
DNA Damage: A Main Determinant of Vascular Aging


Abstract
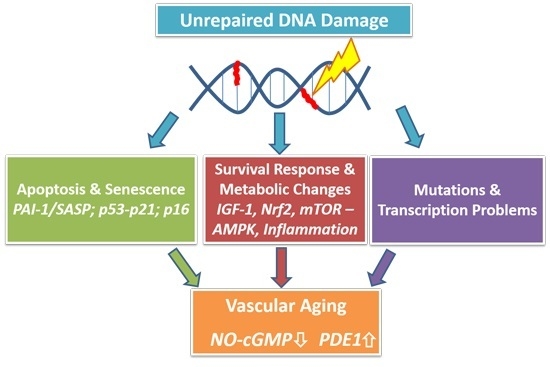
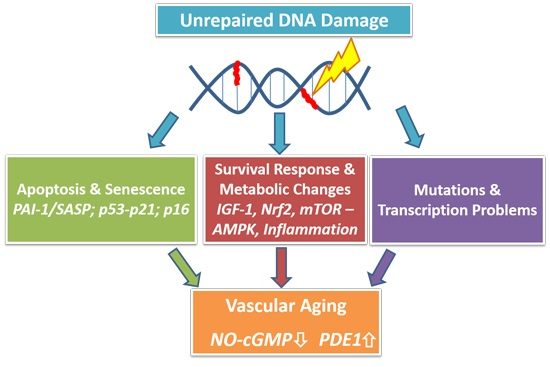
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Genomic Instability and Aging: A Short Outline of the Basic Principles
2.1. DNA Repair Systems
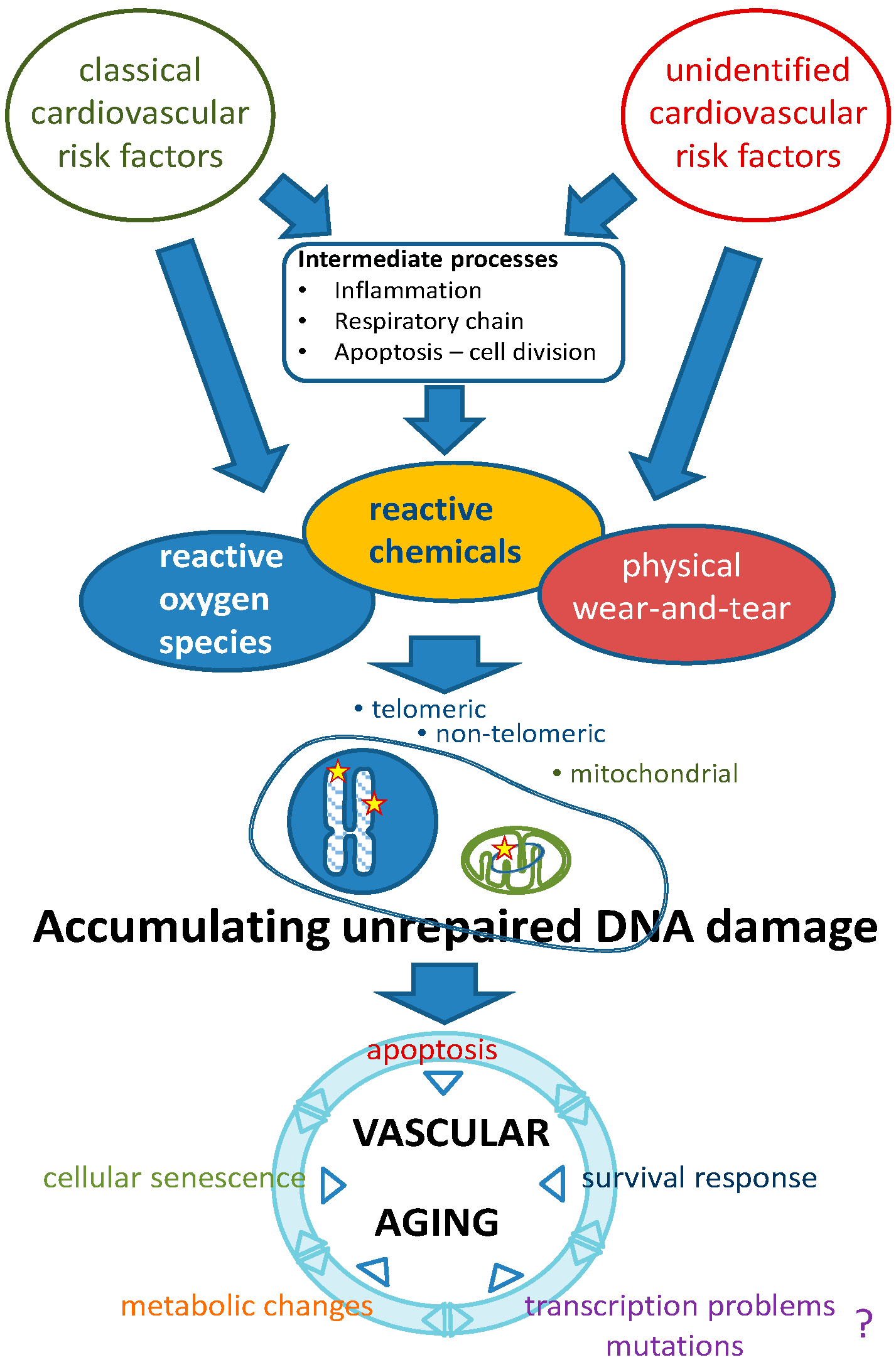
2.2. Aging: The Interplay between Genomic Damage, the Survival Response and Cellular Senescence
3. Genomic Instability as a Causal Factor in Vascular Aging: Evidence in Humans
3.1. Cardiovascular Disease in Progeria Syndromes
3.2. Indicators of a Role of Genomic Instability in the General Population
3.3. Telomere Shortening
3.4. Cellular Senescence and Its Regulators
The Role of Senescent Cells and Plasminogen Activator Inhibitor-1 (PAI-1)-Related Signaling Pathways in Vascular Aging
3.5. Cyclin-Dependent Kinase Inhibitor 2 (CDNK2) A and B
4. Genomic Instability as a Causal Factor in Vascular Aging: Evidence from Animal Models
4.1. Telomerase-Deficient (TERC−/− and TERT−/−) Mice
4.2. Mouse Models of Genomic Instability Associated with Human Progeria
4.3. Mitochondrial DNA Maintenance Defects
4.4. BubR1 Knockout
4.5. Vascular Functional/Pharmacological Changes Due to Genomic Instability
4.5.1. NO-cGMP Signaling
4.5.2. NF-E2-Related Factor-2 (Nrf2) and Antioxidant Pathways
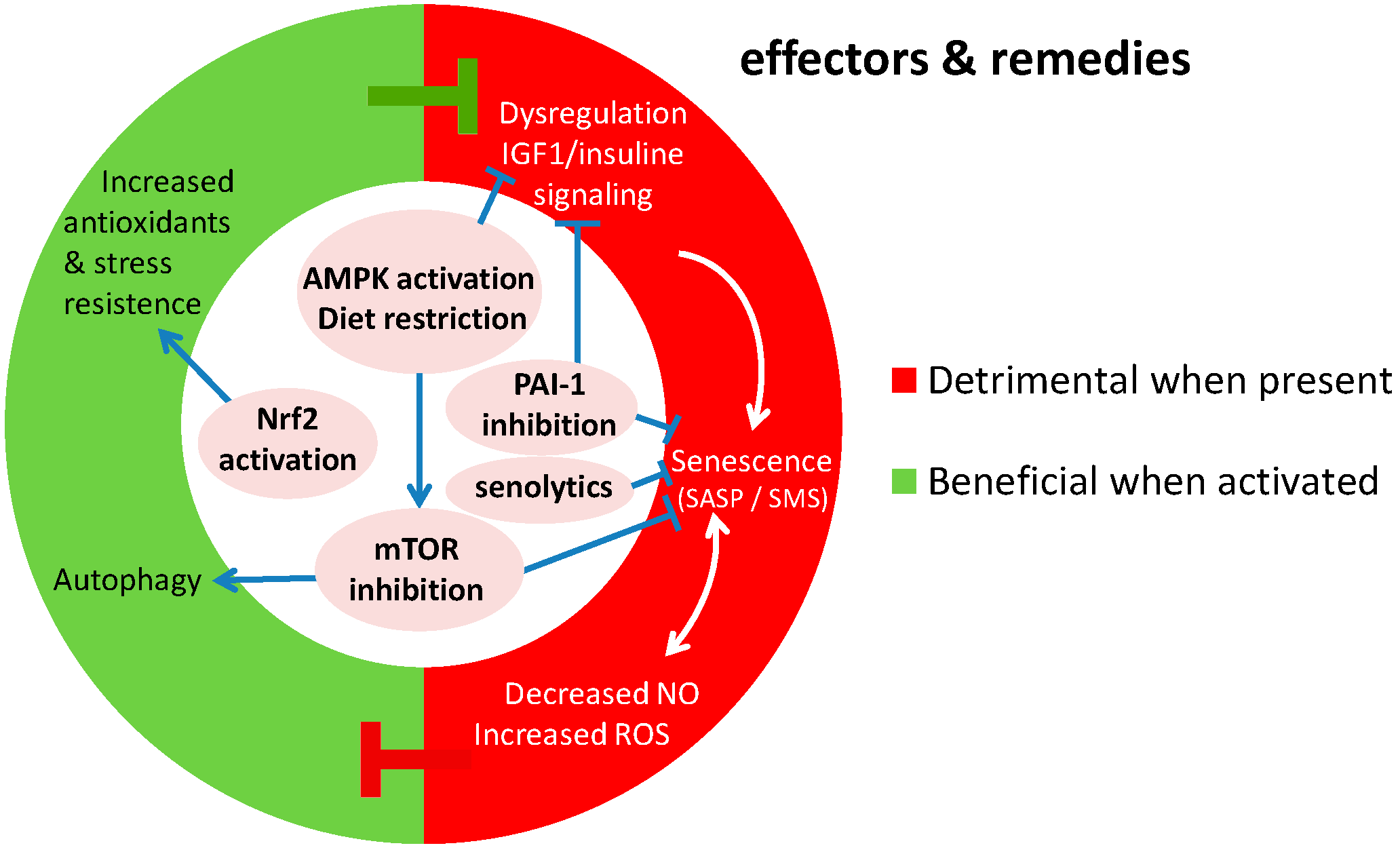
5. Perspectives
5.1. Directions for Future Studies Establishing the Role of Genomic Instability
5.2. Towards New Interventions in Vascular Aging Caused by Genomic Instability
5.2.1. Mtor, Rapamycin and Autophagy
5.2.2. Senolytics and Inhibitors of Senescent Cell Signaling
5.3. Dietary Restriction
5.4. PDE Inhibition
5.5. Reconsideration of Antioxidant Therapies
6. Summary
Abbreviations
| AMPK | adenosine monophosphate-activated protein kinase |
| APE-1/Ref1 | Apurinic/apyrimidinic endonuclease 1/redox factor 1 |
| ApoE | ApoE, Apolipoprotein E |
| ARE | antioxidant response element |
| ATM | ataxia telangiectasia mutated |
| BER | base excision repair |
| CAD | coronary artery disease |
| CDNK2 | cyclin-dependent kinase inhibitor 2 |
| cGMP | cyclic guanosine monophosphate |
| cIMT | carotid intima media thickness |
| CVD | cardiovascular diseases |
| DDB2 | Damage-Specific DNA Binding Protein 2 |
| DNA-PK | DNA-dependent protein kinase |
| DR | dietary restriction |
| eNOS | endothelial nitric oxide synthase |
| EPC | endothelial progenitor cells |
| Ercc1 | excision repair cross-complementation group 1 |
| GH | growth hormone |
| GTF2H | general transcription factor IIH |
| HGPS | Hutchinson-Gilford progeria syndrome |
| HMBG-1 | high mobility group box 1 |
| hMSC | human mesenchymal stem cells |
| HR | homologous recombination |
| IGF1 | insulin-like growth factor 1 |
| IGFBP3 | insulin-like growth factor-binding protein 3 |
| INK-ATTAC mice | genetically modified mice in which cells expressing the cyclin-dependent kinase inhibitor p16INK4A are being removed by apoptosis due to caspase 8 activation |
| LMNA | lamin A gene |
| MMR | mismatch repair |
| MtDNA | mitochondrial DNA |
| mTOR(C1) | mammalian target of rapamycin (complex 1) |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NER | nucleotide excision repair |
| NHEJ | non-homologous end joining |
| NfkB | nuclear factor kappa B |
| NO | nitric oxide |
| Nrf2 | transcription factor NF-E2-related factor-2 |
| PAI-1 | plasminogen activator inhibitor-1 |
| PARP-1 | poly [ADP-ribose] polymerase 1 |
| PDE | phosphodiesterase |
| POLG | polymerase gamma |
| RecQ | Escherichia coli recQ-like helicase |
| ROS | reactive oxygen species |
| SASP | senescence-associated secretory phenotype |
| (s)GC | (soluble) guanylyl cyclase |
| SIRT-1 | sirtuin-1 |
| SMS | senescence-messaging secretome |
| SNP | single nucleotide polymorphism |
| T2DM | type 2 diabetes mellitus |
| TERC | RNA template of telomerase |
| TERT | telomerase reverse transcriptase |
| TRF2 | telomeric repeat-binding factor 2 |
| TTD | trichothiodystrophy |
| VSMC | vascular smooth muscle cell |
| WRN | Werner gene |
| WS | Werner Syndrome |
| XP | xeroderma pigmentosum |
| XRCC3 | gene coding for x-ray repair cross-complementing protein 3 |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.-P.; Fullerton, H.J.; Howard, V.J. Heart disease and stroke statistics-2015 update: A report from the american heart association. Circulation 2015, 131, e29. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.R.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar] [PubMed]
