The epidermal growth factor receptor modulates DNA double-strand break repair by regulating non-homologous end-joining. DNA Repair 2010, 9, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Szumiel, I. Epidermal growth factor receptor and DNA double strand break repair: The cell’s self-defence. Cell Signal. 2006, 18, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Morgan, R.N.; Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther. 2009, 8, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Mandal, M.; Adam, L.; Mendelsohn, J.; Kumar, R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J. Biol. Chem. 1998, 273, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Rodemann, H.P. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother. Oncol. 2005, 76, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef] [PubMed]
- Sakanyan, V.; Angelini, M.; Le Bechec, M.; Lecocq, M.F.; Benaiteau, F.; Rousseau, B.; Gyulkhandanyan, A.; Gyulkhandanyan, L.; Loge, C.; Reiter, E.; et al. Screening and discovery of nitro-benzoxadiazole compounds activating epidermal growth factor receptor (EGFR) in cancer cells. Sci. Rep. 2014, 4, 3977. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.G.; Cao, C.X.; Zhang, H.; Kohn, K.W.; Wold, M.S.; Pommier, Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999, 18, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Kouranti, I.; Peyroche, A. Protein degradation in DNA damage response. Semin. Cell Dev. Biol. 2012, 23, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Sakasai, R.; Teraoka, H.; Tibbetts, R.S. Proteasome inhibition suppresses DNA-dependent protein kinase activation caused by camptothecin. DNA Repair 2010, 9, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Sakanyan, V.; Hulin, P.; de Sousa, R.; Silva, V.; Hambardsumyan, A.; Nedellec, S.; Tomasoni, C.; Loge, C.; Pineau, C.; Roussakis, C.; et al. Activation of EGFR by small compounds through coupling the generation of hydrogen peroxide to stable dimerization of Cu/Zn SOD1. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Amrein, L.; Panasci, L.; Aloyz, R. Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Front. Pharmacol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.C.; Li, H.; Wan, R.Z.; Liu, Z.P. Developments of DNA-dependent Protein Kinase Inhibitors as Anticancer Agents. Mini Rev. Med. Chem. 2014, 14, 884–895. [Google Scholar] [CrossRef]
- Dolman, M.E.; van der Ploeg, I.; Koster, J.; Bate-Eya, L.T.; Versteeg, R.; Caron, H.N.; Molenaar, J.J. DNA-dependent protein kinase as molecular target for radiosensitization of neuroblastoma cells. PLoS ONE 2015, 10, e0145744. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Lobrich, M.; Shiloh, Y.; Chen, D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Kessinger, C.; Kobayashi, J.; Chen, B.P.; Chen, D.J.; Chatterjee, A.; Burma, S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair 2006, 5, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.Y.; Frit, P.; Malivert, L.; Revy, P.; Biard, D.; Salles, B.; Calsou, P. Interplay between Cernunnos-XLF and nonhomologous end-joining proteins at DNA ends in the cell. J. Biol. Chem. 2007, 282, 31937–31943. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Vassin, V.M.; Borowiec, J.A. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 2007, 282, 35910–35923. [Google Scholar] [CrossRef] [PubMed]
- Sakasai, R.; Shinohe, K.; Ichijima, Y.; Okita, N.; Shibata, A.; Asahina, K.; Teraoka, H. Differential involvement of phosphatidylinositol 3-kinase-related protein kinases in hyperphosphorylation of replication protein A2 in response to replication-mediated DNA double-strand breaks. Genes Cells 2006, 11, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2012, 8, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Patridge, E.V.; Eriksson, E.S.; Penketh, P.G.; Baumann, R.P.; Zhu, R.; Shyam, K.; Eriksson, L.A.; Sartorelli, A.C. 7-Nitro-4-(phenylthio)benzofurazan is a potent generator of superoxide and hydrogen peroxide. Arch. Toxicol. 2012, 86, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Chen, Y.; Li, A.S.; Reid, M.B. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am. J. Physiol. Cell Physiol. 2003, 285, C806–C812. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.R.; Zhu, H.; Huang, M.; Chen, Y.; Cai, Y.J.; Miao, Z.H.; Zhang, J.S.; Ding, J. Reactive oxygen species elicit apoptosis by concurrently disrupting topoisomerase II and DNA-dependent protein kinase. Mol. Pharmacol. 2005, 68, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Calsou, P.; Frit, P.; Cayrol, C.; Carter, T.; Salles, B. UV sensitivity and impaired nucleotide excision repair in DNA-dependent protein kinase mutant cells. Nucleic Acids Res. 1998, 26, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Peddi, P.; Loftin, C.W.; Dickey, J.S.; Hair, J.M.; Burns, K.J.; Aziz, K.; Francisco, D.C.; Panayiotidis, M.I.; Sedelnikova, O.A.; Bonner, W.M.; et al. DNA-PKcs deficiency leads to persistence of oxidatively induced clustered DNA lesions in human tumor cells. Free Radic. Biol. Med. 2010, 48, 1435–1443. [Google Scholar] [CrossRef] [PubMed]









© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, V.A.O.; Lafont, F.; Benhelli-Mokrani, H.; Breton, M.L.; Hulin, P.; Chabot, T.; Paris, F.; Sakanyan, V.; Fleury, F. Rapid Diminution in the Level and Activity of DNA-Dependent Protein Kinase in Cancer Cells by a Reactive Nitro-Benzoxadiazole Compound. Int. J. Mol. Sci. 2016, 17, 703. https://doi.org/10.3390/ijms17050703
Silva VAO, Lafont F, Benhelli-Mokrani H, Breton ML, Hulin P, Chabot T, Paris F, Sakanyan V, Fleury F. Rapid Diminution in the Level and Activity of DNA-Dependent Protein Kinase in Cancer Cells by a Reactive Nitro-Benzoxadiazole Compound. International Journal of Molecular Sciences. 2016; 17(5):703. https://doi.org/10.3390/ijms17050703
Chicago/Turabian StyleSilva, Viviane A. O., Florian Lafont, Houda Benhelli-Mokrani, Magali Le Breton, Philippe Hulin, Thomas Chabot, François Paris, Vehary Sakanyan, and Fabrice Fleury. 2016. "Rapid Diminution in the Level and Activity of DNA-Dependent Protein Kinase in Cancer Cells by a Reactive Nitro-Benzoxadiazole Compound" International Journal of Molecular Sciences 17, no. 5: 703. https://doi.org/10.3390/ijms17050703
APA StyleSilva, V. A. O., Lafont, F., Benhelli-Mokrani, H., Breton, M. L., Hulin, P., Chabot, T., Paris, F., Sakanyan, V., & Fleury, F. (2016). Rapid Diminution in the Level and Activity of DNA-Dependent Protein Kinase in Cancer Cells by a Reactive Nitro-Benzoxadiazole Compound. International Journal of Molecular Sciences, 17(5), 703. https://doi.org/10.3390/ijms17050703