- Cooney, M.T.; Dudina, A.L.; Graham, I.M. Value and limitations of existing scores for the assessment of cardiovascular risk: A review for clinicians. J. Am. Coll. Cardiol. 2009, 54, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Black, A.D.; Jablonski, K.L.; Gano, L.B.; Seals, D.R. Aging is associated with greater nuclear nfκb, reduced iκbα, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 2008, 7, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; Brosnan, M.J.; McIntyre, M.; Graham, D.; Dominiczak, A.F. Superoxide excess in hypertension and aging a common cause of endothelial dysfunction. Hypertension 2001, 37, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris Nikolaos Papageorgiou, C.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Roks, A.J. Genomic instability and vascular aging: A focus on nucleotide excision repair. Trends Cardiovasc. Med. 2014, 24, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Hoeijmakers, J.H. Cell biology: Ageing nucleus gets out of shape. Nature 2006, 440, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Campisi, J.; Hoeijmakers, J.; van Steeg, H.; Vijg, J. Aging and genome maintenance: Lessons from the mouse? Science 2003, 299, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Oeseburg, H.; de Boer, R.A.; van Gilst, W.H.; van der Harst, P. Telomere biology in healthy aging and disease. Pflug. Arch. 2010, 459, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, W.P.; Hoeijmakers, J.H.; Pothof, J. Genome integrity in aging: Human syndromes, mouse models, and therapeutic options. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Burtner, C.R.; Kennedy, B.K. Progeria syndromes and ageing: What is the connection? Nat. Rev. Mol. Cell Biol. 2010, 11, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.J.; Martin, G.M.; Schultz, A.L.; Motulsky, A.G. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966, 45, 177–221. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.C.; Gray, M.D.; Oshima, J.; Kamath-Loeb, A.S.; Fry, M.; Loeb, L.A. Werner syndrome protein. I. DNA helicase and dna exonuclease reside on the same polypeptide. J. Biol. Chem. 1998, 273, 34139–34144. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.M.; German, J.; Cunniff, C. Bloom’s Syndrome. In GeneReviews 1993, eBook; Pagon, R.A., Adam, M.P., Ardinger, H.H., et al., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar] [PubMed]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of hutchinson-gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in hutchinson-gilford progeria: Correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Houtsmuller, A.B.; Rademakers, S.; Nigg, A.L.; Hoogstraten, D.; Hoeijmakers, J.H.; Vermeulen, W. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 1999, 284, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Robinson, A.R.; Duensing, A.; van Drunen, E.; Beverloo, H.B.; Weisberg, D.B.; Hasty, P.; Hoeijmakers, J.H.; Niedernhofer, L.J. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 2008, 28, 5082–5092. [Google Scholar] [CrossRef] [PubMed]
- Bergstralh, D.T.; Sekelsky, J. Interstrand crosslink repair: Can XPF-ERCC1 be let off the hook? Trends Genet. 2008, 24, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Kashiyama, K.; Nakazawa, Y.; Pilz, D.T.; Guo, C.; Shimada, M.; Sasaki, K.; Fawcett, H.; Wing, J.F.; Lewin, S.O.; Carr, L.; et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes cockayne syndrome, xeroderma pigmentosum, and fanconi anemia. Am. J. Hum. Genet. 2013, 92, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair 2011, 10, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Borghini, A.; Cervelli, T.; Galli, A.; Andreassi, M.G. DNA modifications in atherosclerosis: From the past to the future. Atherosclerosis 2013, 230, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Borghini, A.; Mercuri, A.; Turchi, S.; Chiesa, M.R.; Piccaluga, E.; Andreassi, M.G. Increased circulating cell-free DNA levels and mtdna fragments in interventional cardiologists occupationally exposed to low levels of ionizing radiation. Environ. Mol. Mutagen. 2015, 56, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Andreassi, M.G.; Piccaluga, E.; Gargani, L.; Sabatino, L.; Borghini, A.; Faita, F.; Bruno, R.M.; Padovani, R.; Guagliumi, G.; Picano, E. Subclinical carotid atherosclerosis and early vascular aging from long-term low-dose ionizing radiation exposure: A genetic, telomere, and vascular ultrasound study in cardiac catheterization laboratory staff. JACC Cardiovasc. Interv. 2015, 8, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Preston, D.L.; Shimizu, Y.; Pierce, D.A.; Suyama, A.; Mabuchi, K. Studies of mortality of atomic bomb survivors. Report 13: Solid cancer and noncancer disease mortality: 1950–1997. Radiat. Res. 2003, 160, 381–407. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Knaapen, M.W.; de Meyer, G.R.; Herman, A.G.; Kockx, M.M. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 2002, 106, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Botto, N.; Masetti, S.; Petrozzi, L.; Vassalle, C.; Manfredi, S.; Biagini, A.; Andreassi, M.G. Elevated levels of oxidative DNA damage in patients with coronary artery disease. Coron. Artery Dis. 2002, 13, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Bhaggoe, U.M.; Leijten, F.; et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012, 126, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, J.J.; Trompet, S.; Deelen, J.; Stott, D.J.; Sattar, N.; Buckley, B.M.; Ford, I.; Heijmans, B.T.; Guchelaar, H.J.; Houwing-Duistermaat, J.J.; et al. Non-homologous end-joining pathway associated with occurrence of myocardial infarction: Gene set analysis of genome-wide association study data. PLoS ONE 2013, 8, e56262. [Google Scholar] [CrossRef] [PubMed]
- Cervelli, T.; Borghini, A.; Galli, A.; Andreassi, M.G. DNA damage and repair in atherosclerosis: Current insights and future perspectives. Int. J. Mol. Sci. 2012, 13, 16929–16944. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Gorenne, I.; Mercer, J.; Figg, N.; Littlewood, T.; Bennett, M. Statins use a novel nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ. Res. 2008, 103, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Mercer, J.; Bennett, M. DNA damage and repair in atherosclerosis. Cardiovasc. Res. 2006, 71, 259–268. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Telomere-related genome instability in cancer. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2005; Volume 70, pp. 197–204. [Google Scholar]
- Deng, Y.; Chang, S. Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab. Investig. 2007, 87, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P. Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J. 1999, 13, 2091–2104. [Google Scholar] [PubMed]
- Polanská, E.; Dobšáková, Z.; Dvořáčková, M.; Fajkus, J.; Štros, M. HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma 2012, 121, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Butt, H.; Atturu, G.; London, N.; Sayers, R.; Bown, M. Telomere length dynamics in vascular disease: A review. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Atturu, G.; Brouilette, S.; Samani, N.; London, N.; Sayers, R.; Bown, M. Short leukocyte telomere length is associated with abdominal aortic aneurysm (AAA). Eur. J. Vasc. Endovasc. Surg. 2010, 39, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere shortening in human coronary artery diseases. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.J.; Boultby, R.; Butler, R.; Thompson, J.R.; Goodall, A.H. Telomere shortening in atherosclerosis. Lancet 2001, 358, 472–473. [Google Scholar] [CrossRef]
- Valdes, A.M.; Andrew, T.; Gardner, J.P.; Kimura, M.; Oelsner, E.; Cherkas, L.F.; Aviv, A.; Spector, T.D. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Fitzpatrick, A.L.; Kronmal, R.A.; Gardner, J.P.; Psaty, B.M.; Jenny, N.S.; Tracy, R.P.; Walston, J.; Kimura, M.; Aviv, A. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am. J. Epidemiol. 2007, 165, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Jeanclos, E.; Krolewski, A.; Skurnick, J.; Kimura, M.; Aviv, H.; Warram, J.H.; Aviv, A. Shortened telomere length in white blood cells of patients with iddm. Diabetes 1998, 47, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Demissie, S.; Levy, D.; Benjamin, E.J.; Cupples, L.A.; Gardner, J.P.; Herbert, A.; Kimura, M.; Larson, M.G.; Meigs, J.B.; Keaney, J.F.; et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the framingham heart study. Aging Cell 2006, 5, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Vasan, R.S.; Demissie, S.; Kimura, M.; Cupples, L.A.; Rifai, N.; White, C.; Wang, T.J.; Gardner, J.P.; Cao, X.; Benjamin, E.J.; et al. Association of leukocyte telomere length with circulating biomarkers of the renin-angiotensin-aldosterone system: The framingham heart study. Circulation 2008, 117, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Strazhesko, I.; Tkacheva, O.; Boytsov, S.; Akasheva, D.; Dudinskaya, E.; Vygodin, V.; Skvortsov, D.; Nilsson, P. Association of insulin resistance, arterial stiffness and telomere length in adults free of cardiovascular diseases. PLoS ONE 2015, 10, e0136676. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Ishikawa, Y.; Takahashi, Y.; Itoh, T.; Minami, Y.; Nakamura, M. Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis 2008, 198, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Brouilette, S.W.; Moore, J.S.; McMahon, A.D.; Thompson, J.R.; Ford, I.; Shepherd, J.; Packard, C.J.; Samani, N.J. West of Scotland Coronary Prevention Study Group. Telomere length, risk of coronary heart disease, and statin treatment in the west of scotland primary prevention study: A nested case-control study. Lancet 2007, 369, 107–114. [Google Scholar] [CrossRef]
- Haycock, P.C.; Heydon, E.E.; Kaptoge, S.; Butterworth, A.S.; Thompson, A.; Willeit, P. Leucocyte telomere length and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2014, 349, g4227. [Google Scholar] [CrossRef] [PubMed]
- Mather, K.A.; Jorm, A.F.; Parslow, R.A.; Christensen, H. Is telomere length a biomarker of aging? A review. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Takeshita, K.; Yamamoto, K.; Ito, M.; Kondo, T.; Matsushita, T.; Hirai, M.; Kojima, T.; Nishimura, M.; Nabeshima, Y.; Loskutoff, D.J.; et al. Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “klotho” mouse. Semin. Thromb. Hemost. 2002, 28, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Aillaud, M.F.; Pignol, F.; Alessi, M.C.; Harle, J.R.; Escande, M.; Mongin, M.; Juhan-Vague, I. Increase in plasma concentration of plasminogen activator inhibitor, fibrinogen, von willebrand factor, factor VIII:C and in erythrocyte sedimentation rate with age. Thromb. Haemost. 1986, 55, 330–332. [Google Scholar] [PubMed]
- Eren, M.; Boe, A.E.; Murphy, S.B.; Place, A.T.; Nagpal, V.; Morales-Nebreda, L.; Urich, D.; Quaggin, S.E.; Budinger, G.R.; Mutlu, G.M.; et al. PAI-1-regulated extracellular proteolysis governs senescence and survival in klotho mice. Proc. Natl. Acad. Sci. USA 2014, 111, 7090–7095. [Google Scholar] [CrossRef] [PubMed]
- Boe, A.E.; Eren, M.; Murphy, S.B.; Kamide, C.E.; Ichimura, A.; Terry, D.; McAnally, D.; Smith, L.H.; Miyata, T.; Vaughan, D.E. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nω-nitro-l-arginine methyl ester-induced hypertension and vascular senescence. Circulation 2013, 128, 2318–2324. [Google Scholar] [CrossRef] [PubMed]
- Tatar, M.; Bartke, A.; Antebi, A. The endocrine regulation of aging by insulin-like signals. Science 2003, 299, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Festa, A.; D’Agostino, R., Jr.; Tracy, R.P.; Haffner, S.M. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: The insulin resistance atherosclerosis study. Diabetes 2002, 51, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Rosito, G.A.; D'Agostino, R.B.; Massaro, J.; Lipinska, I.; Mittleman, M.A.; Sutherland, P.; Wilson, P.W.; Levy, D.; Muller, J.E.; Tofler, G.H. Association between obesity and a prothrombotic state: The framingham offspring study. Thromb. Haemost. 2004, 91, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Olholm, J.; Paulsen, S.K.; Cullberg, K.B.; Richelsen, B.; Pedersen, S.B. Anti-inflammatory effect of resveratrol on adipokine expression and secretion in human adipose tissue explants. Int. J. Obes. (Lond.) 2010, 34, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Nagi, D.K.; Yudkin, J.S. Effects of metformin on insulin resistance, risk factors for cardiovascular disease, and plasminogen activator inhibitor in niddm subjects. A study of two ethnic groups. Diabetes Care 1993, 16, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef] [PubMed]
- Velthuis-te Wierik, E.J.; Meijer, P.; Kluft, C.; van den Berg, H. Beneficial effect of a moderately energy-restricted diet on fibrinolytic factors in non-obese men. Metabolism 1995, 44, 1548–1552. [Google Scholar] [CrossRef]
- Nilsson, L.; Banfi, C.; Diczfalusy, U.; Tremoli, E.; Hamsten, A.; Eriksson, P. Unsaturated fatty acids increase plasminogen activator inhibitor-1 expression in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Juhan-Vague, I.; Kooistra, T.; Declerck, P.J.; Collen, D. Insulin stimulates the synthesis of plasminogen activator inhibitor 1 by the human hepatocellular cell line Hep G2. Thromb. Haemost. 1988, 60, 491–494. [Google Scholar] [PubMed]
- Chen, Y.Q.; Su, M.; Walia, R.R.; Hao, Q.; Covington, J.W.; Vaughan, D.E. Sp1 sites mediate activation of the plasminogen activator inhibitor-1 promoter by glucose in vascular smooth muscle cells. J. Biol. Chem. 1998, 273, 8225–8231. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Siebold, A.P.; Sharpless, N.E. Review: A meta-analysis of gwas and age-associated diseases. Aging Cell 2012, 11, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Menocal, L.; Pham, S.M.; Mateu, D.; St-Pierre, M.; Wei, Y.; Pestana, I.; Aitouche, A.; Vazquez-Padron, R.I. Aging increases p16INK4a expression in vascular smooth-muscle cells. Biosci. Rep. 2010, 30, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Motterle, A.; Pu, X.; Wood, H.; Xiao, Q.; Gor, S.; Ng, F.L.; Chan, K.; Cross, F.; Shohreh, B.; Poston, R.N. Functional analyses of coronary artery disease associated variation on chromosome 9p21 in vascular smooth muscle cells. Hum. Mol. Genet. 2012, 21, 4021–4029. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rivero, G.; Ruiz-Torres, M.P.; Rivas-Elena, J.V.; Jerkic, M.; Diez-Marques, M.L.; Lopez-Novoa, J.M.; Blasco, M.A.; Rodriguez-Puyol, D. Mice deficient in telomerase activity develop hypertension because of an excess of endothelin production. Circulation 2006, 114, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Massip, L.; Garand, C.; Turaga, R.V.; Deschenes, F.; Thorin, E.; Lebel, M. Increased insulin, triglycerides, reactive oxygen species, and cardiac fibrosis in mice with a mutation in the helicase domain of the werner syndrome gene homologue. Exp. Gerontol. 2006, 41, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R.A. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004, 36, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive vascular smooth muscle cell defects in a mouse model of hutchinson-gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [PubMed]
- Weeda, G.; Donker, I.; de Wit, J.; Morreau, H.; Janssens, R.; Vissers, C.J.; Nigg, A.; van Steeg, H.; Bootsma, D.; Hoeijmakers, J.H. Disruption of mouse ercc1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 1997, 7, 427–439. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Spiegelhalter, D.J.; Georgakopoulos, D.; Robinson, J.; Deanfield, J.E. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J. Am. Coll. Cardiol. 1994, 24, 471–476. [Google Scholar] [CrossRef]
- Kuo, L.; Chilian, W.M.; Davis, M.J. Coronary arteriolar myogenic response is independent of endothelium. Circ. Res. 1990, 66, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am. J. Physiol. 1990, 259, H1063–H1070. [Google Scholar] [PubMed]
- Gerhard, M.; Roddy, M.A.; Creager, S.J.; Creager, M.A. Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension 1996, 27, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.C.; Steingrimsdottir, H.; Weber, C.A.; Lehmann, A.R. Mutations in the xeroderma pigmentosum group d DNA repair/transcription gene in patients with trichothiodystrophy. Nat. Genet. 1994, 7, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.R.; Cheng, K.K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.P.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res. 2010, 107, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.K.; Jablonski, S.A.; Sudakin, V.; Hittle, J.C.; Yen, T.J. Human BUBR1 is a mitotic checkpoint kinase that monitors cenp-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 1999, 146, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Baker, D.J.; d'Uscio, L.V.; Mozammel, G.; Katusic, Z.S.; van Deursen, J.M. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke 2007, 38, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W.S. Vascular nitric oxide: Beyond enos. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. Suppl. 1997, 20, 3–10. [Google Scholar]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. Enos uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.L.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar]
- Murohara, T.; Asahara, T.; Silver, M.; Bauters, C.; Masuda, H.; Kalka, C.; Kearney, M.; Chen, D.; Symes, J.F.; Fishman, M.C. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Investig. 1998, 101, 2567. [Google Scholar]
- Freedman, J.E.; Sauter, R.; Battinelli, E.M.; Ault, K.; Knowles, C.; Huang, P.L.; Loscalzo, J. Deficient platelet-derived nitric oxide and enhanced hemostasis in mice lacking the NOSIII gene. Circ. Res. 1999, 84, 1416–1421. [Google Scholar]
- Tai, S.C.; Robb, G.B.; Marsden, P.A. Endothelial nitric oxide synthase a new paradigm for gene regulation in the injured blood vessel. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 405–412. [Google Scholar]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease from marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar]
- Bautista Nino, P.K.; Durik, M.; Danser, A.H.; de Vries, R.; Musterd-Bhaggoe, U.M.; Meima, M.E.; Kavousi, M.; Ghanbari, M.; Hoeijmakers, J.H.; O’Donnell, C.J.; et al. Phosphodiesterase 1 regulation is a key mechanism in vascular aging. Clin. Sci. (Lond.) 2015, 129, 1061–1075. [Google Scholar]
- Yan, C. Cyclic nucleotide phosphodiesterase 1 and vascular aging. Clin. Sci. (Lond.) 2015, 129, 1077–1081. [Google Scholar]
- Rybalkin, S.D.; Rybalkina, I.; Beavo, J.A.; Bornfeldt, K.E. Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ. Res. 2002, 90, 151–157. [Google Scholar]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar]
- He, X.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 47–58. [Google Scholar]
- Warabi, E.; Takabe, W.; Minami, T.; Inoue, K.; Itoh, K.; Yamamoto, M.; Ishii, T.; Kodama, T.; Noguchi, N. Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: Role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 2007, 42, 260–269. [Google Scholar]
- Mylroie, H.; Dumont, O.; Bauer, A.; Thornton, C.C.; Mackey, J.; Calay, D.; Hamdulay, S.S.; Choo, J.R.; Boyle, J.J.; Samarel, A.M.; et al. PKCε-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc. Res. 2015, 106, 509–519. [Google Scholar]
- Ungvari, Z.; Bailey-Downs, L.; Sosnowska, D.; Gautam, T.; Koncz, P.; Losonczy, G.; Ballabh, P.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Vascular oxidative stress in aging: A homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H363–H372. [Google Scholar]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar]
- Jódar, L.; Mercken, E.M.; Ariza, J.; Younts, C.; González-Reyes, J.A.; Alcaín, F.J.; Burón, I.; de Cabo, R.; Villalba, J.M. Genetic deletion of Nrf2 promotes immortalization and decreases life span of murine embryonic fibroblasts. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 247–256. [Google Scholar]
- Zhou, X.; Zhao, L.; Mao, J.; Huang, J.; Chen, J. Antioxidant effects of hydrogen sulfide on left ventricular remodeling in smoking rats are mediated via PI3K/Akt-dependent activation of Nrf2. Toxicol. Sci. 2015, 144, 197–203. [Google Scholar]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar]
- He, X.; Ma, Q. Disruption of Nrf2 synergizes with high glucose to cause heightened myocardial oxidative stress and severe cardiomyopathy in diabetic mice. J. Diabetes Metab. 2012, (Suppl. S7). [Google Scholar] [CrossRef]
- Liao, C.-Y.; Kennedy, B.K. SIRT6, oxidative stress, and aging. Cell Res. 2016, 26, 143–144. [Google Scholar]
- Barnhoorn, S.; Uittenboogaard, L.M.; Jaarsma, D.; Vermeij, W.P.; Tresini, M.; Weymaere, M.; Menoni, H.; Brandt, R.M.; de Waard, M.C.; Botter, S.M. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease xpg deficiency. PLoS Genet. 2014, 10, e1004686. [Google Scholar]
- Heiss, E.H.; Schachner, D.; Werner, E.R.; Dirsch, V.M. Active NF-E2-related factor (Nrf2) contributes to keep endothelial NO synthase (eNOS) in the coupled state role of reactive oxygen species (ROS), eNOS, and heme oxygenase (Ho-1) levels. J. Biol. Chem. 2009, 284, 31579–31586. [Google Scholar]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; von Zglinicki, T.; Passos, J.F. Quantitative assessment of markers for cell senescence. Exp. Gerontol. 2010, 45, 772–778. [Google Scholar]
- Prasher, J.M.; Lalai, A.S.; Heijmans-Antonissen, C.; Ploemacher, R.E.; Hoeijmakers, J.H.; Touw, I.P.; Niedernhofer, L.J. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1−/− mice. EMBO J. 2005, 24, 861–871. [Google Scholar]
- Durik, M.; Seva Pessoa, B.; Roks, A.J. The renin-angiotensin system, bone marrow and progenitor cells. Clin. Sci. 2012, 123, 205–223. [Google Scholar]
- Gelino, S.; Hansen, M. Autophagy—An emerging anti-aging mechanism. J. Clin. Exp. Pathol. 2012, (Suppl. S4). [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar]
- Ward, W.F. The relentless effects of the aging process on protein turnover. Biogerontology 2000, 1, 195–199. [Google Scholar]
- Bergamini, E.; Cavallini, G.; Donati, A.; Gori, Z. The role of macroautophagy in the ageing process, anti-ageing intervention and age-associated diseases. Int. J. Biochem. Cell Biol. 2004, 36, 2392–2404. [Google Scholar]
- Grootaert, M.O.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; de Meyer, G.R.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar]
- Zhang, Y.; Bokov, A.; Gelfond, J.; Soto, V.; Ikeno, Y.; Hubbard, G.; Diaz, V.; Sloane, L.; Maslin, K.; Treaster, S. Rapamycin extends life and health in C57BL/6 mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 119–130. [Google Scholar]
- Adelman, S.J. Sirolimus and its analogs and its effects on vascular diseases. Curr. Pharm. Des. 2010, 16, 4002–4011. [Google Scholar]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar]
- Martinet, W.; de Loof, H.; de Meyer, G.R. Mtor inhibition: A promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis 2014, 233, 601–607. [Google Scholar]
- Reihl, K.D.; Seals, D.R.; Henson, G.D.; LaRocca, T.J.; Magerko, K.; Bosshardt, G.C.; Lesniewski, L.A.; Donato, A.J. Dietary rapamycin selectively improves arterial function in old mice. FASEB J. 2013, 27, 1194–1117. [Google Scholar]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar]
- Marino, G.; Ugalde, A.P.; Salvador-Montoliu, N.; Varela, I.; Quiros, P.M.; Cadinanos, J.; van der Pluijm, I.; Freije, J.M.; Lopez-Otin, C. Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum. Mol. Genet. 2008, 17, 2196–2211. [Google Scholar]
- Reineke, D.C.; Müller-Schweinitzer, E.; Winkler, B.; Kunz, D.; Konerding, M.A.; Grussenmeyer, T.; Carrel, T.P.; Eckstein, F.S.; Grapow, M.T.R. Rapamycin impairs endothelial cell function in human internal thoracic arteries. Eur. J. Med. Res. 2015, 20, 1–8. [Google Scholar]
- Trapp, A.; Weis, M. The impact of immunosuppression on endothelial function. J. Cardiovasc. Pharmacol. 2005, 45, 81–87. [Google Scholar]
- Mischie, A.N.; Nazzaro, M.S.; Fiorilli, R.; de Felice, F.; Musto, C.; Confessore, P.; Parma, A.; Boschetti, C.; Violini, R. Head-to-head comparison of sirolimus-eluting stent versus bare metal stent evaluation of the coronary endothelial dysfunction in the same patient presenting with multiple coronary artery lesions: The credential study. Catheter. Cardiovasc. Interv. 2013, 82, E184–E191. [Google Scholar]
- Habib, A.; Karmali, V.; Polavarapu, R.; Akahori, H.; Cheng, Q.; Pachura, K.; Kolodgie, F.D.; Finn, A.V. Sirolimus-FKBP12.6 impairs endothelial barrier function through protein kinase C-α activation and disruption of the p120-vascular endothelial cadherin interaction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2425–2431. [Google Scholar]
- Jiang, P.; Lan, Y.; Luo, J.; Ren, Y.L.; Liu, D.G.; Pang, J.X.; Liu, J.; Li, J.; Wang, C.; Cai, J.P. Rapamycin promoted thrombosis and platelet adhesion to endothelial cells by inducing membrane remodeling. BMC Cell Biol. 2014, 15, 7. [Google Scholar]
- Clever, Y.P.; Cremers, B.; Speck, U.; Dietz, U.; Bohm, M.; Scheller, B. Influence of a paclitaxel coated balloon in combination with a bare metal stent on restenosis and endothelial function: Comparison with a drug eluting stent and a bare metal stent. Catheter. Cardiovasc. Interv. 2014, 84, 323–331. [Google Scholar]
- Lehle, K.; Birnbaum, D.E.; Preuner, J.G. Predominant inhibition of interleukin-6 synthesis in patient-specific endothelial cells by mtor inhibitors below a concentration range where cell proliferation is affected and mitotic arrest takes place. Transplant. Proc. 2005, 37, 159–161. [Google Scholar]
- Muldowney, J.A., 3rd.; Stringham, J.R.; Levy, S.E.; Gleaves, L.A.; Eren, M.; Piana, R.N.; Vaughan, D.E. Antiproliferative agents alter vascular plasminogen activator inhibitor-1 expression: A potential prothrombotic mechanism of drug-eluting stents. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 400–406. [Google Scholar]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar]
- Schumacher, B.; van der Pluijm, I.; Moorhouse, M.J.; Kosteas, T.; Robinson, A.R.; Suh, Y.; Breit, T.M.; van Steeg, H.; Niedernhofer, L.J.; van Ijcken, W.; et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 2008, 4, e1000161. [Google Scholar]
- Katewa, S.D.; Kapahi, P. Dietary restriction and aging, 2009. Aging Cell 2010, 9, 105–112. [Google Scholar]
- Speakman, J.R.; Mitchell, S.E. Caloric restriction. Mol. Asp. Med. 2011, 32, 159–221. [Google Scholar]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition 1989, 5, 155–171, discussion 172. [Google Scholar]
- McCay, C.M. Effect of restricted feeding upon aging and chronic diseases in rats and dogs. Am. J. Public Health Nations Health 1947, 37, 521–528. [Google Scholar]
- Mair, W.; Dillin, A. Aging and survival: The genetics of life span extension by dietary restriction. Annu. Rev. Biochem. 2008, 77, 727–754. [Google Scholar]
- Kennedy, B.K.; Steffen, K.K.; Kaeberlein, M. Ruminations on dietary restriction and aging. Cell. Mol. Life Sci. 2007, 64, 1323–1328. [Google Scholar]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar]
- Parker, B.; Noakes, M.; Luscombe, N.; Clifton, P. Effect of a high-protein, high-monounsaturated fat weight loss diet on glycemic control and lipid levels in type 2 diabetes. Diabetes Care 2002, 25, 425–430. [Google Scholar]
- Rees, K.; Dyakova, M.; Ward, K.; Thorogood, M.; Brunner, E. Dietary advice for reducing cardiovascular risk. Cochrane Database Syst. Rev. 2013, 3, CD002128. [Google Scholar]
- Csiszar, A.; Labinskyy, N.; Jimenez, R.; Pinto, J.T.; Ballabh, P.; Losonczy, G.; Pearson, K.J.; de Cabo, R.; Ungvari, Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: Role of circulating factors and SIRT1. Mech. Ageing Dev. 2009, 130, 518–527. [Google Scholar]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar]
- Aissa, A.F.; Gomes, T.D.; Almeida, M.R.; Hernandes, L.C.; Darin, J.D.; Bianchi, M.L.; Antunes, L.M. Methionine concentration in the diet has a tissue-specific effect on chromosomal stability in female mice. Food Chem. Toxicol. 2013, 62, 456–462. [Google Scholar]
- Di, L.J.; Byun, J.S.; Wong, M.M.; Wakano, C.; Taylor, T.; Bilke, S.; Baek, S.; Hunter, K.; Yang, H.; Lee, M.; et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat. Commun. 2013, 4, 1449. [Google Scholar]
- Vera, E.; Bernardes de Jesus, B.; Foronda, M.; Flores, J.M.; Blasco, M.A. Telomerase reverse transcriptase synergizes with calorie restriction to increase health span and extend mouse longevity. PLoS ONE 2013, 8, e53760. [Google Scholar]
- Shammas, M.A. Telomeres, lifestyle, cancer, and aging. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 28–34. [Google Scholar]
- Blagosklonny, M.V. Calorie restriction: Decelerating mtor-driven aging from cells to organisms (including humans). Cell Cycle 2010, 9, 683–688. [Google Scholar]
- Fok, W.C.; Zhang, Y.; Salmon, A.B.; Bhattacharya, A.; Gunda, R.; Jones, D.; Ward, W.; Fisher, K.; Richardson, A.; Pérez, V.I. Short-term treatment with rapamycin and dietary restriction have overlapping and distinctive effects in young mice. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 108–116. [Google Scholar]
- Takimoto, E. Controlling myocyte cgmp: Phosphodiesterase 1 joins the fray. Circ. Res. 2009, 105, 931–933. [Google Scholar]
- Lugnier, C. PDE inhibitors: A new approach to treat metabolic syndrome? Curr. Opin. Pharmacol. 2011, 11, 698–706. [Google Scholar]
- Editorial. Deal watch: Intra-cellular therapies and takeda to develop PDE1 inhibitors for schizophrenia. Nat. Rev. Drug Discov. 2011, 10, 329. [Google Scholar]
- Lukowski, R.; Krieg, T.; Rybalkin, S.D.; Beavo, J.; Hofmann, F. Turning on cGMP-dependent pathways to treat cardiac dysfunctions: Boom, bust, and beyond. Trends Pharmacol. Sci. 2014, 35, 404–413. [Google Scholar]
- Brown, K.E.; Dhaun, N.; Goddard, J.; Webb, D.J. Potential therapeutic role of phosphodiesterase type 5 inhibition in hypertension and chronic kidney disease. Hypertension 2014, 63, 5–11. [Google Scholar]
- Kemeny, V.; Molnar, S.; Andrejkovics, M.; Makai, A.; Csiba, L. Acute and chronic effects of vinpocetine on cerebral hemodynamics and neuropsychological performance in multi-infarct patients. J. Clin. Pharmacol. 2005, 45, 1048–1054. [Google Scholar]
- Cai, Y.; Knight, W.E.; Guo, S.; Li, J.D.; Knight, P.A.; Yan, C. Vinpocetine suppresses pathological vascular remodeling by inhibiting vascular smooth muscle cell proliferation and migration. J. Pharmacol. Exp. Ther. 2012, 343, 479–488. [Google Scholar]
- Zhuang, J.; Peng, W.; Li, H.; Lu, Y.; Wang, K.; Fan, F.; Li, S.; Xu, Y. Inhibitory effects of vinpocetine on the progression of atherosclerosis are mediated by Akt/NF-κB dependent mechanisms in apoE−/− mice. PLoS ONE 2013, 8, e82509. [Google Scholar]
- Myung, S.-K.; Ju, W.; Cho, B.; Oh, S.-W.; Park, S.M.; Koo, B.-K.; Park, B.-J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346, f10. [Google Scholar]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.-H.; Chen, C.-H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter new therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar]
- Bruns, D.R.; Drake, J.C.; Biela, L.M.; Peelor Iii, F.F.; Miller, B.F.; Hamilton, K.L.; Cabello-Verrugio, C. Nrf2 signaling and the slowed aging phenotype: Evidence from long-lived models. Oxid. Med. Cell. Longev. 2015, 2015, 732596. [Google Scholar]
- Ramprasath, T.; Vasudevan, V.; Sasikumar, S.; Syed Mohamed Puhari, S.; Saso, L.; Sadasivam Selvam, G. Regression of oxidative stress by targeting eNOS and Nrf2/ARE signaling: A guided drug target for cardiovascular diseases. Curr. Top. Med. Chem. 2015, 15, 857–871. [Google Scholar]
- Bocci, V.; Valacchi, G. Nrf2 activation as target to implement therapeutic treatments. Front. Chem. 2015, 3, 4. [Google Scholar]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bautista-Niño, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.J.; Roks, A.J.M. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. https://doi.org/10.3390/ijms17050748
Bautista-Niño PK, Portilla-Fernandez E, Vaughan DE, Danser AHJ, Roks AJM. DNA Damage: A Main Determinant of Vascular Aging. International Journal of Molecular Sciences. 2016; 17(5):748. https://doi.org/10.3390/ijms17050748
Chicago/Turabian StyleBautista-Niño, Paula K., Eliana Portilla-Fernandez, Douglas E. Vaughan, A. H. Jan Danser, and Anton J. M. Roks. 2016. "DNA Damage: A Main Determinant of Vascular Aging" International Journal of Molecular Sciences 17, no. 5: 748. https://doi.org/10.3390/ijms17050748
APA StyleBautista-Niño, P. K., Portilla-Fernandez, E., Vaughan, D. E., Danser, A. H. J., & Roks, A. J. M. (2016). DNA Damage: A Main Determinant of Vascular Aging. International Journal of Molecular Sciences, 17(5), 748. https://doi.org/10.3390/ijms17050748